
A robust electrochemiluminescence immunoassay for carcinoembryonic antigen detection based on a microtiter plate as a bridge and Au@Pd nanorods as a peroxidase mimic†
Yong
Zhang
ab,
Xuehui
Pang
b,
Dan
Wu
c,
Hongmin
Ma
c,
Zhaoqing
Yan
b,
Jiatao
Zhang
*a,
Bin
Du
*c and
Qin
Wei
*b
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, PR China. E-mail: zhangjt@bit.edu.cn; Fax: +86-10-68918065; Tel: +86-10-68918065
bSchool of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, PR China. E-mail: sdjndxwq@163.com; Fax: +86-531-82765969; Tel: +86-531-82765730
cKey Laboratory of Chemical Sensing and Analysis in Universities of Shandong, Jinan 250022, PR China. E-mail: dubin61@gmail.com; Fax: +86-531-82767370; Tel: +86-531-82767370
First published on 16th November 2015
Abstract
The common drawbacks of most traditional electrochemiluminescence (ECL) immunoassays are the strict storage conditions for the ECL electrode and the steric hindrance caused by bovine serum albumin and antigen. The strict storage conditions require that the modified electrode must be stored at 4 °C before measurement, which may cause the degradation of protein molecules and low reproducibility as the time goes by. The steric hindrance can hinder electron transfer between the electrode and the electrochemical active substance unable to transmit proteins on the electrode surface. The current study takes a 96-well microtiter plate (MTP) as a bridge for analyte pre-treatment and Au@Pd nanorods as a peroxidase mimic to assemble a simple and robust ECL immunoassay. Advantages of such assay include not only high sensitivity but also robust detection circumstance. We demonstrated the method by detecting carcinoembryonic antigen from human serum and obtained a good detection limit of 3 fg mL−1.
Introduction
Electrochemiluminescence (ECL) as a useful analytical technique has been established in the past decade, and is still gaining momentum.1–3 In ECL assays, the chemiluminescent reaction is electrically initiated. The reactive species which can react with one another to produce light are generated from stable precursors (i.e., the ECL-active label) at the surface of an electrode. Besides it can be precisely controlled, and ECL technology has many distinct advantages over other detection methods, such as simple fabrication and high sensitivity with low background.4–7 Therefore, the ECL immunoassay exhibits great potential in biomedical applications. In general, three types of ECL immunoassays, including label-free, sandwich-type and competitive assays, have been developed to date. The principle of a label-free ECL immunoassay is that the luminescent signal can be decreased by the steric hindrance produced from the antibody–antigen (Ab–Ag) immune-complexation. In the other two types, the ECL signals can be altered by the enzymatic signal generated reaction.8–13 However, one of their common drawbacks is that antibody immobilization on the electrodes can increase the steric hindrance, which can hinder electron transfer between the electrode and the electrochemical active substance.14–16 The main reason to hinder electron transfer by the steric hindrance is due to the fact that most of the protein macromolecules are not conductive. Therefore, the density of the antibodies on the surface cannot be too high to minimize steric hindrance.17 In addition, the other common drawback of all such assays is that the modified electrodes have to be stored at 4 °C before measurement. Otherwise, low reproducibility may be caused as the time goes by, due to the degradation of protein molecules. These two drawbacks limit their applications and development.Compared with horse radish peroxidase (HRP), the peroxidase mimic has robust and stable merits. Specially, noble metal nanoparticles (NPs) as a peroxidase mimic has received widespread attention.18 Due to the advantages, including easy preparation, high homogeneity, and biocompatibility with proteins, Au NPs have been regarded as a versatile template for the immobilization of biomolecules and also play an important role in numerous fields of biomedical applications.19–21 In addition, Pd and Ag NPs with superior electrochemical properties can facilitate electron transfer from the redox center of the protein to the electrode surface. So bimetallic NPs such as Au/Ag and Au/Pd also attracted widespread attention especially in the field of immunoassays, and they often showed better electrochemical performance than their monometallic counterparts.22
Cancer biomarkers, which are some kinds of proteins expressed in high abundance in blood under abnormal physiological conditions, can be used for early diagnosis of diseases.23,24 To rapidly detect such biomarkers for point-of-care cancer diagnostics, simple immunoassays are desirable. The colorimetric immunoassay is a developed method for biomarker detection. Probably due to the vulnerable/limited signal amplification strategies applied, the sensitivity of the colorimetric immunoassay is not always satisfactory.25 Therefore, successful detection of cancer biomarkers, which are usually present in very low concentrations, is not easily achievable. Nevertheless, because it can realize high-throughput detection by means of a microtiter plate (MTP), the colorimetric immunoassay is potential to be applied for clinical detection.26
In this study, we present a novel design of using a 96-well MTP as a bridge and Au@Pd nanorods (NRs) as a peroxidase mimic for biomarker detection. After bio-samples are treated in a 96-well MTP to achieve high throughput under a biological environment, they are detected by the ECL assay to achieve high sensitivity under a robust environment. As a proof-of-principle, carcinoembryonic antigen (CEA) was analysed from human serum, and achieved higher sensitivity, higher throughput and better sensitivity compared with the conventional ECL immunoassay.
Experimental
Materials and reagents
All chemicals were used as-received without further processing. Luminol was purchased from TCI Co. Ltd (Hong Kong, China). K2PdCl4, HAuCl4·4H2O and AgNO3 were obtained from Shanghai Chemical Reagent Co., Ltd (Shanghai, China).Cetyltrimethylammonium bromide (CTAB), ascorbic acid (AA), glutaraldehyde (GA) and graphite powder were provided by Shanghai Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). 3,3′,5,5′-Tetramethylbenzidine (TMB) and Nafion (5%) were obtained from Sigma-Aldrich (Beijing, China). Bovine serum albumin (BSA, 96–99%) was purchased from Sigma (USA) and used as received. CEA and CEA antibody (anti-CEA) were purchased from Wanger Biotechnology Co., Ltd (Beijing, China). All other reagents were of analytical grade. Phosphate buffered saline (PBS, 0.1 mol L−1 containing 0.1 mol L−1 NaCl) was used as an electrolyte for all electrochemistry measurements. High-binding polystyrene 96-well MTP was obtained from Greiner Bio-One (Frickenhausen, Germany, cat#: 655061). Deionised water was prepared by using a Millipore Milli-Q system and used throughout.
Apparatus
Double-step potential (DSP) experiments were performed on a CHI 760D electrochemical workstation (Shanghai, China). The ECL intensity was measured with a MPI-ECL Analyzer (Xi'an Remax Electronic High-Tech Ltd) with the voltage of the photomultiplier tube set at 800 V in the process of detection. Electrochemical impedance spectroscopy (EIS) was obtained from the impedance measurement unit (IM6e, ZAHNER Elektrik, Germany). All electrochemical and ECL experiments were carried out in a conventional three-electrode cell with a modified glassy carbon electrode (GCE, diameter 4 mm) as the working electrode, a Pt wire electrode as the counter electrode and an Ag/AgCl electrode as the reference electrode.The UV-Vis absorption spectra of water colloid were recorded by using a plate reader (DNM-9602, Beijing Perlong Medical Instrument Ltd, China). The absorbance of TMB–H2O2 reaction on MTP was read at 650 nm with a plate reader (DNM-9602, Beijing Perlong Medical Instrument Ltd, China). TEM (JEOL JEM 1200EX working at 100 kV) and high-resolution TEM (HRTEM, FEI Tecnai G2 F20 S-Twin working at 200 kV) were utilized to characterize morphology and interfacial lattice details.
Preparation of Au NRs and Au@Pd NRs
Au NRs were synthesized via a typical seed mediated growth method with some modifications.27 Briefly, Au-seed aqueous solution was firstly prepared by adding 0.6 mL of ice-cold 10 mmol L−1 NaBH4 to 10 mL of solution containing 0.10 mol L−1 CTAB and 0.25 mmol L−1 HAuCl4. After vigorous stirring for 30 s, the resulting brownish yellow solution was undisturbed at 25 °C for 30 min. Then, the growth solution was prepared via successively adding 0.35 g KBr, 1.2 mL of 4 mmol L−1 AgNO3, 25 mL of 1 mmol L−1 HAuCl4 and 0.45 mL of 64 mmol L−1 AA to 0.10 mol L−1 CTAB aqueous solution with continuous stirring during the whole procedure. At last, 0.08 mL Au-seeds was added to the growth solution and undisturbed at 25 °C for 12 h. After the color changed from colorless to dark blue, Au NRs were obtained and centrifuged twice at 6000 rpm for 10 min. Finally, Au NRs were dispersed in 4 mL of CTAB (0.1 mol L−1).The obtained Au NRs were used as seeds for the preparation of Au@Pd NRs, which can be simply prepared via the following steps: 300 μL of 0.1 mol L−1 AA, 50 μL of 10 mol L−1 K2PdCl4, and 300 μL of 0.1 mol L−1 NaOH were successively added to 5 mL Au NR solutions under stirring at 25 °C. After 30 min, the Au@Pd NR solution was centrifuged (6000 rpm for 15 min) followed by consecutive washing three times with deionised water and the resulting precipitates were re-dispersed in 2 mL of PBS (pH 7.4) for a later reaction. This pathway produced Au@Pd NRs with an aspect ratio of ∼3.6 and an average length of 30 nm.
Preparation of Au@Pd/luminol-Ab2
A luminol stock solution (0.01 mol L−1) was prepared firstly by dissolving luminol in 0.1 mol L−1 NaOH. Au@Pd NRs and luminol labeled secondary anti-CEA (Au@Pd/luminol-Ab2) were synthesized via incubation with 0.5 mL of luminol stock solution and 0.5 mL of anti-CEA (10 μg mL−1) and the mixture was shaken overnight on a table concentrator in the dark. Following purification by centrifugation, the product was re-dispersed into 2 mL of PBS (pH 7.4). Then 250 μL of 1% BSA solution was added and incubated at room temperature for 0.5 h to block nonspecific binding and avoid the nonspecific adsorption on Au@Pd NRs. After continued purification by centrifugation and re-dispersion into 2 mL of PBS (pH 7.4), Au@Pd/luminol-Ab2 was obtained and stored in the dark at 4 °C.Preparation of NH2-graphene (NH2-G)
Firstly, the graphite oxide (GO) powders were synthesized according to the reported method with some improvements.28 In a typical preparation, the mixture of graphite powder (0.3 g) and KMnO4 (1.8 g) was added into the mixture solution of H2SO4 and H3PO4 at a molar ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After the reaction mixture was heated to 50 °C with constant stirring for 12 h, the mixture solution was poured onto ice (40 mL) containing 0.3 mL of H2O2 (30%). The product was collected by centrifugation at 9000 rpm for 10 min. The remaining precipitate was washed two times with HCl (0.2 mol L−1), ethyl alcohol and diethyl ether, respectively. Finally, the GO powders were obtained after the product was dried in a vacuum at 35 °C and ground in a quartz mortar.
:1. After the reaction mixture was heated to 50 °C with constant stirring for 12 h, the mixture solution was poured onto ice (40 mL) containing 0.3 mL of H2O2 (30%). The product was collected by centrifugation at 9000 rpm for 10 min. The remaining precipitate was washed two times with HCl (0.2 mol L−1), ethyl alcohol and diethyl ether, respectively. Finally, the GO powders were obtained after the product was dried in a vacuum at 35 °C and ground in a quartz mortar.
Secondly, 200 mg of GO powder was added to ethylene glycol (80 mL) under ultra-sonication for 30 min, followed by the addition of 2 mL of NH3·H2O under constant stirring for several minutes. Then, the dark brown solution was transferred into an autoclave for solvothermal reactions at 180 °C for 10 h. The obtained NH2-G precipitate was washed three times with deionised water, and dried in a vacuum at 50 °C.29
Pre-treatment procedure of the measurements
First, each well of MTP was coated with 50 μL of anti-CEA (Ab1, 100 μg mL−1) in PBS buffer (pH 7.4) for 12 h at 4 °C according to a previous report.26 Then, 200 μL of 1% BSA solution was used as a blocking buffer per well to suppress the nonspecific adsorption to the plates for 2 h at 4 °C. At the end of every step above a washing step of using PBS buffer (pH 7.4) was performed. Before the ECL or colorimetric measurements were operated, 50 μL of different concentrations of CEA standards or real samples were added and incubated for 2 h at 4 °C. Lastly, after being washed with PBS (pH 7.4), 50 μL of Au@Pd/luminol-Ab2 were added and incubated for 2 h at 4 °C.Fabrication and measurement of ECL immunoassay
Before use, GCE was polished using a polishing cloth (CHI Inc.) with small particles (1.0 and 0.05 mm) of Al2O3 slurry, rinsed with water, and then ultrasonicated in ethanol and ultra-pure water. Then 10 μL of 2 mg mL−1 NH2-G was added onto the GCE surface. After being dried, 5 μL of Nafion (5%) was added to form a matrix film in order to prevent NH2-G falling off from the electrode surface. Finally, 3 μL of GA solution (2.5%, v/v) was added onto the matrix film for better antibody adsorption. After being washed, the prepared GCE was ready for a later experiment.ECL measurement was operated as follows: after the MTP was pre-treated according to the above pre-treatment procedure, 10 μL of Au@Pd/luminol-Ab2 was pipetted from the upper solution in the well and modified onto the prepared GCE. With no necessary consideration of biological environment, the modified GCE was dried and used as the working electrode. ECL measurement was performed in an ECL cell containing 10 mL of 0.1 mol L−1 PBS (pH 7.4) solution with 1.5 mmol L−1 H2O2. The generation of ECL response resulted from electrochemical reactions of luminol on the surface of the Au@Pd/luminol-Ab2 in the presence of H2O2. ECL signals were generated and recorded using DSP with the parameters: initial potential 0 V, pulse potential 0.9 V, pulse period 30 s and pulse time 0.1 s.12
Measurement of colorimetric assay
Colorimetric measurement was operated as follows: after the MTP was pre-treated according to the above pre-treatment procedure, the upper solution of the unbound Au@Pd/luminol-Ab2 in the well was washed using PBS buffer (pH 7.4). Then, 100 μL of substrate solution containing 1 mol L−1 H2O2 and 4 mmol L−1 TMB in pH 3.2 sodium acetate buffer solution was added and incubated at 37 °C for 10 min, and the absorbance was read at 652 nm for catalysis comparison. For calibration curve detection, after 10 min for color development, the catalytic reaction was stopped by adding 50 μL of 2 mol L−1 H2SO4 to each well and the absorbance of color products was recorded at 452 nm.Results and discussion
Material characterization
To better understand the size distribution of Au@Pd NRs, transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were used for characterization (Fig. 1). Au NRs with an aspect ratio of ∼3.2 and an average length of 30 nm were firstly synthesized and well dispersed (Fig. 1A). After coating Pd NPs, bone-like Au@Pd NRs with a single-crystal structure (Fig. 1B and C) were formed, based on the preferentially adopting epitaxial growth of Pd on the Au surface.30 In this work, Au@Pd NRs of 30 nm (Fig. 1B) were chosen because this was a balanced and appropriate size for combining antibodies, which could be explained by Rubik's cube theory.31 In brief, because the antibody is about 10–15 nm, if Au@Pd NRs are smaller than the size of antibody (the loading substrate and antibody are not in a comparative size), the antibodies cannot be combined well on the substrate due to the limitation of size. In another aspect, if Au@Pd NRs are much larger, for example, over a hundred nanometers, the substrate is many times larger than the antibody in size which may leave over many nonspecific active sites resulting in the inaccuracy of the results. The corresponding UV-vis extinction spectra of the Au NRs (red line) and Au@Pd NRs (blue line) are illustrated in Fig. 1D. Au NRs showed a longitudinal surface plasma resonance (SPR) peak at 682 nm and a transverse SPR band centred at 510 nm. After the Pd shell was coated, the SPR peak of Au NRs was gradually damped suggesting that Pd atoms were grown on the Au NR surface. This is because the conductivity of Pd atoms at optical frequency is much lower than that of Au atoms.30 NH2-G was synthesized according to our previous work29 and well dispersed in solution (see Fig. S1†). | ||
| Fig. 1 (A) TEM image of Au NRs. (B) TEM image of Au@Pd NRs, local zoom (inset). (C) HRTEM image of Au@Pd NRs, the FFT pattern (inset). (D) UV/Vis absorbance of Au NRs (red) and Au@Pd NRs (blue). | ||
Immunoassay fabrication characterization
Scheme 1 illustrates an overview of the fabrication and measurement of the immunoassay. First, Au@Pd NRs and luminol coefficient labelled anti-CEA were synthesized (Scheme 1a). In the meantime, NH2-G was prepared and modified onto GCE (Scheme 1b) for later use. Then, as shown in Scheme 1c, a high-binding polystyrene 96-well MTP was coated with Ab1. After blocking the active sites with BSA, solutions containing CEA standards or real samples with various concentrations were added into the MTP wells. Following that, 50 μL of Au@Pd/luminol-Ab2 with the same concentrations was added. After Ab–Ag immunoreactions, 10 μL unbound Au@Pd/luminol-Ab2 from the upper solution in the well was measured by pipetting onto the GCE prepared previously. Following quick drying at room temperature, the modified GCE was measured by the ECL assay via a luminol–H2O2 system. The ECL signal corresponding to the concentration of unbound Au@Pd/luminol-Ab2 is inversely proportional to the concentration of CEA. For proving the sum of combined and unbound Au@Pd/luminol-Ab2 as consistent, after unbound Au@Pd/luminol-Ab2 was washed off the wells, 100 μL of the substrate solution containing TMB and H2O2 was added. Because of the catalytic effect of Au@Pd NRs on the colorimetric system of TMB–H2O2, different colors in the wells containing various concentrations of Au@Pd/luminol-Ab2 bound to the bottom of the well were visualized. The corresponding absorbance intensity is in direct proportion to the concentration of CEA.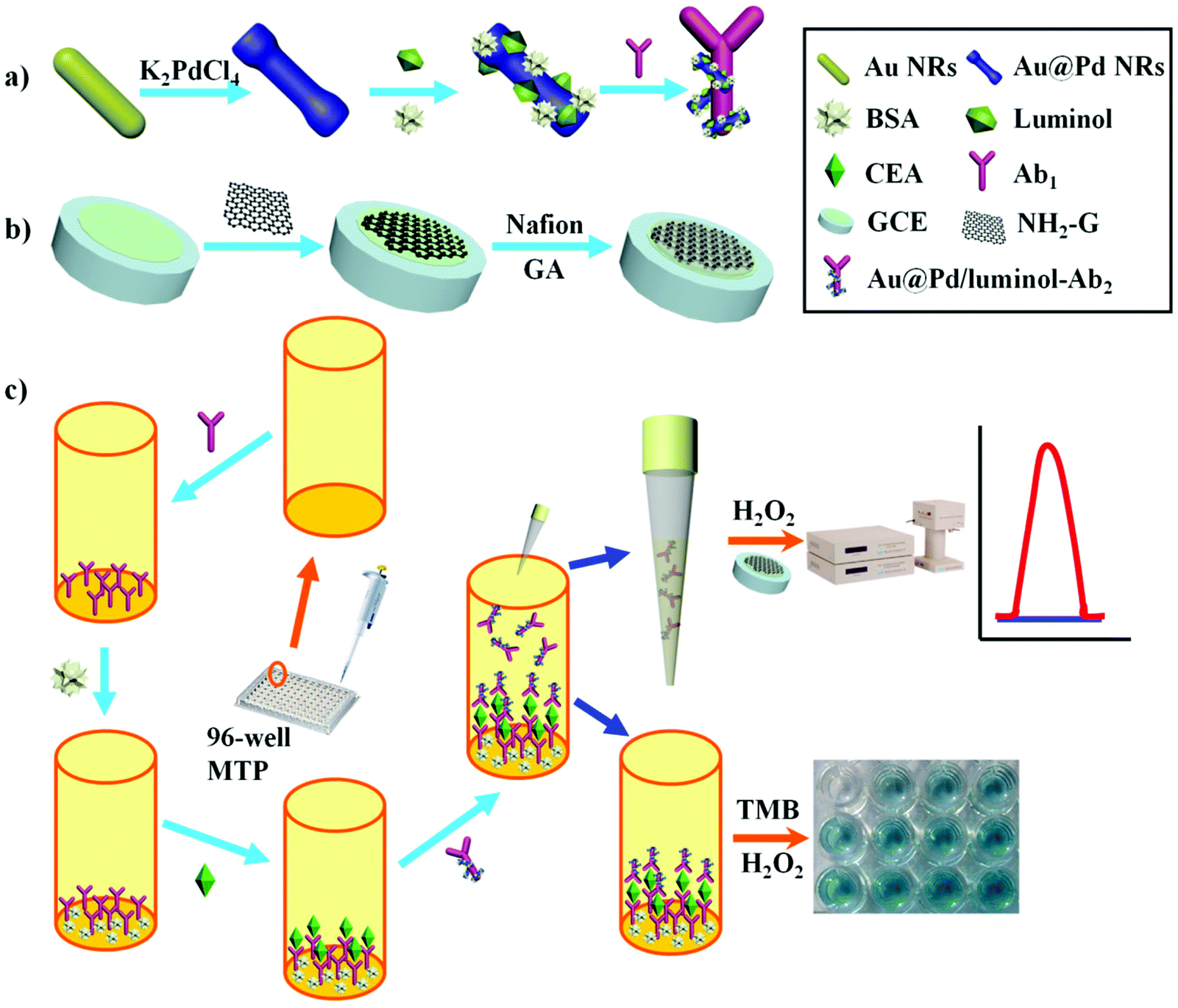 | ||
| Scheme 1 Schematic illustration of the fabrication and detection procedures of ECL immunoassay by means of 96-well MTP: (a) preparation of Au@Pd NRs and Au@Pd/luminol-Ab2. (b) Preparation of NH2-G. (c) Fabrication and measurement of ECL immunoassay and colorimetric assay. | ||
The assembly process of the modified GCE used in the ECL assay was also characterized by EIS. EIS is an effective method to monitor the assembly of the immunoassay and probes the features of the modified electrode surface.32 The EIS profile of bare GCE was almost a straight line (curve a in Fig. 2A), characteristic of the diffusion limiting step of the electrochemical process. Because of its advantages of good conductivity and high surface areas, NH2-G was used as a substrate modified on GCE for ECL detection. As expected, when the NH2-G unites in a matrix with nafion and GA modified electrode, the EIS was similar to that of the bare GCE (curve b in Fig. 2A). One of the possible reasons is the matrix immobilized on the electrode is similar to a conducting wire, which makes it easier for electron transfer.33 The electron transfer resistance increased dramatically after Au@Pd/luminol-Ab2 (curve c in Fig. 2A) was dropped on the modified GCE and increased successively with the increase of Au@Pd/luminol-Ab2 concentration. This is because of the steric hindrance effect by the non-conductive biological molecules.34 It can also be the evidence of good adsorption of Au@Pd/luminol-Ab2 on the modified GCE.
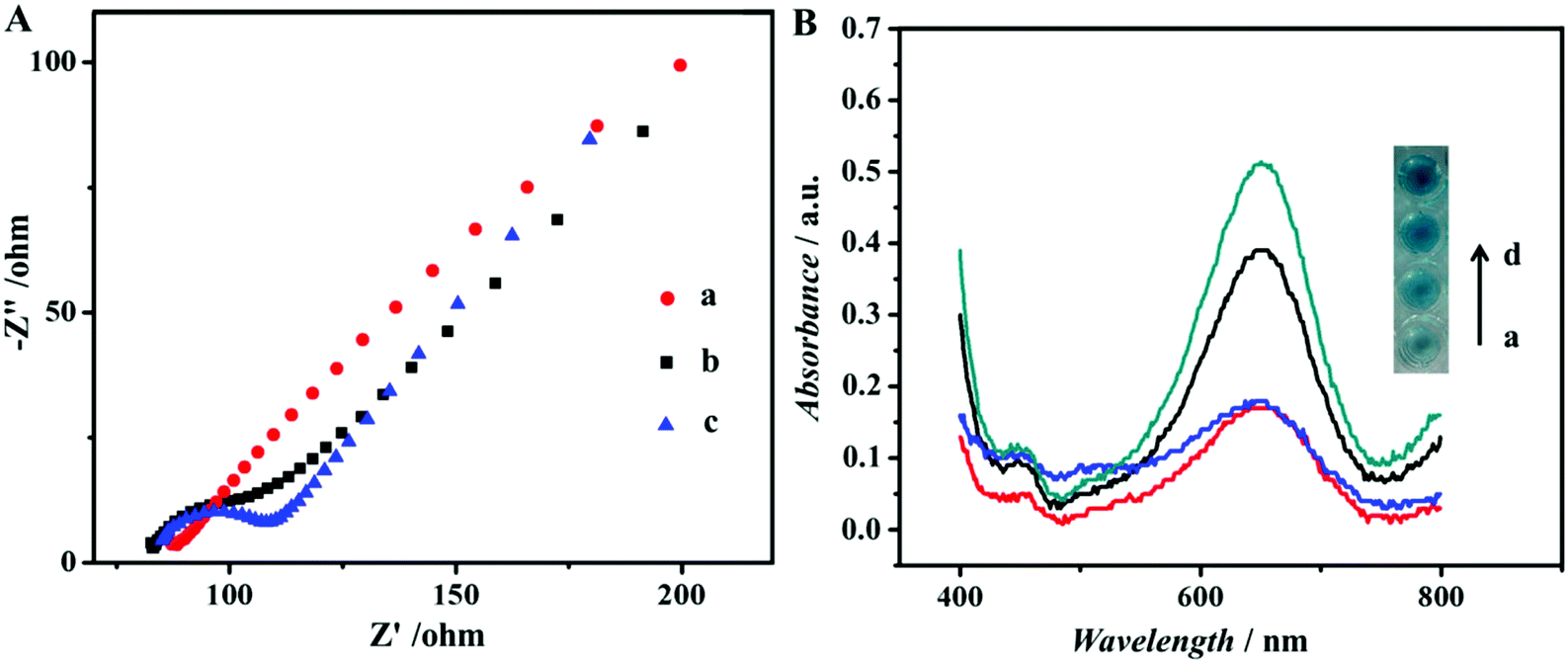 | ||
| Fig. 2 (A) Nyquist plots of the EIS for immobilized step recorded from 0.1 to 105 Hz of (a) bare GCE, (b) NH2-G/GA/nafion/GCE, (c) Au@Pd/luminol-Ab2/GA/nafion/NH2-G/GCE in 1 mmol mL−1 Fe(CN)63−/4− solution. (B) A comparison of colorimetric assay after H2O2 and TMB were added and 10 min for color development: (a) Ab1-CEA, (b) Ab1-CEA/luminol, (c) Ab1-CEA-Au/luminol-Ab2, (d) Ab1-CEA-Au@Pd/luminol-Ab2. | ||
Mechanism and analytical performance of the ECL immunoassay
To better demonstrate that Au@Pd NRs as a peroxidase mimic played an essential role in our strategy, the peroxidase-like catalytic activity of Au@Pd/luminol-Ab2 was studied in the catalysis of TMB oxidation with H2O2, respectively. When Au@Pd/luminol-Ab2 was added (curve d in Fig. 2B), the absorbance intensity was greatly increased after 10 min at 652 nm, a characteristic peak of oxidized TMB. Compared to the minimal change of absorbance intensity of Ab1 and CEA immobilized in the MTP wells (curve a in Fig. 2B), Au@Pd/luminol-Ab2 showed an intrinsic peroxidase-like activity. To exclude the possible catalytic activity of luminol, the luminol for the catalytic TMB–H2O2 reaction was put alone, and no obvious catalytic reaction was observed in this system (curve b in Fig. 2B) as expected, indicating that Au@Pd NRs played the peroxidase mimic role and were crucial for the formation of Au@Pd/luminol-Ab2. In addition, the catalytic activity of Au NRs was compared with Au@Pd NRs. Although under the same conditions Au NRs can catalyse the TMB–H2O2 reaction (curve c in Fig. 2B), the absorbance of the Au NR catalytic system has obviously a lower intensity than the Au@Pd NR catalytic system at 652 nm, probably due to the fact that the catalytic activity of Pd is higher than Au.35To further prove the feasibility of the ECL immunoassay, ECL signals were investigated before and after Au@Pd/luminol-Ab2 modified the prepared GCE. No ECL signal can be seen when the GCE was modified with NH2-G only (curve a in Fig. S2†), whereas a strong ECL signal was observed when the Au@Pd/luminol-Ab2 was introduced to the prepared GCE via the stable cross-linked action of GA to amine groups between NH2-G and antibody (curve b in Fig. S2†). The dramatic increase of the ECL signal is due to the emission of the luminol–H2O2 ECL system catalysed by Au@Pd NRs. The Au@Pd NRs is concentrated close to the electrode surface, so they can be shaped and accurately positioned for an optical measurement system with maximum sensitivity.3 This phenomenon also indicated that as illustrated in Fig. 3A, the ECL intensity decreased gradually with increasing antigen concentrations. Taking into account direct electron transfer and electrogenerated chemiluminescence without the steric hindrance produced by other proteins (i.e. BSA and antigen) on the electrode surface, our immunoassay achieved a promising analytical performance. The standard calibration curve of CEA detection is shown in the inset of Fig. 3A suggesting that the ECL intensity decreased linearly in the range of 0.01 pg mL−1 to 100 ng mL−1 CEA. The linear equation is I = 5893 − 894lgCCEA (unit of CCEA is ng mL−1) with a correlation coefficient of 0.9934. The detection limit is 3 fg mL−1. According to the linear equation, a trace amount of CEA concentration quantitatively detected by our ECL immunoassay, is comparable to other published methods36–39 as shown in Table 1. Our detection limit decreased two or more orders of magnitude.
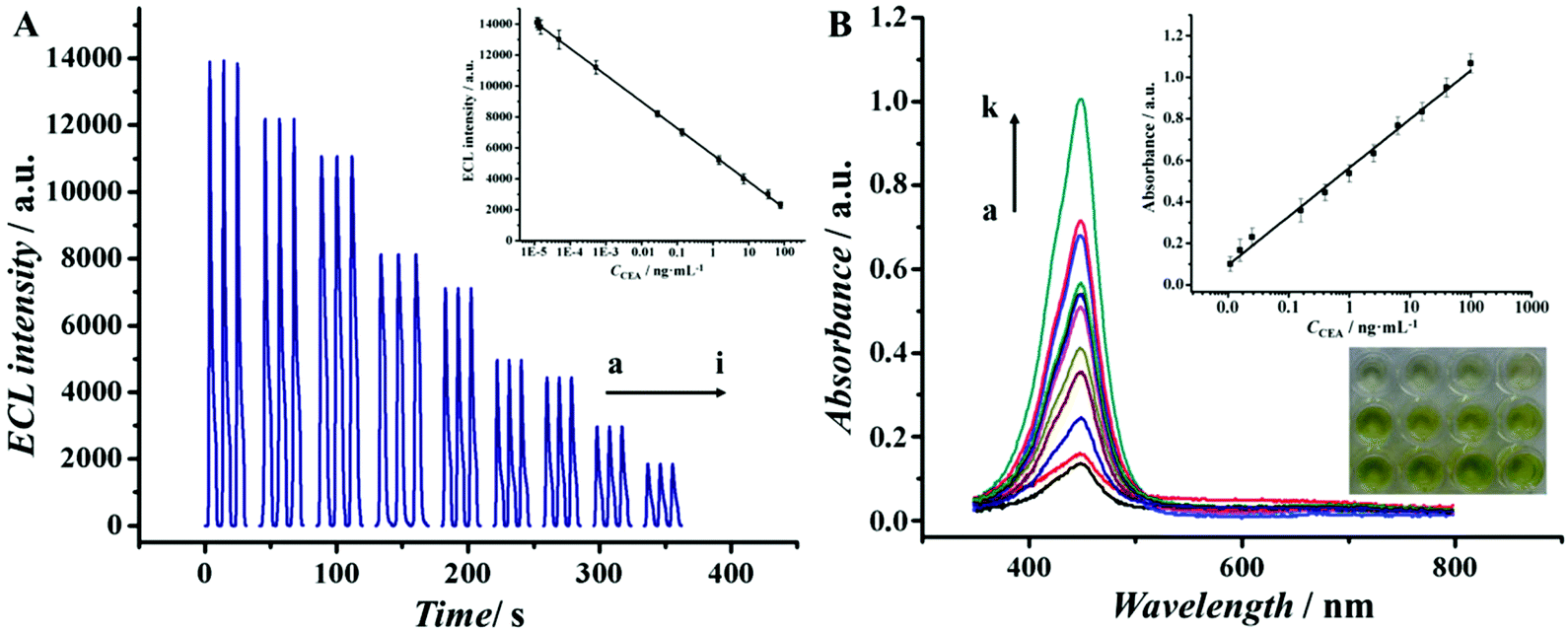 | ||
| Fig. 3 (A) ECL response for different concentrations of CEA: 0, 1 × 10−5, 1 × 10−4, 1 × 10−3, 0.01, 0.1, 1, 10, 100 ng mL−1 (curve a to i). (B) Absorbance for different concentrations of CEA: 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 25, 50, 75, 100 ng mL−1 (curve a to k) after 10 min for color development, and visual detection (inset). Insets of (A) and (B) are calibration curves of the immunoassay for different concentrations of CEA. | ||
Analytical performance of the colorimetric assay
The colorimetric assay in our work can be used to further confirm the feasibility of our strategy. Because the unbound Au@Pd/luminol-Ab2 from the upper solution in the well can be sensitively detected by the ECL assay and the concentration of Au@Pd/luminol-Ab2 is inversely proportional to the concentration of CEA, the combined Au@Pd/luminol-Ab2 in the well via Ab–Ag immunoreactions should be in direct proportion to CEA. Therefore, after washing away the unbound Au@Pd/luminol-Ab2 from the MTP wells, the bound Au@Pd/luminol-Ab2 can be used as signal detection probes to catalyse the TMB oxidation and produce colorimetric signals. Following the same procedure as the conventional ELISA, H2SO4 was used to stop the catalytic reaction where the oxidized TMB was further oxidized to a yellow diimine with the maximum absorption wavelength of 452 nm. So the absorbance intensity in the UV-vis spectra at 452 nm was recorded to quantify the CEA. It can be observed in Fig. 3B that the absorbance at 452 nm increased while CEA concentration increased. Meanwhile, there was a visually observable color change correlated with CEA concentrations. The inset of Fig. 3B showed that the absorption intensity was linear to the concentrations of CEA in the linear range from 0.01 ng mL−1 to 100 ng mL−1 with a detection limit of 3 pg mL−1. Compared with the ECL assay in our work, the colorimetric assay has a narrower linear range and higher detection limit. One possible reason is that the conjugation of Au@Pd NRs with protein molecules caused the decrease of Au@Pd NR catalytic efficiency.Detection condition optimization
The above described optimal conditions were investigated and illustrated in Fig. 4A–E. Firstly, pH values of both ECL assay and colorimetric assay were optimized (Fig. 4A). When the pH values of measurement solution were 7.4 for the ECL assay and 9.2 for the colorimetric assay, these two assays displayed optimal signal intensity. Then, the concentration of NH2-G in our study is an important factor that would affect the performance of the ECL assay, because it is used as a substrate for Au@Pd/luminol-Ab2 loading. As shown in Fig. 4B, when concentrations of NH2-G changed from 1.0 to 2.0 mg mL−1, the ECL intensity increased gradually and reached the maximum at 2.0 mg mL−1. However, with further increasing the concentrations of NH2-G from 2.0 to 3.0 mg mL−1, the ECL intensity decreased slightly. One of the possible reasons is that excessive NH2-G may be stacked on each other.27 Therefore, 2 mg mL−1 was chosen as the best concentration of NH2-G for subsequent experiments. To obtain the optimal concentration of luminol, luminol solutions with different concentrations were used for preparing Au@Pd/luminol-Ab2 and measured by the ECL assay. As illustrated in Fig. 4C, when 0.010 mol L−1 luminol was used, the ECL intensity achieved the maximum. So 0.010 mol L−1 luminol was chosen as the optimal condition. The effect of developing time for the operation circumstance in the colorimetric assay was optimal as 20 min (Fig. 4D) because of the catalytic properties of Au@Pd NRs. Finally, the optimal temperature for operating the colorimetric assay was investigated. Au@Pd NRs were used as a peroxidase mimic and no consideration of enzyme inactivation was needed, as the colorimetric assay was almost not influenced by temperature (Fig. 4F).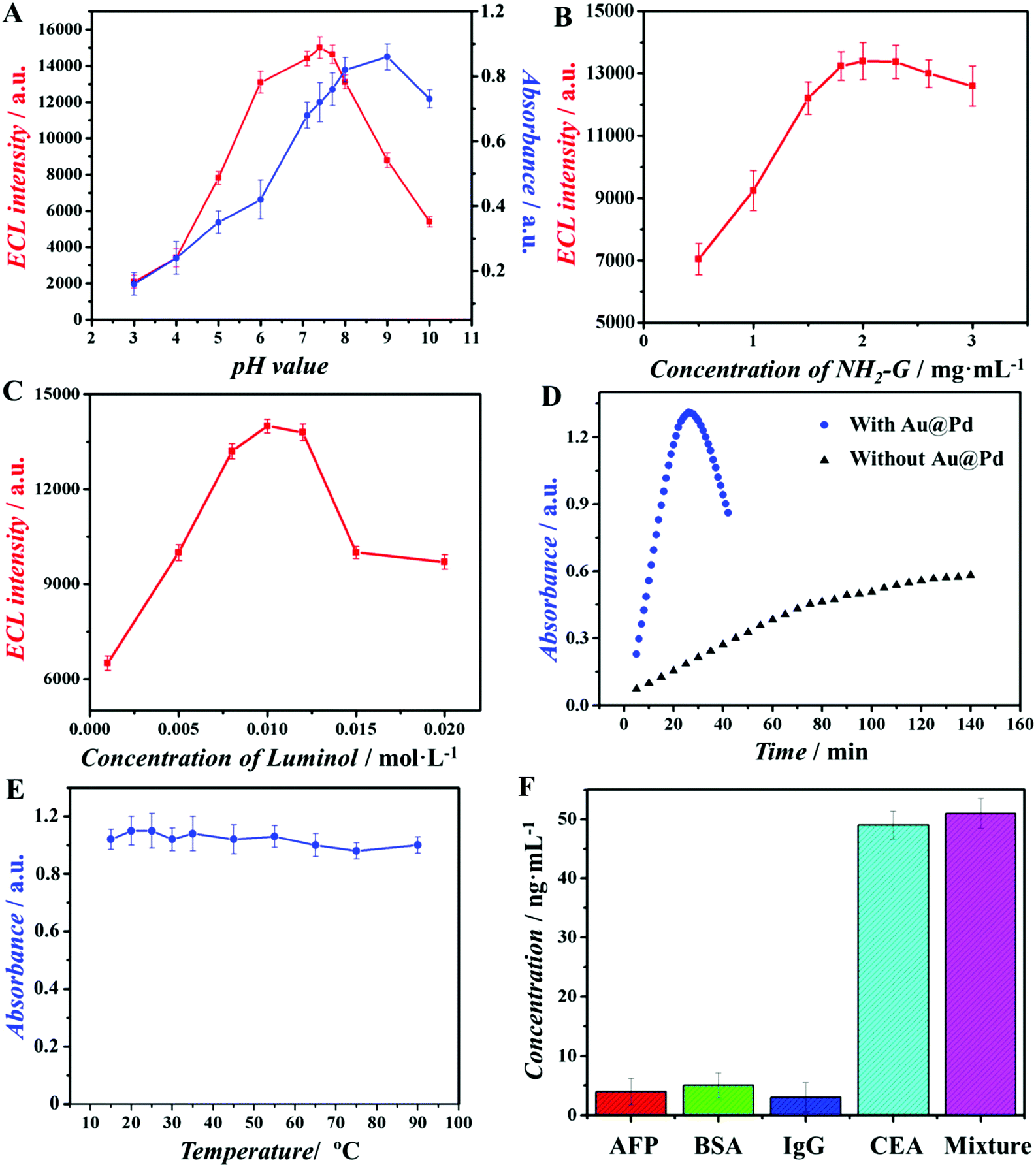 | ||
| Fig. 4 Optimization of the experimental parameters: (A) pH value on the response of the ECL (red) and colorimetric (blue) assay; (B) the amount of NH2-G on the response of the ECL assay; (C) the amount of luminol on the response of the ECL assay; (D) time of color development in the colorimetric assay; (E) temperature of color development in the colorimetric assay. (F) A comparison of ECL assay for detecting CEA with those of different interfering species: 50 ng mL−1 AFP, 50 ng mL−1 BSA, 50 ng mL−1 IgG, 50 ng mL−1 CEA and mixture (50 ng mL−1 CEA, 50 ng mL−1 AFP, 50 ng mL−1 BSA and 50 ng mL−1 IgG). | ||
Specificity, reproducibility and stability of the immunoassay
Since the antibody lifetime is not an issue, our ECL assay demonstrated impressive stability and reproducibility. In this study, after the modified GCE was stored under room temperature for four weeks, the ECL response decreased only ∼1.5% of its initial value. Such good stability may be attributed to three reasons. The first is that the modified GCE need not consider the biological activity loss of proteins; the second is that Au@Pd NRs as a peroxidase mimic has robust and stable merits; the last is the large specific surface area and good electron transfer ability of NH2-G modified on the electrode surface. The relative standard derivation (RSD) of the replicate measurements of CEA at 10 ng mL−1 within a day (intra-day precision, n = 5) was 0.5%; the RSD over 7 days (inter-day precision, n = 5) was 1.2%, which showed good reproducibility.40To characterize the specificity of the immunoassay, various interfering species, including AFP, BSA and IgG, were used in the MTP wells for immunoreactions via the ECL assay. Results indicated that these proteins showed negligible interference in the determination of CEA (Fig. 4F), indicating good selectivity of our immunoassay. This could be explained by the fact that Au@Pd NR is a kind of noble metal nanomaterial, which may prevent the binding affinity loss of biomolecules. Therefore, the immunoassay in this study can be used for the detection of CEA in serum samples.
Application in analysis of samples
To evaluate the feasibility of our immunoassay for clinical diagnosis, the concentration of CEA in human plasma was analysed. The human plasma samples were diluted 10 times with 0.1 mol L−1 PBS buffer (pH 7.4) before analysis. Then, CEA was spiked in with different concentrations (0.1, 0.5, 1.0, 10.0 and 100.0 ng mL−1), and analysed by the ECL assay. Results showed that the recovery was in the range of 100–112% and RSD was in the range of 2.5–3.5% (Table 2). These results indicated that the proposed ECL immunoassay could be suitable for the clinical determination of CEA levels in human plasma.| Initial human plasma (ng mL−1) | The addition content (ng mL−1) | The detection content (ng mL−1) | RSD/%, n = 5 | Recovery/% |
|---|---|---|---|---|
| 0.3 | 0.1 | 0.150 | 3.5 | 112 |
| 0.3 | 0.5 | 0.540 | 3.2 | 102 |
| 0.3 | 1.0 | 1.13 | 3.3 | 110 |
| 3.5 | 10.0 | 10.43 | 2.8 | 101 |
| 12.1 | 100.0 | 101.4 | 2.5 | 100 |
Conclusions
In summary, we provide a novel strategy for preparing a robust ECL immunoassay by means of MTP and Au@Pd NRs. Due to the good catalytic properties of Au@Pd NRs and the direct electron transfer for ECL without the steric hindrance produced by BSA and antigen compared with the traditional ECL immunoassay on the electrode surface, our ECL immunoassay has three advantages over conventional methods for CEA detection: low detection limit, and robust conditions for measurement and high-throughput. Importantly, our strategy opened a new window for the application of nanomaterials in the immunoassay field and can be broadly applied to many other similar technologies, such as electrochemical immunoassay, photoelectrochemical immunoassay, etc.Acknowledgements
This study was supported by the Natural Science Foundation of China (No. 21375047, 21377046), the Natural Science Foundation of Shandong Province (No. ZR2013BL003) and the Natural Science Foundation of UJN (No. XKY1305). QW thanks the Special Foundation for Taishan Scholar Professorship of Shandong Province and UJN (No. ts20130937).References
- Y. Dong, T. Gao, Y. Zhou and J. Zhu, Anal. Chem., 2014, 86, 11373 CrossRef CAS PubMed.
- A. W. Knight, TrAC, Trends Anal. Chem., 1999, 18, 47 CrossRef CAS.
- X. Yin, S. Dong and E. Wang, TrAC, Trends Anal. Chem., 2004, 23, 432 CrossRef CAS.
- S. Deng, J. Lei, Y. Liu, Y. Huang and H. Ju, Chem. Commun., 2013, 49, 2106 RSC.
- A. Devadoss, C. Dickinson, T. E. Keyes and R. J. Forster, Anal. Chem., 2011, 83, 2383 CrossRef CAS PubMed.
- X. Jiang, Y. Chai, H. Wang and R. Yuan, Biosens. Bioelectron., 2014, 54, 20 CrossRef CAS PubMed.
- Y. Zhang, F. Lu, Z. Yan, D. Wu, H. Ma, B. Du and Q. Wei, Microchim. Acta, 2015, 182, 1421 CrossRef CAS.
- H. Dai, Y. Wang, X. Wu, L. Zhang and G. Chen, Biosens. Bioelectron., 2009, 24, 1230 CrossRef CAS PubMed.
- Y. Dong, W. Tian, S. Ren, R. Dai, Y. Chi and G. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 1646 CAS.
- T. Huang, Q. Meng and G. Jie, Biosens. Bioelectron., 2015, 66, 84 CrossRef CAS PubMed.
- C. Li, J. Lin, Y. Guo and S. Zhang, Chem. Commun., 2011, 47, 4442 RSC.
- D. Tian, C. Duan, W. Wang and H. Cui, Biosens. Bioelectron., 2010, 25, 2290 CrossRef CAS PubMed.
- H. Xia, L. Li, Z. Yin, X. Hou and J. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 696 CAS.
- W. Zhao, J. Xu and H. Chen, Chem. Soc. Rev., 2015, 44, 729 RSC.
- N. J. Ronkainen, H. B. Halsall and W. R. Heineman, Chem. Soc. Rev., 2010, 39, 1747 RSC.
- F. Li, Y. Yu, H. Cui, D. Yang and Z. Bian, Analyst, 2013, 138, 1844 RSC.
- B. Lu, M. R. Smyth and R. O'Kennedy, Analyst, 1996, 121, 29 RSC.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060 RSC.
- Y. Yan, Q. Liu, K. Wang, L. Jiang, X. Yang, J. Qian, X. Dong and B. Qiu, Analyst, 2013, 138, 7101 RSC.
- P. Si, Y. Huang, T. Wang and J. Ma, RSC Adv., 2013, 3, 3487 RSC.
- J. Wang, Microchim. Acta, 2012, 177, 245 CrossRef CAS.
- H. Zhang and N. Toshima, Catal. Sci. Technol., 2013, 3, 268 CAS.
- G. Fan, X. Ren, C. Zhu, J. Zhang and J. Zhu, Biosens. Bioelectron., 2014, 59, 45 CrossRef CAS PubMed.
- J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122 RSC.
- J. Zeng, S. Fan, C. Zhao, Q. Wang, T. Zhou, X. Chen, Z. Yan, Y. Li, W. Xing and X. Wang, Chem. Commun., 2014, 50, 8121 RSC.
- Z. Gao, L. Hou, M. Xu and D. Tang, Sci. Rep., 2014, 4, 1 Search PubMed.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414 CrossRef CAS PubMed.
- X. Zhou, X. Huang, X. Qi, S. Wu, C. Xue, F. Y. C. Boey, Q. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842 CAS.
- J. Li, X. Li, Y. Zhang, R. Li, D. Wu, B. Du, Y. Zhang, H. Ma and Q. Wei, RSC Adv., 2015, 5, 5432 RSC.
- N. Li, Y. Wang, Y. Li, W. Cao, H. Ma, D. Wu, B. Du and Q. Wei, Sens. Actuators, B, 2014, 202, 67 CrossRef CAS.
- X. Ren, D. Wu, Y. Wang, Y. Zhang, D. Fan, X. Pang, Y. Li, B. Du and Q. Wei, Biosens. Bioelectron., 2015, 72, 156 CrossRef CAS PubMed.
- X. Wei, C. Xiao, K. Wang and Y. Tu, J. Electroanal. Chem., 2013, 702, 37 CrossRef CAS.
- G. Jie, L. Wang, J. Yuan and S. Zhang, Anal. Chem., 2011, 83, 3873 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, F. Zheng, R. Ren and S. Zhang, Chem. Commun., 2014, 51, 907 RSC.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044 CrossRef CAS PubMed.
- C. Gao, M. Su, Y. Wang, S. Ge and J. Yu, RSC Adv., 2015, 5, 28324 RSC.
- X. Li, R. Wang and X. Zhang, Microchim. Acta, 2011, 172, 285 CrossRef CAS.
- J. Ji, L. He, Y. Shen, P. Hu, X. Li, L. Jiang, J. Zhang, L. Li and J. Zhu, Anal. Chem., 2014, 86, 3284 CrossRef CAS PubMed.
- D. Wang, Y. Li, Z. Lin, B. Qiu and L. Guo, Anal. Chem., 2015, 87, 5966 CrossRef CAS PubMed.
- Z. Sun, L. Deng, H. Gan, R. Shen, M. Yang and Y. Zhang, Biosens. Bioelectron., 2013, 39, 215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an02053k |
| This journal is © The Royal Society of Chemistry 2016 |