
Mass spectrometry of modified RNAs: recent developments
Collin
Wetzel
and
Patrick A.
Limbach
*
Rieveschl Laboratories for Mass Spectrometry, Department of Chemistry, PO Box 210172. and University of Cincinnati, Cincinnati, OH 45221-0172, USA. E-mail: Pat.Limbach@uc.edu; Fax: +1 (513) 556-9239; Tel: +1 (513) 556-1871
First published on 21st October 2015
Abstract
A common feature of ribonucleic acids (RNAs) is that they can undergo a variety of chemical modifications. As nearly all of these chemical modifications result in an increase in the mass of the canonical nucleoside, mass spectrometry has long been a powerful approach for identifying and characterizing modified RNAs. Over the past several years, significant advances have been made in method development and software for interpreting tandem mass spectra resulting in approaches that can yield qualitative and quantitative information on RNA modifications, often at the level of sequence specificity. We discuss these advances along with instrumentation developments that have increased our ability to extract such information from relatively complex biological samples. With the increasing interest in how these modifications impact the epitranscriptome, mass spectrometry will continue to play an important role in bioanalytical investigations revolving around RNA.
1. Introduction
Ribonucleic acids (RNA) serve crucial biological roles at the heart of the Central Dogma of molecular biology, where deoxyribonucleic acid (DNA) is transcribed into messenger RNA (mRNA) that is finally translated into proteins. RNAs that do not code for a protein are referred to as non-coding RNAs (ncRNA). A large number of ncRNAs have been discovered and studied including ribosomal (rRNA), transfer (tRNA), long non-coding (lncRNA), small nuclear (snRNA), small nucleolar (snoRNAs), small interfering (siRNAs) and micro (miRNA).A common feature of nearly all these types of RNAs is that they can undergo a variety of chemical modifications that occur posttranscriptionally.1,2 These RNA modifications are enzymatically introduced to either the ribose or nucleobase of nucleosides after RNA has been transcribed from the genomic DNA.3–5 Such modifications are extremely diverse, with over 100 now characterized and catalogued.6,7Fig. 1 contains structures for several posttranscriptionally modified nucleosides that illustrate the diversity of chemical modifications possible. Such modifications can alter canonical and non-canonical RNA–RNA interactions resulting in structures and function beyond those found for the canonical nucleosides adenosine, cytidine, uridine and guanosine.5,8 The types and levels of modification vary significantly among these various ncRNAs and mRNAs. While most analyses of modified RNAs have focused on the more heavily modified tRNAs and rRNAs,9–13 all RNAs seem to contain at least some chemical modifications and require methods for their analysis. More recently, it has been found that at least some of these modifications are regulated and reversible, leading to the concept of an epitranscriptome, whose modification status can influence gene expression in a manner similar to what is already known about the epigenome.14,15
 | ||
| Fig. 1 Selected examples of the variety of chemical modifications found in naturally occurring nucleosides. | ||
2. Mass spectrometric analysis of modified nucleosides
There are a significant number of analytical methods that have been used to detect and identify chemical modifications in RNA. Such methods include reverse transcriptase polymerase chain reaction (RT-PCR), thin layer chromatography (TLC), electrophoresis, microarrays, next-generation sequencing and mass spectrometry.15–23 As shown over 20 years ago by McCloskey,24,25 the primary utility of using mass spectrometry for such measurements is that this method provides a direct readout on any chemical modifications that change the mass of the canonical nucleoside. Remarkably, with the exception of pseudouridine (ψ) modifications, all other described modified nucleosides result in this desired mass shift that enables direct MS detection.2.1. Sample prep for MS analysis
Modified RNAs are analyzed using matrix-assisted laser desorption/ionization (MALDI) or electrospray ionization (ESI) sources due to the well-documented advantages of these ionization methods. Regardless of the ionization method used, sample preparation remains a key step for successful mass spectrometric analysis of modified oligoribonucleotides and RNAs.A key concern with ionization of oligoribonucleotides and RNAs is cation adduction, which can lead to charge neutralization and loss of ion signal. Cations are adducted because oligonucleotides possess a negatively charged phosphodiester backbone at neutral pH. In solution, Coulombic repulsion from adjacent charges is reduced by the presence of solvent. However, once solvent is removed, neutralization of the backbone charges occurs through cation adduction. Cation adducts, except H+ or NH4+, are undesirable as they reduce the sensitivity by spreading the ion species in multiple mass values, cause peak broadening and result in possible spectral interferences making data more difficult to interpret. Thus, a significant and general key to efficiently ionizing oligonucleotides is the need to reduce or eliminate the presence of cation adducts from the sample or solution prior to analysis.26–28
One area of concern is the effect of sample preparation on modification structure.29,30 A recent example is the work from the Suzuki lab demonstrating that the modified nucleoside N6-threonylcarbamoyladenosine (t6A), which is commonly found in tRNAs, is actually present in vivo as the cyclic form (ct6A) and that sample preparation often leads to hydrolysis and ring opening prior to mass spectrometry detection.31 In addition, it has previously been assumed that modified nucleosides are polar, with few exceptions. However, it was recently found that geranylated RNA, which is lipophilic, can be detected by mass spectrometry provided appropriate sample analysis steps are used.32 These examples illustrate the care that is necessary when preparing and analyzing biological samples by mass spectrometry as an understanding of the types and chemistries of modified RNAs will become ever more important in the future as technology enables more and more sensitive characterization of such samples.
After appropriate sample preparation, modified RNAs can be analyzed at three different regimes: the monomer (i.e., nucleotide/nucleoside) level, as smaller oligonucleotides (e.g., endonuclease digestion products in a bottom-up approach) or as intact RNAs (via a top-down approach). Recent examples of analytical methods for each of these regimes will be discussed below.
2.2. Modified nucleosides
One of the easiest measurements to conduct on modified RNAs is to enzymatically digest intact RNAs into constituent ribonucleosides, which are amenable to separation and analysis by liquid chromatography tandem mass spectrometry (LC-MS/MS). The key recent development in this regime is the focus on quantitative measurements at both high precision and accuracy. Dedon, Begley and coworkers have created a highly effective targeted LC-MS/MS protocol for the quantitative measurement of modified ribonucleosides.28,33 They have demonstrated the utility of this protocol for analyzing changes in posttranscriptional modifications in Saccharomyces cerevisiae that result from environmental stress.34,35While the Dedon approach typically uses only a single internal standard for quantitative analysis, other researchers have explored options for including a greater number of internal standards. Carell and co-workers have reported the synthesis of multiple modified ribonucleosides, all of which can incorporate one or more isotopic labels.36,37 An alternative strategy was established by Helm and co-workers, wherein standards for modified ribonucleosides are generated by sample culturing in isotopically labeled medium.38 The added advantage of this latter approach is that the diversity of standards is only limited by the organism's inherent modification machinery.
Taking advantage of new gas-phase separation technology, Fabris and co-workers have eliminated upfront nucleoside separation by LC and instead used ion mobility coupled to mass spectrometry for the direct analysis of modified nucleosides.39 The potential power of this rapid approach for direct transcriptome-wide detection of modified nucleosides was recently reported by Rose,40 and one can anticipate future developments that allow structural isomers to be more clearly differentiated in these non-chromatographic approaches.
2.3. Bottom-up modification mapping
Bottom-up RNA modification mapping uses one or more enzymes, typically an endoribonuclease (RNase), which can digest a larger RNA into smaller oligoribonucleotides that can be conveniently analyzed by MALDI- or LC-MS/MS.41,42 These analyses take advantage of the advent of genomic sequencing, where ncRNA species can be inferred by analyzing an organism's genome using motif recognizing software.43 In this manner, the genomic sequence of the RNA of interest is already known, thus the experimental goals are to simply place modified nucleosides into their appropriate sequence location.We have recently developed several different analytical protocols that are based on bottom-up RNA modification mapping. A multiple enzyme “digest and go” strategy, outlined in Fig. 2, was used to create a modification map for the complete set of tRNAs isolated from the bacterial organism Lactococcus lactis.44 The key to this approach is the use of multiple endonucleases to increase mapping coverage as well as high quality MS/MS data. An alternative strategy is to use one or more RNAs of known modification status as a reference for comparison against RNAs of unknown modification status.45,46 This CARD (comparative analysis of RNA digests) approach is most suitable when comparing wild-type against mutant strains or healthy versus diseased samples.
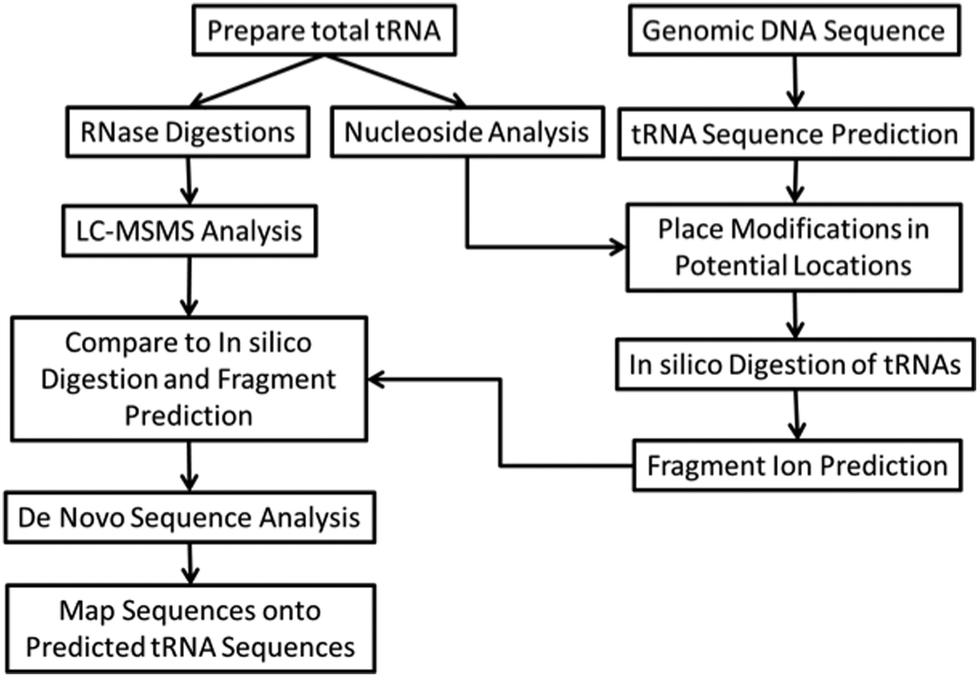 | ||
| Fig. 2 An experimental scheme for mapping modifications on tRNA sequences from total tRNA by a bottom-up strategy is shown. This scheme takes advantage of pre-existing tRNA gene sequences and matches modified nucleosides with MS/MS-based sequence information to map modifications onto the known tRNA sequences. | ||
The bottom-up approach has also been enhanced by use of metabolically labeled RNAs that can be used for the identification of modified oligonucleotides47 or for the relative quantification of changes in RNA modification status.48,49Fig. 3 illustrates the former, wherein the base composition of the RNase digestion product can be determined based on the m/z measurement and then constrained by the number of carbons or nitrogens present, which is determined by the mass shift induced by the metabolic label.47 Alternatively, metabolic labeling to deplete heavy isotopes simplifies detection and identification of larger molecular weight RNase digestion products, and has been used to improve the CARD approach for identification of modified RNAs (Fig. 4).50
 | ||
| Fig. 3 Ultrahigh resolution time-of-flight mass spectrometry analysis of oligoribonucleotide [m62A][m62A]CCUG > p. A. TOF MS spectrum shows both the light (m/z 986.657) and heavy (m/z 998.119) corresponding to monoisotopic masses of 1975.314 and 1998.238 Da, respectively. B. Tandem MS spectra of the light oligoribonucleotide. C. Tandem MS spectra of the heavy oligoribonucleotide. The predominant fragment ions are highlighted. Figure reproduced with permission from S. P. Waghmare and M. J. Dickman, Characterization and quantification of RNA post-transcriptional modifications using stable isotope labeling of RNA in conjunction with mass spectrometry analysis, Anal. Chem., 2011, 83, 4894. Copyright 2011 American Chemical Society. | ||
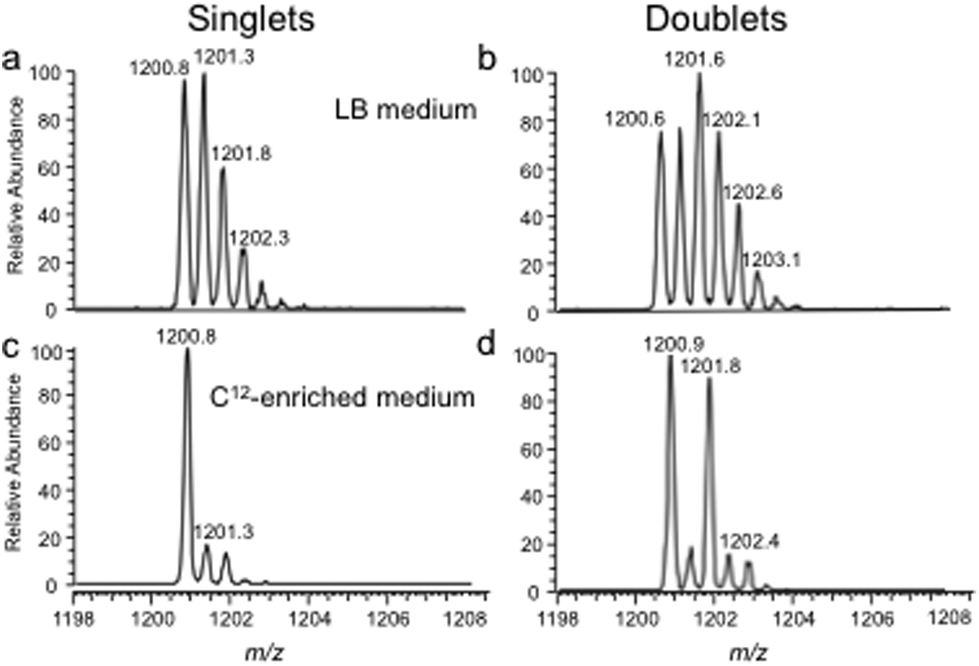 | ||
| Fig. 4 Improvements in singlet and doublet identification using 12C-enriched medium as illustrated with the doubly-charged E. coli total tRNA RNase T1 digestion product A[ms2i6A]AACCGp (MW 2403.4 Da). A. Mass spectrum from sample grown in LB medium and labeled with 16O during RNase T1 digestion. B. Same sample as in (A) except labeled with both 16O and 18O during RNase T1 digestion. C. Mass spectrum obtained when sample grown in 12C-enriched medium and labeled with 16O during RNase T1 digestion. D. Same sample as in (C) except labeled with both 16O and 18O during RNase T1 digestion. Singlet and doublet identifications are simplified in (C) and (D), respectively, by use of 12C-enriched medium. Figure reproduced with permission from C. Wetzel, S. Li and P. Limbach, Metabolic de-isotoping for improved LC-MS characterization of modified RNAs, J. Am. Soc. Mass Spectrom., 2014, 25, 1114. | ||
2.4. Top-down modification mapping
Top-down sequencing methods can also be used to map modifications onto large RNA sequences.51,52 In this regime, the RNA of interest is first isolated and purified, then subsequently dissociated in the mass spectrometer using one or more activation methods (see below). Unlike bottom-up approaches, a top-down method can provide nearly complete sequence and modification mapping coverage for any particular RNA, although specialized mass spectrometry instrumentation is required. Breuker and co-workers have recently identified optimized ESI conditions that improve MS (and subsequent MS/MS) ion yields for top-down approaches.53 As impressively illustrated by this group, top-down modification mapping offers a powerful alternative to bottom-up approaches and can be used for both synthetically modified as well as biological modified RNAs.3. Developments in techniques and instrumentation
During the past several years, the most significant instrumental advances in the analysis of modified nucleosides have arisen due to improvements in tandem mass spectrometry (MS/MS) and new chromatographic techniques. Moreover, several other technologies including ion mobility and droplet-based sample introduction reveal promising future directions for the field.3.1. Chromatography
While there are multiple examples of using MALDI-MS for the identification of modified RNAs,41,54 the more popular approach has been to combine upfront liquid chromatography separation with ESI-MS.55 It was shown very early that ion pair reverse phase liquid chromatography (IP-RP-LC) is most effective for hyphenated LC-MS (and LC-MS/MS) analysis of oligonucleotides.56,57 The most common mobile-phase contains an ion pairing component (typically triethylamine, TEA) along with an additive, such as hexafluoroisopropanol (HFIP), that improves ionization.58 One challenge of the TEA/HFIP mobile phase for oligoribonucleotides is the carryover effects of TEA, along with the background signal (in negative polarity) from HFIP. These concerns have led to a general consensus in the field that LC-MS of oligonucleotides requires a dedicated system.To combat this general consensus, multiple labs have investigated alternatives to the TEA/HFIP mobile phase.58–60 One challenge faced by investigators seeking optimal LC conditions for on-line separation and MS analysis is the impact of oligonucleotide sample on the optimization of LC conditions. For example, Gong and McCullagh reported that hexylamine was the best ion pairing reagent for separation of synthetic oligonucleotides (Fig. 5).59 In contrast, Erb and Oberacher reported that TEA/HFIP is the best system for quantitative analysis by LC-MS, while cyclohexyldimethylammonium acetate (CycHDMAA) should replace TEA as the ion pairing reagent for qualitative analyses.58
 | ||
| Fig. 5 Effect of ion pairing reagent concentration on MS signal sensitivity and the amount of adduct ions formed with oligonucleotides using different IP-HIFP buffers (3 replicates). LC conditions: mobile phase A (MPA) – IP reagent (5–30 mM) and 100 mM HFIP, mobile phase B – MPA in 80% MeOH, 4% MeOH in 5 min, 0.2 mL min−1, column temperature 45 °C, injected 1 μL except 3 μL for TEA. Figure reproduced with permission from L. Gong and J. S. O. McCullagh, Comparing ion-pairing reagents and sample dissolution solvents for ion-pairing reversed-phase liquid chromatography/electrospray ionization mass spectrometry analysis of oligonucleotides, Rapid Commun. Mass Spectrom., 2014, 28, 339. | ||
Bartlett and co-workers have conducted one of the most comprehensive studies of alternatives to the TEA/HFIP mobile phase,60 and have reported appropriate experimental conditions for optimal oligoribonucleotide separation and ionization (Fig. 6).61 Among their key findings was the development of a model that would predict optimal ion pairing reagents based on sample hydrophobicity. Tripropylamine (TPA), diisopropylamine (DIPA), dimethylbutylamine (DMBA) and diisopropylethylamine (DIEA) were found to be better ion pairing reagents than TEA for samples of moderate (TPA) to high (DMBA, DIEA) hydrophobicity. An added advantage of these findings is that the amount of HFIP counterion can be reduced when using these alternatives.61
 | ||
| Fig. 6 Comparison of the base peak ion intensity of siRNA with alkylamines and various concentrations of HFIP: (a) antisense strand and (b) sense strand. Experiments were performed in triplicate, and data are presented as the mean ± the standard deviation. Figure reproduced with permission from A. C. McGinnis, E. C. Grubb and M. G. Bartlett, Systematic optimization of ion-pairing agents and hexafluoroisopropanol for enhanced electrospray ionization mass spectrometry of oligonucleotides, Rapid Commun. Mass Spectrom., 2013, 27, 2655. | ||
Another recent development is the use of ammonium acetate-based mobile phases for oligoribonucleotide separations.48 While such mobile phases were among the earliest explored for LC-MS, they were rapidly replaced by the TEA/HFIP method due to the latter's significant gains in sensitivity. However, for analyses that are not sample limited, these ammonium acetate compositions can reduce any instrumentation-related carry-over, providing practitioners with more flexibility in instrumentation operation.
3.2. Tandem mass spectrometry
The analysis of modified RNAs by tandem mass spectrometry has become routine thanks to numerous fundamental studies by several research groups.62–64 Sequence-specific information can be generated by applying energy to intact RNA oligomers in the gas phase, leading to predictable backbone fragmentation, which can locate modifications. More specifically, when activated using collision induced dissociation (CID), oligoribonucleotides typically fragment along the phosphodiester backbone to generate two separate mass ladders. During CID, the most labile fragmentation pathways generate complementary c-type and y-type fragment ions, with w-type and a-base fragment ions also generated although at much lower abundance.62 The presence of CID-generated mass ladders can be exploited to locate modifications within the oligoribonucleotide sequence through the unique mass shift generated by the posttranscriptional modification. Alternatives to CID include higher-energy collisional dissociation (HCD), which yields similar types of fragment ions as CID albeit at slightly greater amounts,65 and electron dissociation detachment (EDD), which predominantly generates d-type and w-type fragment ions.53As the number of MS/MS investigations of modified RNAs has increased, modification-specific fragmentation patterns have been observed in several cases.63,66 Labile modified nucleosides can yield strong neutral loss pathways that dominate the MS/MS spectrum (Table 1). Fig. 7 illustrates the effect of labile modifications on mass spectral data. Importantly, these non-standard fragmentation pathways can be exploited to generate modification-specific tandem mass spectrometry assays.67–69
 | ||
| Fig. 7 A. Electrospray mass spectrum corresponding to the peak seen in the extracted ion chromatogram for m/z 817.6 of the RNaseT1digest of 5 μg of 16S rRNA from H37Rv M. tuberculosis. The mass spectral data are consistent with the doubly charged ion that would be expected for CC[mG]CG. The other m/z values in this mass spectrum correspond to additional RNase T1 digestion products from 16S rRNA. B. Collision-induced dissociation (CID) mass spectrum of the RNase T1 digestion product at m/z 817.6 shown in panel A. C. CID mass spectrum of the fragment with m/z 735.1 shown in panel B. The observed sequence informative fragments correspond to the expected fragmentation pattern of oligonucleotide CC[mG]CG, which has an mG-base loss. The absence of mG is depicted as [G] in the sequence representation. Sequence-informative fragment ions are labeled following the nomenclature of McLuckey et al.82 Figure reproduced from S. Y. Wong, B. Javid, B. Addepalli, G. Piszczek, M. B. Strader, P. A. Limbach and C. E. Barry 3rd, Functional role of methylation of G518 of the 16S rRNA 530 loop by GidB in Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2013, 57, 6311. | ||
| Ion | Neutral LOSS (Da) |
|---|---|
| PO3H2 | 80.974 |
| [m7G] | 165.065 |
| [k2C] | 144.090 |
| [Q] | 115.064 |
| [oQ] | 131.058 |
| [t6A] | 162.064 |
3.3. Ion mobility mass spectrometry
A promising instrumental advancement, which is underutilized in the mass spectrometry analysis of modified RNAs, is ion mobility spectrometry (IMS), which adds an extra dimension of separation in the gas phase. IMS can separate ions by shape and charge density, referred to as a collisional cross section. Fabris and colleagues have already demonstrated the utility of IMS-MS for the gas-phase separation and analysis of modified nucleosides.39,40 Further, gas phase mobility mass spectrometry has been used to elucidate structural changes of several intact non-modified microRNAs, RNA complexes, as well as changes in HIV viral RNA assembly.70–72 However, the integration of IMS with MS for the characterization of modified oligoribonucleotides has not yet been reported.3.4. New ionization methods
As mentioned earlier in this review, the most popular ionization methods for modified oligoribonucleotides and RNAs are MALDI and ESI. Recently we investigated droplet-based sample introduction, using induction based fluidics (IBF), as a means of generating ESI-like data from oligonucleotides.73 It appears that droplet-based sample introduction may retain the benefits of ESI, while reducing sampling bias for oligoribonucleotide mixtures. A different alternative has been the use of plasma-based ionization sources for the detection of oligoribonucleotides.74,75 Although their utility is limited to very specific applications, detection based on the presence of phosphate can introduce selectivity into the analysis. Finally, despite the numerous advances in ambient ionization sources for mass spectrometry,76 there are no significant reports of their use in the analysis of modified RNAs.3.5. Advances in software and bioinformatics
One of the more substantial advances in the area of mass spectrometry of modified RNAs has been in the generation of new software tools and bioinformatics resources. While the number of publicly available RNA programs is significantly smaller than those available in the field of proteomics,77 they are necessary to manage the large datasets, such as those generated by LC-MS/MS approaches.Schürch and colleagues, building upon their advances in understanding CID fragmentation of modified RNAs, created the OMA-/OPA-suite of software for simplifying interpretation of MS/MS data.78 This software can take inputted RNA sequences, calculate expected MS/MS sequence ladders and then compare the calculated ladders against experimental data to score the strength of the match. A different strategy was used by Sample et al., who created RoboOligo to handle de novo sequence analysis of modified oligoribonucleotides.79 This software program builds up potential oligoribonucleotide sequences with user-selected modified nucleosides based on the input MS/MS data. A useful feature of this software is the ability to map modifications onto an intact RNA in an automated fashion. When these new tools are added to the existing software resources,54,80 the interested analyst has multiple options available to simplify the interpretation of MS/MS data obtained from modified RNAs.
4. Future outlook
RNA-based mass spectrometry has advanced significantly over the past several years. The combination of improved analytical methods and protocols combined with advances in software for analysis of MS/MS data greatly simplifies the challenge of characterizing samples that can contain numerous posttranscriptionally modified nucleosides. While RNA-based mass spectrometry still lags the field of proteomics, many of the analytical improvements that first arose within proteomics are now a common feature for characterizing modified RNAs. Yet, despite these advances, there remain areas where improvements would be valued.As noted earlier in this review, a common challenge for more widespread acceptance of using mass spectrometry for the characterization of modified RNAs is that posed by ion pairing reagents, which can often leave residual contamination within the HPLC system. New developments in HPLC stationary phases or separation modalities that reduce the requirement for an ion pairing reagent would be welcomed. Moreover, additives that can improve ionization efficiency without background effects – especially if combined with less problematic ion pairing reagents such as ammonium acetate – would also benefit this field.
One can anticipate that mass spectrometry will become even more important as additional findings are revealed about the epitranscriptome and the biological importance of modifications becomes better defined.81 While mass spectrometry cannot compete with the throughput available using next-generation sequencing technologies, those technologies cannot match the ability of mass spectrometry to identify nearly all of the modifications present within a single sample. However, should proteomics-like pipelines for RNA modification mapping be developed, then mass spectrometry can provide an important role in validating findings from sequencing approaches at throughputs appropriate for transcriptome-level analysis.
Acknowledgements
Financial support for this work is provided by the National Institutes of Health (GM058843), the National Science Foundation (CHE1507357), the Defense Threat Reduction Agency (HDTRA1-15-1-0033) and the University of Cincinnati.References
- T. Carell, C. Brandmayr, A. Hienzsch, M. Müller, D. Pearson, V. Reiter, I. Thoma, P. Thumbs and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 7110–7131 CrossRef CAS PubMed.
- M. Helm and J. D. Alfonzo, Chem. Biol., 2014, 21, 174–185 CrossRef CAS PubMed.
- I. Moukadiri, M. J. Garzon, G. R. Bjork and M. E. Armengod, Nucleic Acids Res., 2013, 42, 2602–2623 CrossRef PubMed.
- P. C. Thiaville, D. Iwata-Reuyl and V. de Crécy-Lagard, RNA Biol., 2014, 11, 1529–1538 CrossRef PubMed.
- R. J. L. Giegé, in DNA and RNA modification enzymes: comparative structure, mechanism, functions, cellular interactions and evolution, ed. H. Grosjean, Landes Bioscience, Austin, TX, 2009, p. 18 Search PubMed.
- W. A. Cantara, P. F. Crain, J. Rozenski, J. A. McCloskey, K. A. Harris, X. Zhang, F. A. P. Vendeix, D. Fabris and P. F. Agris, Nucleic Acids Res., 2011, 39, D195–D201 CrossRef CAS PubMed.
- M. Machnicka, K. Milanowska, O. Osman, E. Purta, M. Kurkowska, A. Olchowik, W. Januszewski, S. Kalinowski, S. Dunin-Horkawicz, K. Rother, M. Helm, J. Bujnicki and H. Grosjean, Nucleic Acids Res., 2012, D262–267 Search PubMed.
- J. A. Doudna, Nat. Struct. Biol., 2000, 7, 954–956 CrossRef CAS PubMed.
- H. Akashi, Curr. Opin. Genet. Dev., 2001, 11, 660–666 CrossRef CAS PubMed.
- K. Fujishima and A. Kanai, Front. Genet., 2014, 5, 142 Search PubMed.
- R. Guymon, S. C. Pomerantz, P. F. Crain and J. A. McCloskey, Biochemistry, 2006, 45, 4888–4899 CrossRef CAS PubMed.
- M. D. Ermolaeva, Curr. Issues Mol. Biol., 2001, 3, 91–97 CAS.
- K. A. Dittmar, E. M. Mobley, A. J. Radek and T. Pan, J. Mol. Biol., 2004, 337, 31–47 CrossRef CAS PubMed.
- L. Fu, C. Guerrero, N. Zhong, N. Amato, Y. Liu, S. Liu, Q. Cai, D. Ji, S. Jin, L. Niedernhofer, G. Pfeifer, G. Xu and Y. Wang, J. Am. Chem. Soc., 2014, 136, 3 CrossRef.
- Y. Fu, D. Dominissini, G. Rechavi and C. He, Nat. Rev. Genet., 2014, 15, 293–306 CrossRef CAS PubMed.
- T. Yokogawa, Y. Kumazawa, K. Miura and K. Watanabe, Nucleic Acids Res., 1989, 17, 2623–2638 CrossRef CAS PubMed.
- D. Kanduc, Prep. Biochem., 1994, 24, 167–174 CrossRef CAS.
- Y. Kumazawa, T. Yokogawa, E. Hasegawa, K. Miura and K. Watanabe, J. Biol. Chem., 1989, 264, 13005–13011 CAS.
- Y. Kumazawa, T. Yokogawa, H. Tsurui, K. Miura and K. Watanabe, Nucleic Acids Res., 1992, 20, 2223–2232 CrossRef CAS PubMed.
- M. Schaefer, T. Pollex, K. Hanna and F. Lyko, Nucleic Acids Res., 2009, 37, e12 CrossRef PubMed.
- P. B. Cattenoz, R. J. Taft, E. Westhof and J. S. Mattick, RNA, 2013, 19, 257–270 CrossRef CAS PubMed.
- C.-X. Song, C. Yi and C. He, Nat. Biotechnol., 2012, 30, 1107–1116 CrossRef CAS PubMed.
- U. Birkedal, M. Christensen-Dalsgaard, N. Krogh, R. Sabarinathan, J. Gorodkin and H. Nielsen, Angew. Chem., Int. Ed., 2015, 54, 451–455 CAS.
- J. A. McCloskey and S. Nishimura, Acc. Chem. Res., 1977, 10, 403–409 CrossRef CAS.
- J. A. Kowalak, S. C. Pomerantz, P. F. Crain and J. A. McCloskey, Nucleic Acids Res., 1993, 21, 4577–4584 CrossRef CAS PubMed.
- C. M. Castleberry, L. P. Rodicio and P. A. Limbach, Curr. Protoc. Nucleic Acid Chem, 2008, ch. 10, Unit 10 12 Search PubMed.
- W. D. Van Dongen and W. M. A. Niessen, Bioanalysis, 2011, 3, 541–564 CrossRef CAS PubMed.
- W. M. Cai, Y. H. Chionh, F. Hia, C. Gu, S. Kellner, M. E. McBee, C. S. Ng, Y. L. J. Pang, E. G. Prestwich, K. S. Lim, I. Ramesh Babu, T. J. Begley and P. C. Dedon, in Methods Enzymol, ed. H. Chuan, Academic Press, 2015, vol. 560, pp. 29–71 Search PubMed.
- H. Kasai, K. Murao, S. Nishimura, J. G. Liehr, P. F. Crain and J. A. McCloskey, Eur. J. Biochem., 1976, 69, 435–444 CrossRef CAS.
- P. F. Crain, in Methods Enzymol, Academic Press, 1990, vol. 193, pp. 782–790 Search PubMed.
- K. Miyauchi, S. Kimura and T. Suzuki, Nat. Chem. Biol., 2013, 9, 105–111 CrossRef CAS PubMed.
- C. Dumelin, Y. Chen, A. Leconte, Y. Chen and D. Liu, Nat. Chem. Biol., 2012, 8, 913–919 CAS.
- D. Su, C. T. Y. Chan, C. Gu, K. S. Lim, Y. H. Chionh, M. E. McBee, B. S. Russell, I. R. Babu, T. J. Begley and P. C. Dedon, Nat. Protocols, 2014, 9, 828–841 CAS.
- C. T. Chan, M. Dyavaiah, M. S. DeMott, K. Taghizadeh, P. C. Dedon and T. J. Begley, PLoS Genet., 2010, 6, e1001247 CAS.
- P. C. Dedon and T. J. Begley, Chem. Res. Toxicol., 2014, 27, 330–336 CrossRef CAS PubMed.
- C. Brandmayr, M. Wagner, T. Brückl, D. Globisch, D. Pearson, A. C. Kneuttinger, V. Reiter, A. Hienzsch, S. Koch, I. Thoma, P. Thumbs, S. Michalakis, M. Müller, M. Biel and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 11162–11165 CrossRef CAS PubMed.
- D. Globisch, D. Pearson, A. Hienzsch, T. Brückl, M. Wagner, I. Thoma, P. Thumbs, V. Reiter, A. C. Kneuttinger, M. Müller, S. A. Sieber and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 9739–9742 CrossRef CAS PubMed.
- S. Kellner, J. Neumann, D. Rosenkranz, S. Lebedeva, R. F. Ketting, H. Zischler, D. Schneider and M. Helm, Chem. Commun., 2014, 50, 3516–3518 RSC.
- R. Quinn, M. Basanta-Sanchez, R. E. Rose and D. Fabris, J. Mass Spectrom., 2013, 48, 703–712 CrossRef CAS PubMed.
- R. E. Rose, R. Quinn, J. L. Sayre and D. Fabris, RNA, 2015, 21, 1361–1374 CrossRef CAS PubMed.
- S. Douthwaite and F. Kirpekar, Methods Enzymol., 2007, 425, 1–20 Search PubMed.
- K. W. Gaston and P. A. Limbach, RNA Biol., 2014, 11, 1568–1585 CrossRef PubMed.
- T. M. Lowe and S. R. Eddy, Nucleic Acids Res., 1997, 25, 955–964 CrossRef CAS PubMed.
- P. Puri, C. Wetzel, P. Saffert, K. W. Gaston, S. P. Russell, J. A. Cordero Varela, P. van der Vlies, G. Zhang, P. A. Limbach, Z. Ignatova and B. Poolman, Mol. Microbiol., 2014, 93, 944–956 CrossRef CAS PubMed.
- S. Li and P. Limbach, Anal. Chem., 2012, 84, 8607–8613 CrossRef CAS PubMed.
- S. Li and P. Limbach, Analyst, 2013, 138, 1386–1394 RSC.
- S. P. Waghmare and M. J. Dickman, Anal. Chem., 2011, 83, 4894–4901 CrossRef CAS PubMed.
- A. M. Popova and J. R. Williamson, J. Am. Chem. Soc., 2014, 136, 2058–2069 CrossRef CAS PubMed.
- M. Taoka, Y. Nobe, M. Hori, A. Takeuchi, S. Masaki, Y. Yamauchi, H. Nakayama, N. Takahashi and T. Isobe, Nucleic Acids Res., 2015, 43, e115 CrossRef PubMed.
- C. Wetzel, S. Li and P. Limbach, J. Am. Soc. Mass Spectrom., 2014, 25, 1114–1123 CrossRef CAS PubMed.
- M. Taucher, B. Ganisl and K. Breuker, Int. J. Mass Spectrom., 2011, 304, 91–97 CrossRef CAS PubMed.
- T. Y. Huang, J. Liu and S. A. McLuckey, J. Am. Soc. Mass Spectrom., 2010, 21, 890–898 CrossRef CAS PubMed.
- M. Taucher and K. Breuker, Angew. Chem., Int. Ed., 2012, 51, 11289–11292 CrossRef CAS PubMed.
- R. Matthiesen and F. Kirpekar, Nucleic Acids Res., 2009, 37, e48 CrossRef PubMed.
- A. C. McGinnis, B. Chen and M. G. Bartlett, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 76–94 CrossRef CAS PubMed.
- K. J. Fountain, M. Gilar and J. C. Gebler, Rapid Commun. Mass Spectrom., 2003, 17, 646–653 CrossRef CAS PubMed.
- C. Wetzel and P. Limbach, J. Proteomics, 2011, 75, 3450–3464 CrossRef PubMed.
- R. Erb and H. Oberacher, Electrophoresis, 2014, 35, 1226–1235 CrossRef CAS PubMed.
- L. Gong and J. S. O. McCullagh, Rapid Commun. Mass Spectrom., 2014, 28, 339–350 CrossRef CAS PubMed.
- B. Chen, S. F. Mason and M. G. Bartlett, J. Am. Soc. Mass Spectrom., 2013, 24, 257–264 CrossRef CAS PubMed.
- A. C. McGinnis, E. C. Grubb and M. G. Bartlett, Rapid Commun. Mass Spectrom., 2013, 27, 2655–2664 CrossRef CAS PubMed.
- T. Huang, A. Kharlamova, J. Liu and S. McLuckey, J. Am. Soc. Mass Spectrom., 2008, 19, 1832–1840 CrossRef CAS PubMed.
- A. Nyakas, S. Stucki and S. Schürch, J. Am. Soc. Mass Spectrom., 2011, 22, 875–886 CrossRef CAS PubMed.
- A. Nyakas, R. P. Eberle, S. R. Stucki and S. Schürch, J. Am. Soc. Mass Spectrom., 2014, 25, 1155–1166 CrossRef CAS PubMed.
- M. Kullolli, E. Knouf, M. Arampatzidou, M. Tewari and S. J. Pitteri, J. Am. Soc. Mass Spectrom., 2014, 25, 80–87 CrossRef CAS PubMed.
- N. A. Hari Y, S. R. Stucki and S. Schürch, Chimia, 2014, 68, 164–167 CrossRef PubMed.
- B. Addepalli and P. A. Limbach, J. Am. Soc. Mass Spectrom., 2011, 22, 1363–1372 CrossRef CAS PubMed.
- C. Wetzel and P. Limbach, Analyst, 2013, 138, 6063–6072 RSC.
- S. Y. Wong, B. Javid, B. Addepalli, G. Piszczek, M. B. Strader, P. A. Limbach and C. E. Barry 3rd, Antimicrob. Agents Chemother., 2013, 57, 6311–6318 CrossRef CAS PubMed.
- K. Takebayashi, K. Hirose, Y. Izumi, T. Bamba and E. Fukusaki, J. Biosci. Bioeng., 2013, 115, 332–338 CrossRef CAS PubMed.
- D. Fabris, Biopolymers, 2009, 91(4), iii–iv CrossRef CAS PubMed.
- B. C. Legiewicz M, K. B. Turner, D. Fabris, T. E. Hamm, D. Rekosh, M. L. Hammarskjöld and S. F. Le Grice, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5 CrossRef PubMed.
- R. L. Ross, A. D. Sauter Jr. and P. A. Limbach, J. Mass Spectrom., 2015, 50, 1175–1179 CrossRef CAS PubMed.
- R. N. Easter, K. K. Kroning, J. A. Caruso and P. A. Limbach, Analyst, 2010, 135, 2560–2565 RSC.
- B. Catron, J. A. Caruso and P. A. Limbach, J. Am. Soc. Mass Spectrom., 2012, 23, 1053–1061 CrossRef CAS PubMed.
- C. C. Hsu and P. C. Dorrestein, Curr. Opin. Biotechnol., 2015, 31, 24–34 CrossRef CAS PubMed.
- H. Nakayama, N. Takahashi and T. Isobe, Mass Spectrom. Rev., 2011, 30, 1000–1012 CrossRef CAS PubMed.
- A. Nyakas, L. C. Blum, S. R. Stucki, J. L. Reymond and S. Schürch, J. Am. Soc. Mass Spectrom., 2013, 24, 249–256 CrossRef CAS PubMed.
- P. J. Sample, K. W. Gaston, J. D. Alfonzo and P. A. Limbach, Nucleic Acids Res., 2015, 43, e64–e64 CrossRef PubMed.
- H. Nakayama, M. Akiyama, M. Taoka, Y. Yamauchi, Y. Nobe, H. Ishikawa, N. Takahashi and T. Isobe, Nucleic Acids Res., 2009, 37, e47 CrossRef PubMed.
- K. D. Meyer and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2014, 15, 313–326 CrossRef CAS PubMed.
- S. A. McLuckey, G. J. Van Berker and G. L. Glish, J. Am. Soc. Mass Spectrom., 1992, 3, 60–70 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |