
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A FRET-based biosensor for the detection of neutrophil elastase†
C.
Schulenburg
,
G.
Faccio
 *,
D.
Jankowska
,
K.
Maniura-Weber
and
M.
Richter‡
*
*,
D.
Jankowska
,
K.
Maniura-Weber
and
M.
Richter‡
*
Department of Biointerfaces, Empa - Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen (CH), Switzerland. E-mail: greta.faccio@empa.ch; michael.richter@igb.fraunhofer.de; Tel: +41 58 765 7262, +49 9421 187 353
First published on 27th January 2016
Abstract
The direct and specific detection of biomarkers is crucial as it can allow monitoring the state of a tissue or wound as well as the progression of the inflammatory process. Neutrophil elastase (NE) plays an important role in many biological processes. It is involved in inflammatory diseases and is enriched in inflamed tissues, in wound exudate, and in the sputum of cystic fibrosis patients. In order to detect NE, we designed a NE-specific protein sensor whose fluorescence features are altered upon selective cleavage by NE at physiological concentration. The biosensor consists of two fluorescent GFP-derived proteins connected by a short peptide linker containing a NE-specific recognition sequence. Due to the close proximity of the two fluorescent proteins, Förster resonance energy transfer (FRET) occurs in the uncleaved form and, upon cleavage by NE, a decrease of FRET signal is observed. In this study, the construct design, the influence of the linker length, as well as the specificity for NE are described. Furthermore, the influence of biosensor immobilization on the functionality is analysed. By engineering the recognition sequence of the linker, the presented system can potentially be easily adapted to detect other proteases such as cathepsin, caspases or matrix metalloproteases.
Neutrophil elastase (NE, EC 3.4.21.37) is a serine protease involved in the degradation of the extracellular matrix, collagen, and in the activation of matrix-metalloproteases.1 It is associated with numerous inflammatory diseases. Clinical studies have evidenced how an elevated concentration of NE correlates to acute lung injury and acute respiratory distress syndrome,2 with chronic or aggressive periodontitis,3 how it is involved in infection by Salmonella enterica serovar Typhimurium,4 and in the development of cystic fibrosis.4,5 Aiming at contrasting the progression of these clinical conditions, NE is investigated as a major target for novel inhibitory molecules to be used as drugs. Thus, monitoring of NE activity under physiological conditions is crucial.6–9 Currently, NE activity is measured using (1) chromogenic peptide substrates such as N-methoxysuccinyl-Ala-Ala-Pro-Val-pNA or MeOSuc-Ala-Ala-Pro-Val-pNA where p-nitroanilide (pNA) is released and detected upon NE action, (2) fluorogenic peptide substrates such as N-methoxysuccinyl-Ala-Ala-Pro-Val-7-amido-4-methylcoumarin, or (3) by ELISA. Here, we describe a protein-based fluorescent substrate to detect NE activity. The potential of using FRET-based biosensors to monitor protease activity has been investigated in 2004 by Felber and colleagues.10 Since then, FRET-based biosensors are widely used to monitor various biological phenomena, such as protein–protein interactions, protein–molecule interactions, and enzymatic activity in vitro as well as in vivo.11–18 Concerning protease-sensing, a FRET-sensor for thrombin19 and caspase20 have been reported. Multiple of these biosensors consist of two fluorescent proteins, connected via a protein or a short peptide linker conferring affinity. Due to the close proximity of the two proteins, Förster resonance energy transfer (FRET) can occur. In our work, the linker contains an amino acid sequence specifically recognized by NE, and cleavage of this sequence by NE thus results in the dissociation of the fluorescent proteins and decrease in FRET (Fig. 1b). The biosensor described here was assembled by fusing the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP) through peptide linkers of different length (8–24 residues) to give variants I to IV (Fig. 1).
 | ||
| Fig. 1 Schematic representation of the neutrophil elastase-specific biosensor. (a) The FRET pair is the fusion of a yellow and a cyan fluorescent protein through a peptide linker containing a NE recognitions sequence. Upon treatment with neutrophil elastase (PDB ID: 3Q76, active site residues in red), the FRET pair dissociates with a consequent decrease in FRET signal. The NE recognition sequence is shown in bold letters. (b) The four biosensor variants developed in this study are differing only in the length of the linker region. | ||
We suggest this biosensor as a tool to monitor NE activity within screening systems aiming at the engineering and development of NE biosensors for use in healthcare.
In order to confer specificity for human NE, the peptide linker was designed to contain the Phe-Ile-Arg-Trp motif that has been recently identified as a selective NE-recognition sequence.21 To facilitate future modification of the linker sequence and confer specificity for a different protease, two restriction enzyme recognition sites were introduced on the gene level, namely a SpeI site upstream to the linker region and an AgeI site between the linker and YFP.
The linker regions were designed not to carry any typical NE substrate residues such as Val and Ala, as these are also recognized by other proteases, e.g. proteinase 3 and cathepsin G.22In silico analysis confirmed the absence of additional protease sites and only the NE site at position 250 in biosensor IV was identified by PROSPER within the unstructured linker region.23 In particular, no recognition sequence for matrix metalloprotease 2, 3 or 9 was found. However, a low-probability recognition site for cathepsin K was identified within the unstructured linker region in position 234 (Asp-Glu-Leu-Tyr ↓ Lys-Thr-Ser-Gly).
All four variants were recombinantly produced in Escherichia coli and subsequently purified to homogeneity. The FRET ratio of all four variants was calculated as the ratio between the fluorescence emission at 528 nm and 485 nm. The FRET ratio of all variants was 1 and decreased to 0.7 upon treatment with NE in PBS buffer (Table 1†). This indicated dissociation of the two fluorescent proteins as well as no influence of the linker length on the distance of the two fluorescent protein domains. To verify that NE cleaves in the designed linker sequence, SDS PAGE and N-terminal sequencing was performed. The intact biosensors I–IV have a ∼65 kDa molecular mass that, if cleaved by NE only at the FIRW site present in the linker region, parts in the two components YFP and CFP of ∼33 kDa each. The SDS PAGE analysis of the biosensor proteins before and after incubation with NE indicated complete cleavage of the biosensor proteins I–IV under the tested conditions and the production of protein species migrating as a single band between 25 and 35 kDa (Fig. 2b, inset). N-terminal sequencing of the NE-treated biosensor I indicated the presence of two N-termini with sequence (M)SKGEELFTGV and RWGGGGSGGG corresponding to the N-terminus of CFP and the inserted NE-cleavage site before YFP, respectively.
 | ||
| Fig. 2 (a) Decrease of FRET ratio as a function of time upon NE addition. The data were fit to a single exponential equation. Inset: emission spectra of biosensor III with excitation at 428 nm after a 30 min incubation in the presence (red dashed line) or absence (continuous line) of NE. (b) Initial velocity of the NE-catalyzed reaction was measured as a function of the biosensor concentration in PBS buffer, pH 6.2 at room temperature (● = biosensor I, ▲ = biosensor II, ◆ = biosensor III, ■ = biosensor IV). Data were fit to the Michael–Menten equation. Inset: SDS PAGE of the four biosensors before (−) and after (+) incubation with neutrophil elastase. Molecular weight markers are reported in kDa. | ||
To analyse the impact of additives such as salts, proteins or small molecules such as vitamins, the FRET efficiency was determined in Dulbecco's Modified Eagle Medium (DMEM) and in a complex artificial wound exudate solution (AWE). Compared to reactions carried out in PBS buffer, the presence of salts did neither affect the FRET ratio of the non-cleaved constructs, nor did influence NE activity on all protein substrates in DMEM medium (Table 1†). In AWE, the FRET ratio of all four variants was slightly higher, than in PBS or DMEM, most likely caused by the interaction of the components of the serum contained in AWE on the biosensor. In a protein-rich medium such as AWE no cleavage by NE was observed. This might be explainable by the presence of NE inhibitors, e.g. the secretory leukocyte protease inhibitor24 that naturally targets NE, or by the interaction of the biosensor molecules with serum proteins that might shield the linker region. Similarly, the presence of bovine serum albumin (BSA) at a high concentration, or sera from different origin, negatively affected the efficacy of the cleavage of the NE biosensor (Fig. 2†).
The rate of substrate cleavage was determined in PBS buffer and DMEM using ∼20 ng ml−1 (<1 mU) NE, simulating the physiological concentration reported for wounds, e.g.<1 μg ml−1 and <10 mU.25,26 The reaction was initiated by adding NE and the emitted fluorescence was monitored in a 450–600 nm range upon excitation at 428 nm. Over time the emission of the donor CFP at 485 nm increased, whereas the emission of the acceptor YFP at 528 nm decreased (Fig. 2a). Consequently, the FRET ratio decreases and follows a first order kinetic as a function of time (Fig. 2a). Upon cleavage of the biosensors by NE, the YFP emission peak was shifted from 518 nm to the characteristic 528 nm of the protein in its free form in solution (Fig. 2a, inset). Initial velocity was fitted to the Michael–Menten equation to obtain the kobs and KM values (Fig. 2b). All biosensors were cleaved at a comparable rate by NE in both PBS and DMEM (Table 1). NE recognizes and cleaves all four substrates with similar efficiency. The difference in length of the four linkers carrying the recognition sequence Phe-Ile-Arg-Trp did not thus affect the kinetics of the cleavage by NE.
| Sensor | k obs (s−1) | K M (μM) | k obs/KM (M−1 s−1) | |
|---|---|---|---|---|
| PBS | DMEM | PBS | ||
| I | 16.8 ± 2.4 | 33.6 ± 6.6 | 0.7 ± 0.2 | 2.4 × 107 |
| II | 7.8 ± 0.6 | 13.8 ± 1.8 | 0.7 ± 0.2 | 1.1 × 107 |
| III | 25.2 ± 3.6 | 29.4 ± 3.6 | 0.7 ± 0.2 | 3.6 × 107 |
| IV | 27.0 ± 5.4 | 42 ± 10.8 | 0.7 ± 0.2 | 3.9 × 107 |
Specific protease biosensors can be applied to in vitro diagnostics as a soluble component or in immobilized form of an activity assay. In order to evaluate the effect of immobilization on the efficiency of the biosensors, the four variants were immobilized through their C-terminal His-tag to a nickel-coated surface. Treatment of the immobilized protein biosensors with NE led to the separation into two protein species of which one is released in solution with maximum emission at 485 nm, and one immobilized with maximum of emission at 520 nm upon excitation at 428 nm. The latter protein could be released by applying an imidazole containing buffer (Fig. 3). Treatment with NE of the immobilized biosensor led to a drastic decrease of apparent FRET ratio from ∼1.1 of the uncleaved sensor to ∼0.5 upon excitation of the sole donor protein (Fig. 3b, Fig. 3†). Similarly, a decrease in donor fluorescence due to cleavage by NE could be detected upon immobilization of the biosensor via the His-tag to superparamagnetic microparticles (Fig. 4†).
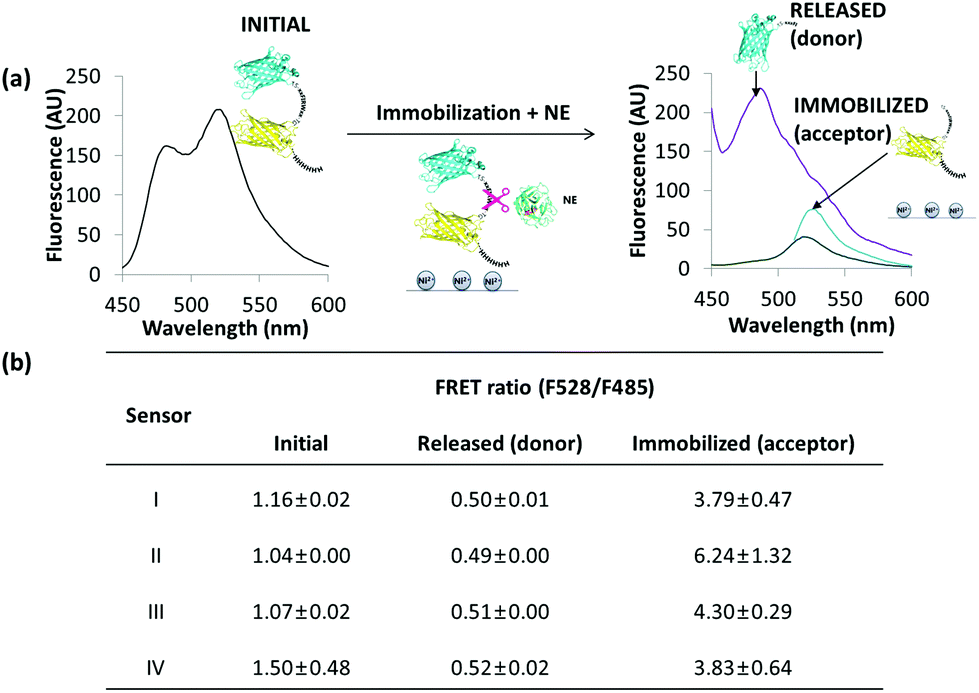 | ||
| Fig. 3 (a) Fluorescence emission spectra of the NE-biosensor I before immobilization to a nickel coated plate (left), and after treatment with neutrophil elastase (right), and corresponding FRET and apparent FRET ratio values (b). The immobilization was carried out in PBS pH 6.3 and elution was performed at a 100 mM imidazole concentration. All spectra were recorded with the proteins in solutions, and after elution of the immobilized fraction with imidazole. The immobilized and released biosensor protein after treatment with NE where excited at 428 nm (purple and green curves, respectively), and the latter also at 485 nm (blue curve). | ||
NE cleaves the substrates selectively in the designed motif, independently of the length of the linker, and in the range of physiological concentrations. The linker length had also no influence on the cleavage rate and on the substrate recognition itself, e.g. the KM values are comparable. However, in comparison to the KM values published for other NE substrates (Table 2), the protein-based biosensor reported here is recognized with a up to 500-fold higher specificity. In a medical diagnostic context, this might allow the early detection of changes in NE activity and thus a prompter response in medical treatment or the regular monitoring of the progression of pathologies such as acute lung injury and acute respiratory distress syndrome.
| Substrate | Detection | K M (μM) | k cat (s−1) | k cat/KM (M−1 s−1) | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|
| Tos-A-ONp | Direct (chromogenic) | 35 | 120 | 3.4 × 106 | 10% DMSO, 0.1 M phosphate, 0.25 M NaCl, pH 7 | 27 |
| Tos-A-ONp | Direct (chromogenic) | 364 | 4500 | 1.2 × 107 | 10% DMSO, 0.1 M phosphate, 0.25 M NaCl, pH 7, 0.35 mM decanol | 27 |
| MeO-Suc-AAPV-NA | Direct (chromogenic) | 140 | 17 | 1.2 × 105 | 0.1 M HEPES buffer, 0.5 M NaCl, 9.8% DMSO, pH 7.5, 25 °C. | 28 |
| Ac-AAPV-NA | Direct (chromogenic) | 310 | 8.1 | 2.7 × 104 | 0.1 M HEPES buffer, 0.5 M NaCl, 9.8% DMSO, pH 7.5, 25 °C. | 28 |
| Biosensors I–IV | Direct (fluorogenic) | 0.7 | 13–42 (kobs) | n.d. | PBS buffer pH 6.2, 25 °C | This study |
In conclusion, the reported FRET-based biosensor is functional in complex media like DMEM and might thus be applicable in diverse diluted body fluids like would liquid, blood or urine. Future studies will address its reactivity with other proteases and aim at improving its recognition by NE in non-diluted biological fluids. The protein-based sensor can be produced in large amounts in recombinant form, and easily purified. The possibility of immobilization in 96-well plates provides a starting point for the development of screening platforms for the high throughput analysis of biological samples and for further biosensor development through protein engineering. The retained activity of the biosensor in immobilized form makes it suitable for incorporation into sensing devices such as strips or electrodes.
Acknowledgements
This work was financed by the Swiss Confederation and funded by Nano-Tera.ch within the Nanotera project “Fabrication of fluorescence biosensors in a Textile Dressing for Non-invasive Lifetime Imaging-based Wound Monitoring”, FLUSITEX (RTD 2013) that was scientifically evaluated by the SNSF. We thank Eike Müller for the support with CLSM imaging. Cindy Schulenburg and Greta Faccio equally contributed to this work.Notes and references
- Y. Okada and I. Nakanishi, FEBS Lett., 1989, 249, 353–356 CrossRef CAS PubMed.
- J. Grommes and O. Soehnlein, Mol. Med., 2010, 17, 293–307 Search PubMed.
- N. Nizam, P. Gümüş, J. Pitkänen, T. Tervahartiala, T. Sorsa and N. Buduneli, Inflammation, 2014, 37, 1771–1778 CrossRef CAS PubMed.
- N. Gill, R. B. R. Ferreira, L. C. Antunes, B. P. Willing, I. Sekirov, F. Al-Zahrani, M. Hartmann and B. B. Finlay, PLoS One, 2012, 7, e49646 CAS.
- V. Papayannopoulos, D. Staab and A. Zychlinsky, PLoS One, 2011, 6, e28526 CAS.
- S. Gehrig, J. Duerr, M. Weitnauer, C. J. Wagner, S. Y. Graeber, J. Schatterny, S. Hirtz, A. Belaaouaj, A. H. Dalpke, C. Schultz and M. A. Mall, Am. J. Respir. Crit. Care Med., 2014, 189, 1082–1092 CrossRef PubMed.
- H. Mikumo, I. Inoshima, S. Ogata, Y. Mizuta, K. Suzuki, T. Yokoyama, N. Hamada and Y. Nakanishi, Eur. Respir. J., 2014, 44 Search PubMed.
- Y. Tsai, H. Yu, W. Chang, F. Liu, Z. Huang and T. Hwang, Sci. Rep., 2015, 5 CAS , 8347.
- J. A. Caruso, S. Akli, L. Pageon, K. K. Hunt and K. Keyomarsi, Oncogene, 2014, 34, 3556–3567 CrossRef PubMed.
- L. M. Felber, S. M. Cloutier, C. Kündig, T. Kishi, V. Brossard, P. Jichlinski, H. J. Leisinger and D. Deperthes, BioTechniques, 2004, 36(5), 878–885 CAS.
- C. Chung, R. Makino, J. Dong and H. Ueda, Anal. Chem., 2015, 87, 3513–3519, DOI:10.1021/acs.analchem.5b00088.
- H. Chen, C. S. Bernard, P. Hubert, L. My and C. Zhang, FEBS J., 2014, 281, 1241–1255 CrossRef CAS PubMed.
- D. Hamers, V. V. van, J. Borst and J. Goedhart, Protoplasma, 2014, 251, 333–347 CrossRef CAS PubMed.
- Z. Wei, H. Chen, C. Zhang and B. Ye, PLoS One, 2014, 9, e92330 Search PubMed.
- Q. Tran and M. VerMeer, PLoS One, 2014, 9, e89669 Search PubMed.
- R. Moussa, A. Baierl, V. Steffen, T. Kubitzki, W. Wiechert and M. Pohl, J. Biotechnol., 2014, 191, 250–259 CrossRef CAS PubMed.
- S. Banerjee, W. K. Versaw and L. R. Garcia, PLoS One, 2015, 10, e0141128 Search PubMed.
- J. L. Vinkenborg, T. J. Nicolson, E. A. Bellomo, M. S. Koay, G. A. Rutter and M. Merkx, Nat. Methods, 2009, 6, 737–740 CrossRef CAS PubMed.
- B. Zhang, Biochem. Biophys. Res. Commun., 2004, 323, 674–678 CrossRef CAS PubMed.
- B. Angres, H. Steuer, P. Weber, M. Wagner and H. Schneckenburger, Cytometry, Part A, 2009, 75A, 420–427 CrossRef CAS PubMed.
- A. J. O'Donoghue, Y. Jin, G. M. Knudsen, N. C. Perera, D. E. Jenne, J. E. Murphy, C. S. Craik and T. W. Hermiston, PLoS One, 2013, 8, e75141 Search PubMed.
- A. Heinz, M. C. Jung, G. Jahreis, A. Rusciani, L. Duca, L. Debelle, A. S. Weiss, R. H. H. Neubert and C. E. H. Schmelzer, Biochimie, 2012, 94, 192–202 CrossRef CAS PubMed.
- J. Song, H. Tan, A. J. Perry, T. Akutsu, G. I. Webb, J. C. Whisstock and R. N. Pike, PLoS One, 2012, 7, e50300 CAS.
- S. Doumas, A. Kolokotronis and P. Stefanopoulos, Infect. Immun., 2005, 73, 1271–1274 CrossRef CAS PubMed.
- N. J. Trengove, M. C. Stacey, S. Macauley, N. Bennett, J. Gibson, F. Burslem, G. Murphy and G. Schultz, Wound Repair Regen., 1999, 7, 442–452 CrossRef CAS PubMed.
- F. Grinnell and M. Zhu, J. Invest. Dermatol., 1994, 103, 155–161 CrossRef CAS PubMed.
- D. S. Jackson, A. D. Brown, R. J. Schaeper and J. C. Powers, Arch. Biochem. Biophys., 1995, 323, 108–114 CrossRef CAS PubMed.
- K. Nakajima, J. Powers, B. Ashe and M. Zimmerman, J. Biol. Chem., 1979, 254(10), 4027–4032 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and further characterization. See DOI: 10.1039/c5an01747e |
| ‡ Current address: Fraunhofer Institute for Interfacial Engineering and Biotechnology IGB, Bio-, Electro and Chemocatalysis BioCat, Straubing Branch, Schulgasse 11a, 94315 Straubing (DE). |
| This journal is © The Royal Society of Chemistry 2016 |