
Polymer-coated micro-optofluidic ring resonator detector for a comprehensive two-dimensional gas chromatographic microsystem: μGC × μGC–μOFRR
William R.
Collin†
ab,
Kee W.
Scholten†
cb,
Xudong
Fan
db,
Dibyadeep
Paul
eb,
Katsuo
Kurabayashi
eb and
Edward T.
Zellers
*abcf
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI, 48109-1055, USA. E-mail: ezellers@umich.edu
bCenter for Wireless Integrated MicroSensing and Systems (WIMS2), University of Michigan, Ann Arbor, MI 48109-2122, USA
cApplied Physics Program, University of Michigan, Ann Arbor, MI, USA 48109-1040
dDepartment of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109- 2110, USA
eDepartment of Mechanical Engineering, University of Michigan, Ann Arbor, MI 48109-2125, USA
fDepartment of Environmental Health Sciences, Univ. of Michigan, Ann Arbor, MI 48109-2029, USA
First published on 6th November 2015
Abstract
We describe first results from a micro-analytical subsystem that integrates a detector comprising a polymer-coated micro-optofluidic ring resonator (μOFRR) chip with a microfabricated separation module capable of performing thermally modulated comprehensive two-dimensional gas chromatographic separations (μGC × μGC) of volatile organic compound (VOC) mixtures. The 2 × 2 cm μOFRR chip consists of a hollow, contoured SiOx cylinder (250 μm i.d.; 1.2 μm wall thickness) grown from a Si substrate, and integrated optical and fluidic interconnection features. By coupling to a 1550 nm tunable laser and photodetector via an optical fiber taper, whispering gallery mode (WGM) resonances were generated within the μOFRR wall, and shifts in the WGM wavelength caused by transient sorption of eluting vapors into the PDMS film lining the μOFRR cylinder were monitored. Isothermal separations of a simple alkane mixture using a PDMS coated 1st-dimension (1D) μcolumn and an OV-215-coated 2nd- dimension (2D) μcolumn confirmed that efficient μGC × μGC–μOFRR analyses could be performed and that responses were dominated by film-swelling. Subsequent tests with more diverse VOC mixtures demonstrated that the modulated peak width and the VOC sensitivity were inversely proportional to the vapor pressure of the analyte. Modulated peaks as narrow as 120 ms and limits of detection in the low-ng range were achieved. Structured contour plots generated with the μOFRR and a reference FID were comparable.
Introduction
Research over the past decade or so on Si-microfabricated gas chromatographic microsystems (μGC) has led to several improvements in design and operation that have moved us closer to low-cost, low-power instrumentation capable of analyzing the components of airborne volatile organic compound (VOC) mixtures at low concentrations in near-real time.1–10 Such air monitoring capabilities are not possible with stand-alone sensors or sensor arrays.11 Unfortunately, the maximum lengths and minimum diameters of μGC separation columns are subject to practical constraints which, in turn, limit the complexity of VOC mixtures that can be reliably analyzed by such microsystems.Microscale comprehensive two-dimensional gas chromatography (μGC × μGC), implemented using Si and/or glass micromachined components, represents one promising approach to overcome these limitations. As in bench scale GC × GC systems,12,13 in μGC × μGC a first-dimension (1D) μcolumn is connected through a (micro-scale) thermal or pneumatic modulator to a shorter second-dimension (2D) μcolumn that has retention properties differing from those of the 1D μcolumn. As the peak from each mixture component elutes from the 1D μcolumn it is re-injected piecewise into the 2D μcolumn at a rate high enough to maintain the 1D elution sequence. Ideally, then, the peak capacity is increased significantly over that provided by a one-dimensional separation column of similar length, and both the resolution and detectability of the eluting peaks can be improved.12,13
Thermal modulation, which offers certain advantages over pneumatic modulation, entails continuous, rapid thermal cycling of the mid-point modulation device during the course of an analysis: cooling to trap peak segments from the 1D μcolumn and then heating to remobilize/reinject them into the 2D μcolum.14,15 Kim et al. developed the first microfabricated thermal modulator (μTM).16 It contained a series of two spiral, Pyrex-capped, deep-reactive-ion-etched (DRIE) Si microchannel sections (stages) with independent thin-metal-film heaters. Mounted just above a compact stack of thermoelectric coolers (TEC), this μTM could be heated to ≥250 °C and then cooled to ≤−20 °C in rapid succession. By virtue of the focusing effect exerted on the eluting analytes, the modulated peak segments could be compressed, leading to commensurate improvements in resolution and detectability.
Recently, this type of device was used to perform GC × GC separations with conventional capillary columns17,18 and μGC × μGC separations with microfabricated 1D and 2D columns,19 but in all cases using a conventional, bench-scale flame ionization detector (FID). Due to nature of the modulation process, the short length of the 2D μcolumn, and the relatively high linear velocity of the carrier gas, the peaks generated at the outlet of the separation module can be very narrow. Therefore, a detector with a low dead volume and short response time, such as an FID, is required. For ultimate application in field or clinical settings, a more compact, portable detector is needed.
Whiting et al., were the first to describe a GC × GC separation using microfabricated separation and detection components.20 High-aspect-ratio DRIE-Si separation columns were used with a conventional high-pressure, pneumatic modulation system to separate a 4-VOC mixture in just a few seconds; an array of polymer coated cantilever sensors was used for detection. Other multi-dimensional separation subsystems made using microfabricated columns and various sample manipulation and sensing technologies have been reported recently that embody alternative approaches to enhancing peak capacity in GC microsystems.21,22 However, there has yet to be a report of a μGC × μGC system in which all critical components were microfabricated.
We recently introduced the microfabricated optofluidic ring resonator (μOFRR) sensor and demonstrated it as a μGC detector.23 It was modeled after the OFRR sensors developed by Fan et al. from thinned glass capillaries.24 The μOFRR sensing structure consists of a hollow, wide-bore, vertical SiOx cylinder with an expanded midsection grown, and subsequently etched free, from a Si mold. Resonant whispering gallery modes (WGM) are generated in the cylinder wall by coupling to a tunable laser with an optical fiber taper placed beside the μOFRR cylinder. The evanescent field of the WGM extends into the interior of the cylinder, and a shift in resonant wavelength, λWGM, will occur from changes in the optical properties (e.g., the refractive index, RI) at the inner surface according to the following expression:24 ΔλWGM = 2πrΔneff/m, where r is the radius of the μOFRR, m is an integer specifying the mode number, and neff is the effective RI that takes into account the mode distribution in the air, wall, surface layer and the interior fluid. Transient shifts in λWGM result from swelling and RI changes of a thin polymer film lining the cylinder due to reversible sorption of vapor passing through the cylinder. Initial tests of a PDMS-coated μOFRR connected downstream from a single μGC column showed remarkably fast responses and low detection limits under typical operating conditions.23 These results suggested that this device might have sufficiently high sensitivity and sufficiently rapid response times to serve as the detector for μGC × μGC analyses.
Here, we report on preliminary performance characterization tests of a μGC × μGC separation module with a polymer-coated μOFRR sensor installed as the detector. Fig. 1 shows a block diagram of the analytical components all of which were microfabricated. After describing the materials and methods employed, results are presented from a series of μGC × μGC–μOFRR analyses of three VOC mixtures under different isothermal conditions. The factors affecting the responses from the μOFRR sensor are explored. The inherent tradeoff between resolution and sensitivity attributable to the volatility of the analytes is highlighted, and it is shown that adequately rapid responses are achievable for most analytes. The prospects of using μOFRRs and μOFRR arrays in portable μGC × μGC instrumentation are considered.
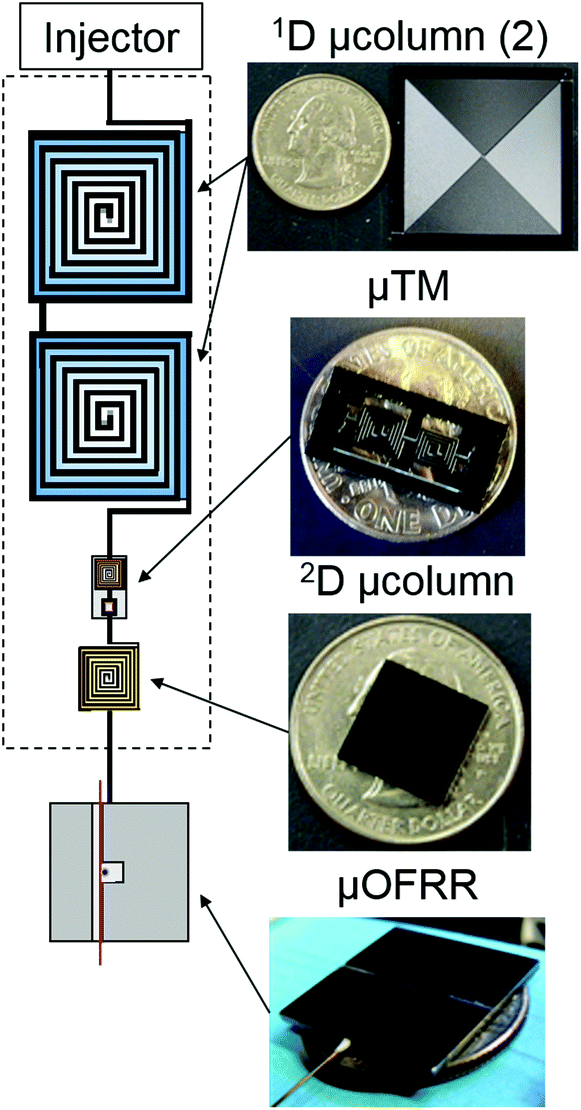 | ||
| Fig. 1 Illustration depicting the four separate microcomponents of the μGC × μGC–μOFRR sub system and their interconnection. Photographs to the right show the μcolumns and μOFRR with US quarters for scale, and the μTM with a US dime for scale. | ||
Experimental methods
Materials
The test compounds 1,4-dioxane (DOX), 4-methyl-2-pentanone (PON), toluene (TOL), cyclopentanone (CPN), hexanal (HAL), n-heptane (C7), n-octane (C8), n-nonane (C9), n-decane (C10), ethylbenzene (ETB), m-xylene (XYL), and cumene (CUM) as well as all other solvents used were >98% pure (Sigma-Aldrich, Milwaukee, WI) and used without further purification. The PDMS (OV-1) and poly(trifluoropropylmethyl)siloxane (PTFPMS, OV-215) polymers used as stationary phases or sensor coatings were obtained from Ohio Valley Specialty Chemicals (Marietta, OH).Device descriptions and preparations
The μTM fabrication, mounting configuration, and operation have been described previously.16–19 Briefly, the Si chip (1.3 × 0.6 cm; Fig. 1) contains a Pyrex-sealed DRIE-Si μchannel (250 × 140 μm cross section) arranged in two thermally isolated convolved square-spiral segments, 4.2 cm (upstream) and 2.8 cm (downstream) long, separated by a 1.0 mm segment. Each stage, as well as each rim, has a Ti/Pt meander-line heater patterned on the Pyrex channel cap. RTDs are patterned in close proximity to the heaters to measure the temperature of each location. Two nominally identical μTM devices were used in the course of this study.Fluidic connections between the μTM and upstream/downstream μcolumns were made through ∼5 cm sections of deactivated fused silica capillary (250 μm i.d., upstream; 100 μm i.d., downstream) inserted into expansion ports on the chip and sealed with epoxy (Hysol 1C, Rocky Hill, CT). The device was wire-bonded, heater side up, to a custom printed circuit board (PCB) Two small Si spacer chips were positioned under the heaters and held in place with photoresist. The assembly was inverted and then carefully placed on two additional Si chips positioned on the top surface of the TEC, with thermal grease ensuring thermal contact. A plastic enclosure was then secured around the μTM through which a blanketing stream of dry air was passed during operation to prevent atmospheric water condensation on the device.
Each μcolumn consisted of a DRIE-Si convolved square spiral channel with an anodically bonded Pyrex cap, the basic design and fabrication of which have also been described previously.25–27 The 1D separation stage assembled for this study consisted of two 3 m-long, series-coupled μcolumns (3.1 × 3.1 cm chips, 250 × 140 μm channel cross-section) wall-coated with a PDMS stationary phase (Fig. 1). The 2D separation stage consisted of a single 0.5 m-long μcolumn (1.2 × 1.2 cm chip, 46 × 150 μm cross-section) wall-coated with OV-215 (Fig. 1). Fluidic connections to the μTM were made through ∼5 cm segments of fused silica capillary (250 μm i.d. for 3 m μcolumns, 100 μm i.d. for 0.5 m μcolumns) epoxied into expansion ports in the Si chips, and attached through fused silica press-fit connectors.
The μOFRR structure and fabrication have been described in detail.23,28 The μOFRR cylinder is 250 μm i.d. and has a 1.2 μm thick SiOx wall. The internal cavity of the cylinder extends completely through the center of the 2 × 2 cm, 520 μm thick Si chip. The μOFRR resonator protrudes vertically 80 μm from an annular trench etched into the substrate and has a 30 μm tall toroidal expansion region at the midsection, with a maximum diameter ≅300 μm. Backside DRIE was used to create both a tapered expansion port along the underside of the chip for capillary insertion, and a narrower microfluidic channel connecting the capillary port and the μOFRR inlet aperture. A final front-side DRIE step created an optical-fiber alignment channel running laterally across the surface tangential to the μOFRR cylinder.23
A PDMS stationary phase was deposited and cross-linked separately on the inner walls of the 1D μcolumns and the μTM by known methods,16,27 producing estimated PDMS film thicknesses of 0.20 and 0.30 μm, respectively. A 0.08 μm thick film of OV-215 was deposited on the wall of the 2D μcolumn and cross linked by the same methods, following pretreatment with (3,3,3-trifluoropropyl)methylcyclotrisiloxane to promote adhesion by the OV-215.19 To coat the inner wall of the μOFRR, the resonator cavity was filled with a toluene solution of PDMS and the solvent was evaporated by placing the device in a vacuum chamber for 10 min. The PDMS film thickness was estimated from the solution concentration to be ∼0.3 μm assuming uniform deposition on the cavity. Following PDMS deposition, the backside fluidic channel was sealed with a 2 × 2 cm Pyrex coverplate using UV curable glue (NOA 81, Norland Optical, Cranbury, NJ). A short section of fused-silica capillary (250 μm i.d.) was then inserted into the tapered expansion port and sealed with epoxy to provide fluidic connection to the upstream μcolumns.
System integration
The two 3 m 1D μcolumns were bonded to individual carrier PCBs with epoxy and connected using a press-fit union. A polyimide thin-metal-film heater pad (Omega Engineering, Inc., Stamford, CT) was affixed to the 2D μcolumn with thermal grease and polyimide tape, with a fine-wire thermocouple inserted between them to monitor temperature. The μTM was connected between the 1D and 2D μcolumns using press-fit unions.The μGC × μGC subsystem was placed inside the oven of a bench scale GC (Agilent 6890, Agilent Technologies, Palo Alto, CA). The temperature of the oven determined the temperature of the 1D μcolumns as well as the ambient of the TEC. The temperature of the 2D μcolumn was further controlled by the heater pad and was set higher than that of the oven. The outlet capillary of the 2D μcolumn was fed through the wall of the oven and connected to the μOFRR or connected directly to the FID with a press-fit union to generate reference chromatograms under the same conditions as used with the μOFRR. The FID is considered to have no dead volume and to provide virtually instantaneous responses to eluting analytes.
An optical fiber (SMF-28, Corning Inc., Corning, NY) was drawn over a hydrogen flame and a 1.4 cm segment was tapered down to an outer diameter of ∼1 μm. The fiber was positioned in the on-chip alignment channel using a Vernier micrometer such that the thinnest part of the fiber contacted the expanded section of the μOFRR. The fiber was secured in place using a UV curable adhesive applied on the far left and right sides of the chip. This assembly, as well as a photodiode (InGaAs PIN, Marktech Optoelectronics, Latham, NY) and a fiber splice (Fiberlok II, 3M, Saint Paul, MN), were mounted on the 3D-printed mounting fixture depicted in Fig. 2. One end of the optical fiber terminated at the photodiode and the other was inserted into the fiber splice for easy connection to the external laser. This arrangement provided a stable, robust platform for the sensor and allowed for interconnecting the fluidics without needing to worry about the optics.
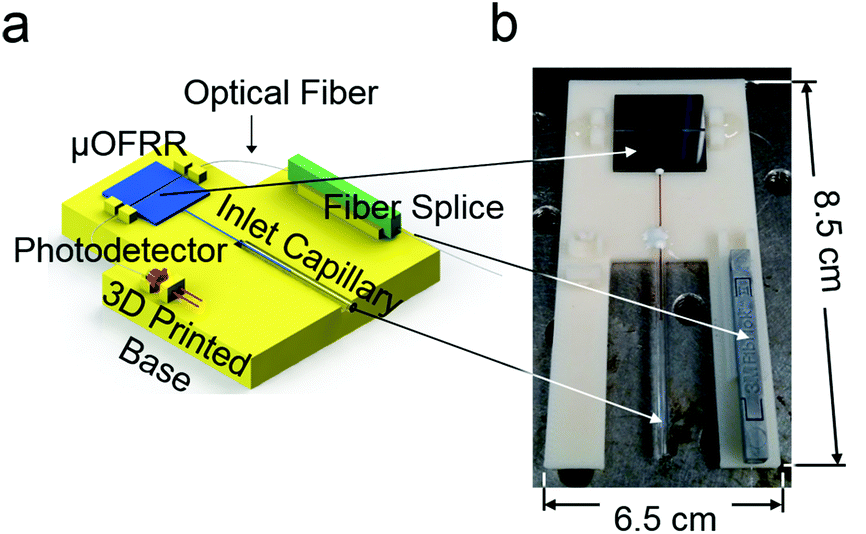 | ||
| Fig. 2 (a) Diagram of the 3-D-printed mounting fixture for the μOFRR sensor, photodetector and fiber splice; (b) photograph of the assembly with the photodetector removed. | ||
The entire μOFRR assembly was placed inside a small custom-made chamber equipped with a thermocouple and resistive heater which was maintained at 25 °C. The optical source was a 1550 nm fiber-coupled laser (CQF939/251, Philips, Amsterdam, NE); both the laser and the photodiode were connected to a DAQ card and controlled by custom-developed LabVIEW software. Two separate μOFRRs were used in the study: after completing the analysis of the n-alkane mixture, an optical fiber broke on the first device and it was replaced with a second, nominally identical device for subsequent tests.
System testing
A test atmosphere of a mixture of C7–C10 vapors was generated in a 10 L FlexFilm® bag (SKC Inc., Eighty Four, PA) pre-filled with N2 into which liquid samples of each mixture component were injected and allowed to evaporate. The injected volumes were ∼40 μL corresponding to nominal vapor concentrations ranging of ∼250 to 1300 parts-per-million (ppm) by volume. Test atmospheres of 7- and 11-component VOC mixtures were generated similarly, but more precisely, for subsequent analyses. The 7-VOC mixture contained 1,4-dioxane, 4-methyl-2-pentanone, toluene, C8, ethylbenzene, 3-heptanone, and C9. The 11-VOC mixture contained the same 7 components in addition to cyclopentanone, hexanal, m-xylene, and cumene. For these test atmospheres, 40.0 μL of each neat liquid was injected, except for cyclopentanone, hexanal, and 3-heptanone, for which 80.0 μL was injected. The resulting concentrations ranged from 550 to 2200 ppm. The VOC air concentrations were verified post-hoc by a single point calibration of each compound with the FID reference detector. For all analyses, samples were drawn by a small diaphragm pump through a 100 μL sample loop via a 6-port valve maintained at 30 °C, and then injected into the 1D μcolumn through a 10 cm segment of deactivated fused-silica capillary for (modulated) separation and detection.The μTM was operated as described previously;18,19 temperature was modulated between a minimum, Tmin, of about −20 °C and a maximum, Tmax, of 180 °C, with a 500 ms offset between heating of the first and second stages. A modulation period, Pm, of 7 s was used for the n-alkane tests and a Pm of 5 s was used for the other vapor mixtures. The longer Pm was used in an effort to reach a lower Tmin by increasing the μTM cooling time. The shorter Pm was used to increase the modulation rate.
A custom Visual C# program was used to control the timing of the applied voltages and to read the temperature sensors of the μTM via a DAQ card (NI USB-6212, National Instruments, Austin, TX). For the μOFRR, the laser was swept over a wavelength range of 330 pm at a rate between 26 and 56 hertz, while the output of the photodiode was monitored. Resonant wavelength was defined as the wavelength at the output minimum and was calculated and recorded in real time by a peak finding algorithm in the LabVIEW software. OriginPro 9.1 (OriginLab, Northampton, MA) and GC Image (Rev 2.2, Zoex, Houston, TX) were used for chromatographic data processing and display of 2-D chromatograms, respectively. The FID was operated at 250 °C with a data sampling rate of 200 Hz. Chromatographic data were collected by ChemStation software (Rev. B.01.01, Agilent Technologies, Santa Clara, CA).
Results and discussion
Alkane mixture
The raw μGC × μGC–μOFRR chromatogram showing the isothermal separation and detection of C7–C10 is presented in Fig. 3. The total elution time was ∼25 min due to the low column temperatures and low flow rate. In all cases, vapor exposure resulted in λWGM shifting to longer wavelengths, which indicates an increase in the effective RI of the PDMS film. Since the difference between any of the n-alkane RI values (Table 1) and that of the PDMS (n = 1.404) is small, and C7 and C8 have RI values lower than that of PDMS, evidently film swelling dominates the net responses. This follows from the nominal PDMS film thickness of 300 nm being much less than the penetration depth of the evanescent field of the 1550 nm WGM. In this so-called “thin-film” regime,23,29 any polymer swelling would increase the fraction of the probed interior volume occupied by the polymer. The observation of reversible red shifts λWGM is consistent with previous reports on polymer-coated (μ)OFRR sensors.23,24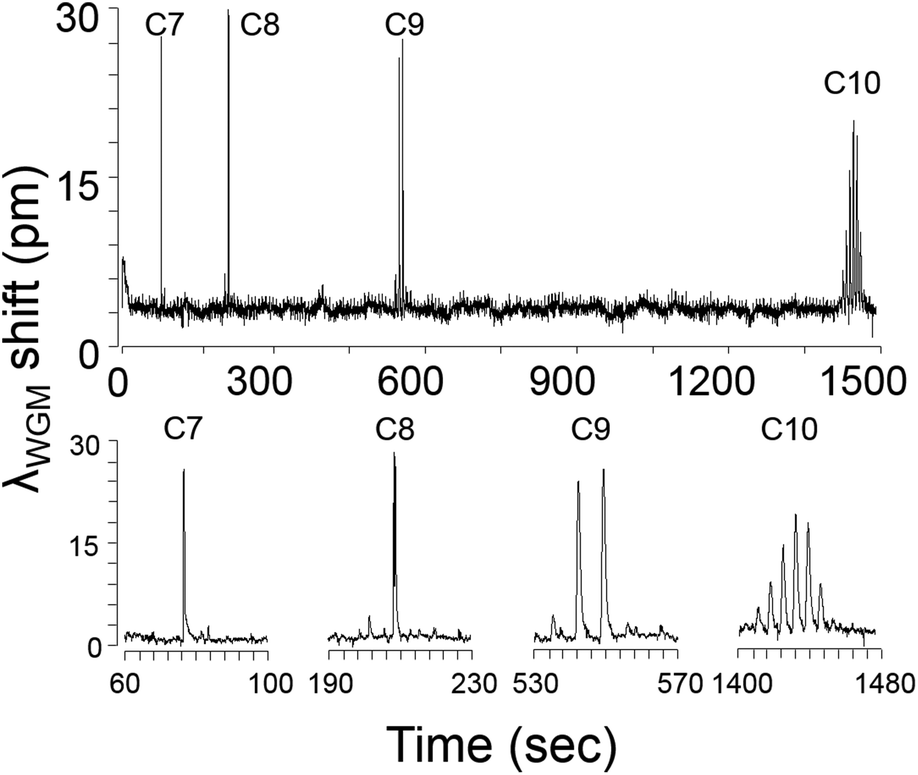 | ||
| Fig. 3 Raw μGC × μGC–μOFRR chromatogram of C7–C10. Enlarged views of the modulated peaks for each analyte are shown beneath the full trace. Conditions: 1D μcolumns (oven), 30 °C; 2D μcolumn, 50 °C; μOFRR, 25 °C; Pm, 7 s; He carrier gas, 1.5 mL min−1. | ||
The modulation number, MN, is the number of modulations per peak, and it is one variable affected by the operating conditions of any μGC × μGC separation. It is primarily a function of the width of the peak eluting from the 1D μcolumn and the selected Pm value, but can also be affected by the detector response speed. Early eluting peaks are invariably narrower and hence have lower MN values. For effective μGC × μGC analyses it is generally recommended to adjust conditions to get MN values of 3–4 for as many peaks as possible.30 Higher MN values provide diminishing returns, and temperature programming is typically used to decrease the retention time (tR) and peak width of less-volatile mixture components. The MN values for the n-alkanes increased from 1 for C7, to 6 for C10 (see enlarged traces in Fig. 3). Peak shapes were relatively symmetric, though some tailing was evident in all cases. For C10, the baseline was barely recovered between successive modulated peaks.
Table 1 presents the values of the full-width-at-half-maximum (fwhm) of the largest modulated peak for each alkane. This variable is a function of the efficiency of remobilization from the μTM, the retention time on the 2D μcolumn, and the kinetics of sorption and desorption into and out of the PDMS interface film in the μOFRR. All of these factors are affected by the vapor pressure (pv) of each analyte, primarily through its influence on the desorption rates from the PDMS films in the μTM and the μOFRR, and to a lesser extent through its contribution to chromatographic band broadening in the (polar) 2D μcolumn. Consistent with the expected trend, the fwhm values increased from 340 ms for C7, to 2000 ms for C10.
A rough estimate of the sensitivity of the μOFRR to each alkane was determined by summing the areas of all modulated peaks (in pm s) and dividing by the injected mass (in ng). The latter was taken as the product of the test atmosphere concentration and the sample loop volume, but since the volumes of injected compounds used to establish the test atmosphere were not carefully measured, and there was no independent verification of the resulting air concentrations, we present only relative values here. The relative sensitivities increased from C7 to C10, with ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:5.6:13, respectively, in fairly good agreement the corresponding ratios of partition coefficients in PDMS among these alkanes reported in the literature.31,32
:2.5:5.6:13, respectively, in fairly good agreement the corresponding ratios of partition coefficients in PDMS among these alkanes reported in the literature.31,32
These results illustrate a phenomenon common to VOC sensors relying on reversible physisorption: peak width and sensitivity both increase with decreasing analyte pv value. Since the resolution between two peaks is inversely proportional to the average peak width, there is an inherent tradeoff between peak-area sensitivity and chromatographic resolution.1
VOC mixtures
Fig. 4 shows the raw μGC × μGC chromatograms with the μOFRR and the FID for the 7-VOC mixture comprising compounds from several different functional group classes (see Fig. 4 caption for operating conditions). Compounds 1–3 had MN values of 1 with both detectors, while for compounds 4–7 the second modulated peak is more apparent with the FID than with the μOFRR. This is due to differences in detector sensitivity and response speed: the faster, more sensitive FID captured the smaller modulated peaks in the two cases where they were not apparent from the μOFRR trace. Note that peak 1 (1,4-dioxane) in the FID trace suffered from breakthrough in the modulator and, therefore, appears broad and truncated, whereas for the μOFRR run it was captured and remobilized efficiently. As shown, the tR values aligned precisely between the two runs with the two detectors. This, notwithstanding the differences in relative magnitudes of the pair of peaks for those compounds with MN = 2, separated by the 5 s modulation period, that occurred because of slight differences in the onset of μTM heating relative to the elution of a peak from the 1D μcolumn.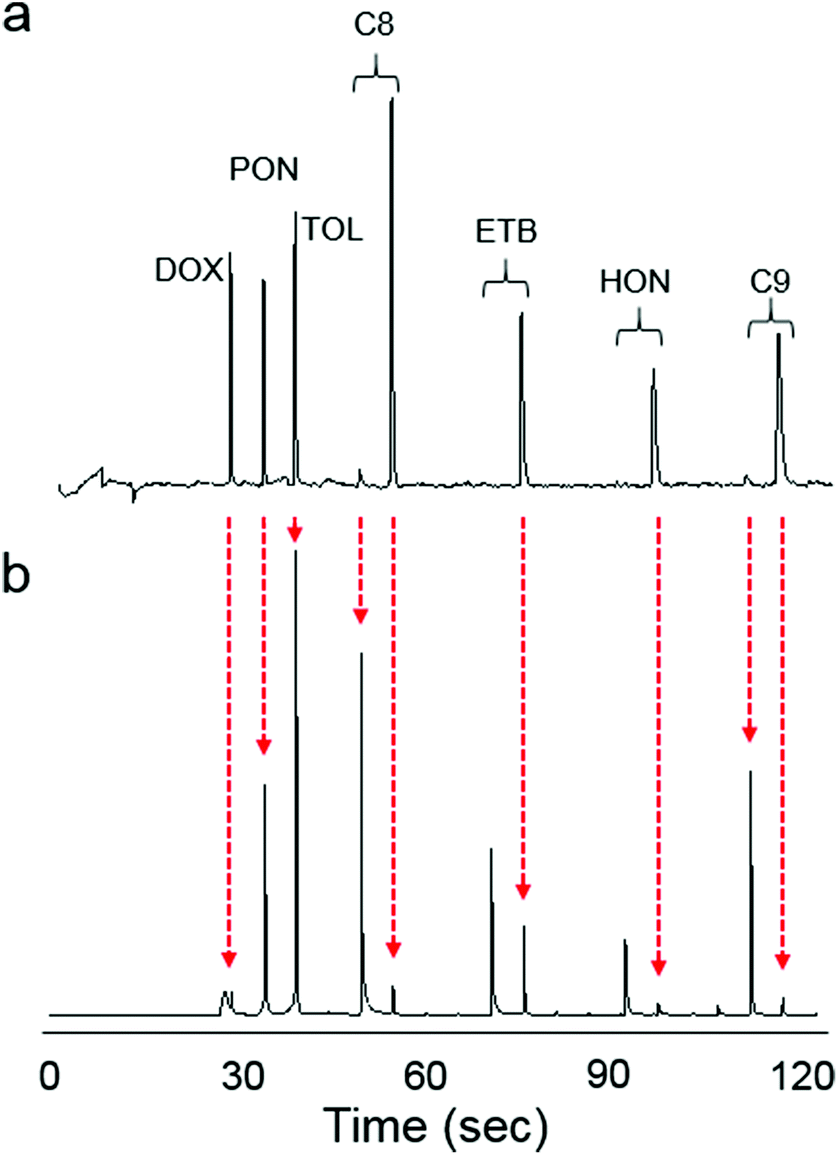 | ||
| Fig. 4 Raw chromatograms of the 7-VOC mixture with (a) μGC × μGC–μOFRR and (b) μGC × μGC–FID. Vertical, dashed red arrows show the time registration of the corresponding peaks between the two runs. Conditions: 1D μcolumns, 50 °C; 2D μcolumn, 80 °C; μOFRR, 25 °C; Pm, 5 s; He carrier gas, 2.5 mL min−1. | ||
Fig. 5a shows the inverse proportionality between pv and fwhm for the 7-VOC mixture with both the μOFRR and the reference FID. All fwhm values from the μOFFR were larger than the corresponding fwhm values with the FID, and the slope of the line for the μOFRR in Fig. 5a is ∼5.5 times larger than that for the FID. The (shallower) slope of the FID curve reflects the influence of upstream (i.e., non-detector) factors on the peak width. Specific values of pv, fwhm, and the fwhm ratios are listed in Table 2. The trends in fwhm values with the μOFRR are consistent with those observed for the n-alkanes in Table 1.
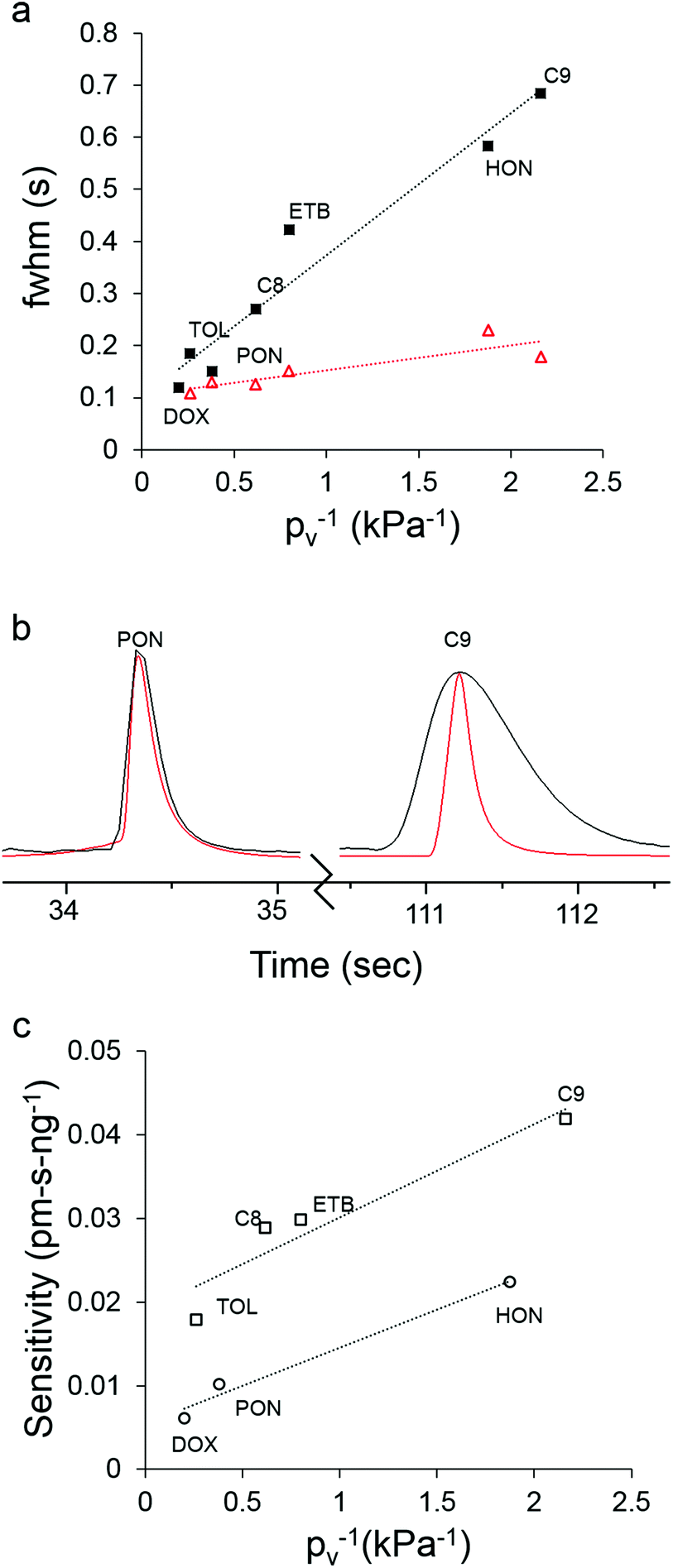 | ||
| Fig. 5 (a) Plot of analyte pv−1vs. fwhm of the largest modulated peak for the 7-VOC mixture with the μOFRR (filled squares) and FID (unfilled triangles), and the corresponding best-fit regression lines (note: the 1,4-dioxane peak is missing from the FID data due to μTM breakthrough); (b) superimposed chromatograms from the μOFRR (black) and FID (red) for 4-methyl-2-pentanone (left, pv = 2.63 kPa) and C9 (right; pv = 0.46 kPa); (c) plot of analyte pv−1vs. peak-area sensitivity (sum of all modulated peaks) for the 7-VOC mixture with the μOFRR, and the corresponding best-fit regression lines for the polar (circles) and non-polar (squares) compounds. For conditions, see Fig. 4. | ||
| Compound | RIc |
p
vd (kPa) |
7-VOC mixturea | 11-VOC mixtureb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| fwhm (s) | fwhm (s) | Sensitivity (pm s ng−1) | |||||||||
| μOFRR | FID | Ratio | Sensitivity (pm s ng−1) | LODe (ng) | μOFRR | FID | Ratio | ||||
| a He flow rate = 2.5 mL min−1. b He flow rate = 1.5 mL min−1. c @ 25 °C, ref. 33. d @ 25 °C, ref. 34. e LOD calculated as 3 × (mass injected)/(signal-to-noise ratio) of tallest modulated peak. f μTM breakthrough. g Data not collected. | |||||||||||
| 1,4-Dioxane | 1.422 | 4.97 | 0.12 | naf | na | 0.006 | 15 | 0.22 | 0.17 | 1.3 | 0.007 |
| Toluene | 1.494 | 3.84 | 0.19 | 0.11 | 1.7 | 0.018 | 8 | 0.32 | 0.15 | 2.1 | 0.021 |
| 4-Methyl-2-pentanone | 1.400 | 2.63 | 0.15 | 0.13 | 1.2 | 0.010 | 12 | 0.60 | na | na | 0.010 |
| n-Octane | 1.394 | 1.62 | 0.27 | 0.13 | 2.1 | 0.029 | 7 | 0.48 | 0.16 | 3.0 | 0.027 |
| Cyclopentanone | 1.437 | 1.50 | —g | — | — | — | — | 0.49 | 0.34 | 1.4 | 0.008 |
| Ethylbenzene | 1.493 | 1.25 | 0.42 | 0.15 | 2.8 | 0.030 | 11 | 0.79 | 0.22 | 3.6 | 0.037 |
| Hexanal | 1.404 | 1.20 | — | — | — | — | — | 0.45 | 0.31 | 1.5 | 0.003 |
| m-Xylene | 1.494 | 1.11 | — | — | — | — | — | 0.78 | 0.25 | 3.1 | 0.030 |
| Cumene | 1.491 | 0.60 | — | — | — | — | — | 1.30 | 0.33 | 3.9 | 0.040 |
| 3-Heptanone | 1.406 | 0.53 | 0.59 | 0.23 | 2.6 | 0.022 | 19 | 1.01 | 0.51 | 2.0 | 0.017 |
| n-Nonane | 1.406 | 0.46 | 0.69 | 0.18 | 3.8 | 0.042 | 16 | 1.24 | 0.24 | 5.2 | 0.041 |
In Fig. 5b, the largest modulated peaks from the μOFRR and FID are superimposed for 4-methyl-2-pentanone and C9. The ordinate scales were adjusted to so that the two peak heights matched (note: the fwhm is independent of the magnitude of the peak, as long as the peak shape is approximately Gaussian). For the more volatile 4-methyl-2-pentanone (pv = 2.63 kPa) the fwhm value of the μOFRR peak was 150 ms, just 15% larger than the 130 ms fwhm value of the FID peak. For the less volatile C9 (pv = 0.46 kPa), the fwhm of the μOFRR peak was 690 ms, nearly 4 times larger than the 180 ms fwhm of the FID peak. These data depict quite clearly the extent to which analyte volatility affects the response speed of the μOFRR. The smallest fwhm value observed with the μOFRR was 120 ms, for 1,4-dioxane. Unfortunately, as noted above, this compound did not yield a Gaussian peak with the FID so no comparison could be made. Regardless, these data demonstrate that the μOFRR is capable of resolving very narrow peaks for compounds of relatively high volatility.
Fig. 5c shows the inverse proportionality between pv and peak-area sensitivity for the 7-VOC mixture with the μOFRR. The systematic differences in sensitivities between the non-polar and polar subsets can be ascribed to differences in vapor-PDMS affinity (i.e., partition coefficient). Of course, peak-height sensitivity, from which limits of detection (LOD) are derived, is generally increased significantly by thermal modulation; but small shifts in the timing of the modulation relative to the elution of the peak can lead to large changes in the distribution of heights among the modulated peaks for a given analyte. This reduces the reliability of LOD estimates when using manual initiation of injections and modulator heating as we did in this study. Regardless, LODs were calculated on the basis of the responses obtained, just to get rough estimates of detectability. These ranged from 7 ng (C8) to 15 ng (C9) for the nonpolar compounds and 12 ng (4-methyl-2-pentanone) to 18 ng (3-heptanone) for the polar compounds. Thus, sensitive detection is easily achievable using the μGC × μGC–μOFRR.
The 11-VOC mixture analyses were performed under the same conditions as the 7-VOC analyses, with the exception that the He carrier gas flow rate was decreased to 1.5 mL min−1 to increase the time spent by the analytes on the 2D μcolumn. The values of fwhm and sensitivity for each compound are presented in Table 2, for comparison with the corresponding values measured with the 7-VOC set at the higher flow rate. Sensitivities were quite similar for the compounds common to both data sets, whereas fwhm values for the 11-VOC set were approximately twice those for the 7-VOC set, and the μOFRR:FID fwhm ratios were also larger, both because of the lower flow rate. Interestingly, the fwhm ratios for the polar analytes were consistently lower than those of the non-polar analytes of similar vapor pressure; undoubtedly due to the lower extent of partitioning of the former into the PDMS interface film. Nonetheless, all 11 compounds were well resolved and eluted in ∼3 min.
The 2-D contour plots in Fig. 6a and b, generated from the 11-VOC separations with the μOFRR and FID, respectively, show that the two detectors yielded comparable performance. Several of the peak contours from the μOFRR are broader along the y axis, reflecting the larger fwhm values from that detector, and the μOFRR contours from several of the later eluting compounds are narrower along the x axis, reflecting the smaller MN values. Features appearing on the far left side of Fig. 6a are artifacts from the initial temperature stabilization of the laser source, which did not affect the analysis. The small peak to the right of 4-methyl-2-pentanone in both plots was traced to a residual impurity in the bag used to prepare the test atmosphere. Both plots show the expected longer 2D tR values for the polar compounds, as well as reasonably good use of the available chromatographic space. Notably, the 2D separation markedly improved the resolution of the cluster of peaks with 1D tR values in the range of 50–65 s, many of which would otherwise partially overlap (i.e., with only a 1D separation).
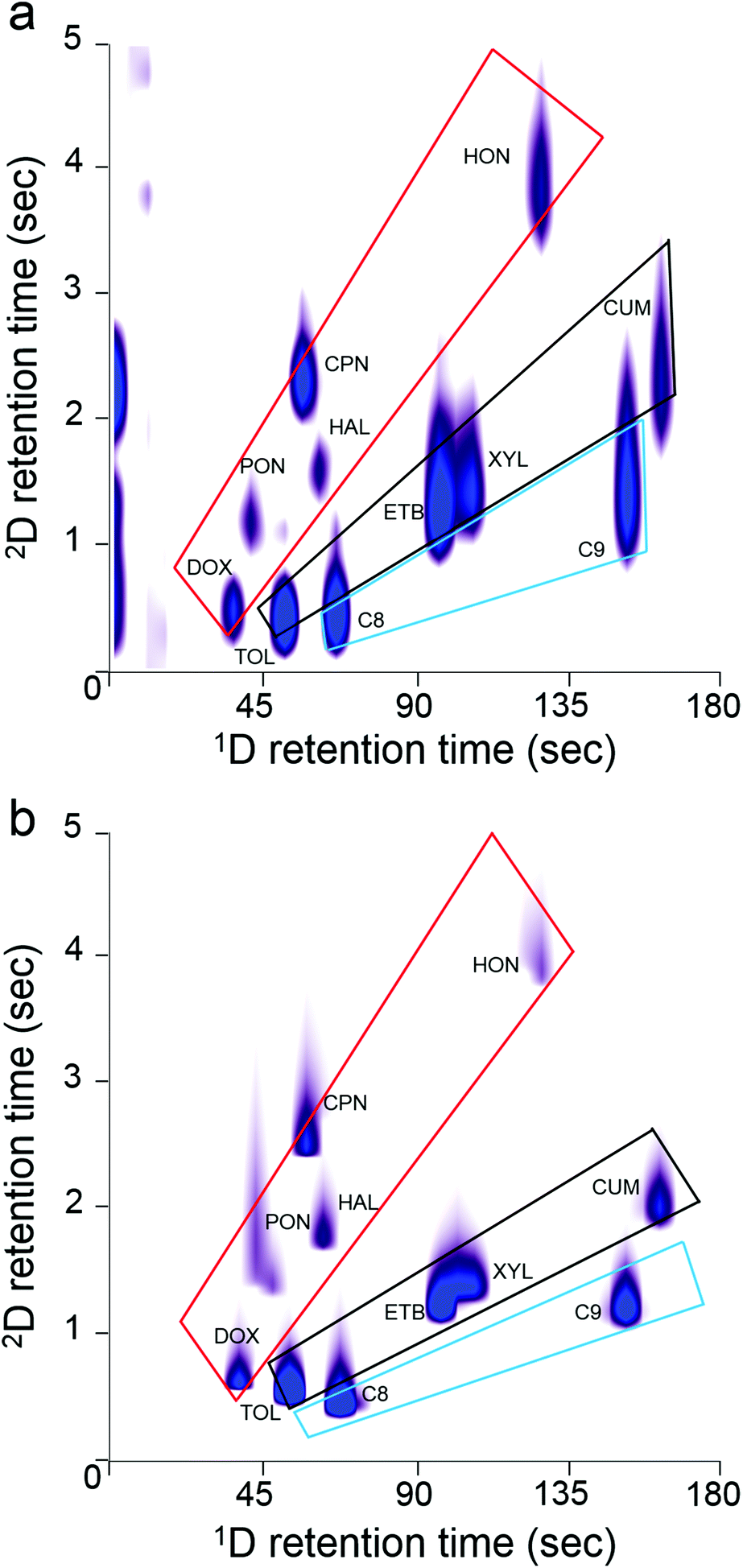 | ||
| Fig. 6 2-D contour plots of the 11-VOC mixture with (a) μOFRR detection and (b) FID. Overlayed boxes are visual guides to the structure of each chromatogram: alkanes (blue), aromatics (black), and oxygenates (red) occupy the segregated zones indicated. Conditions: 1D μcolumns, 50 °C; 2D μcolumn, 80 °C; Pm, 5 s; He carrier gas, 1.5 mL min−1. | ||
One hallmark of GC × GC is the “structure” of the contour plot, in which the peaks from members of different functional group classes align along segregated zones. Given the simplicity of the 11-VOC mixture, there are only three such zones, one each for alkanes, aromatics, and “oxygenates” (i.e., ketones, aldehydes, ethers). As shown, both plots exhibit similar structural zones, but the boundaries are a bit sharper with the FID, due to higher degree of resolution afforded by this detector. Still, the μOFRR plot retains all of the key aspects of the FID plot. (Note: in both plots the 2D tR values for C10 and cumene are shorter than expected due to a common phenomenon called “wraparound”, which occurs when analyte tR values are longer than the Pm and they elute, not during the modulation period in which they should, but in the next period. Thus, although the C10 and cumene 2D tR values appear to be between 1 and 3 s, they are actually between 6 and 9 s).
Conclusions
From the results of this preliminary study, we conclude that the PDMS-coated μOFRR can, indeed, serve as an effective detector for μGC × μGC, and that the thermally modulated μGC × μGC–μOFRR represents a promising new technology for analyzing airborne VOC mixtures. This is the first instance of comprehensive two-dimensional gas chromatographic analysis using a subsystem in which all core analytical components were microfabricated.Perhaps the most prominent finding from this study was the critical dependence of the μOFRR response time on the analyte pv value, through its influence on the rate of desorption of a vapor from the polymer interface film on the μOFRR cylinder. This is a feature common to all VOC sensors employing sorptive interfaces, but it takes on more significance with μGC × μGC because of the narrowness of the modulated peaks that need to be resolved. For the most volatile VOCs tested here the fwhm values of the μOFRR peaks were comparable to those of an (ideal) FID, but for the least volatile compounds tested here they were several-fold larger.
Although the μOFRR peak widths were sufficiently narrow to permit effective separations, their dependence on pv−1 represents a potentially limiting factor for this application. Using thinner polymer films or operating a slightly elevated temperature would reduce this problem, but both would be accompanied by losses in sensitivity. Ramping the temperature of the μOFRR over the course of an analysis would be a better solution, and its feasibility to address this issue is currently being explored.
The LODs we estimated from the response data were in the low-ng range, indicating a useful level of detectability among all analytes tested; however, the use of manual coordination of injection and modulation functions rendered the LODs quite variable. We believe this can be easily addressed by automatically synchronizing injection and modulation triggers and thereby generating more reproducible modulated-peak intensity profiles.
We are currently working on incorporating a micro-preconcentrator/focuser to complete the microsystem, and to permit autonomous air monitoring in the field. The integration of the μOFRR with embedded optical fiber waveguide and miniaturized ancillary components, demonstrated here, constitutes an enabling step toward such a fieldable unit. Although scrubbed ambient air could be used as the carrier gas with this microsystem,1,3,5,23 the inevitable loss of chromatographic efficiency incurred at the relatively high separation flow rates employed here argues strongly for retaining He as the carrier gas. This option is facilitated by the availability of small He canisters and regulators. On-going efforts are being directed toward the use of nanoparticle interface films instead of polymer films,35 and the development of μOFRR arrays that can provide response patterns for analyte identification.
Acknowledgements
The authors gratefully acknowledge funding provided by Grant ECCS 1307154 and Grant ECCS 1128157 from the National Science Foundation (NSF). Additional funding was provided by a grant from Agilent Technologies. Devices were fabricated in the Lurie Nanofabrication Facility, a member of the National Nanotechnology Infrastructure Network, which is supported by the NSF.References
- C.-J. Lu, W. H. Steinecker, W.-C. Tian, M. C. Oborny, J. M. Nichols, M. Agah, J. A. Potkay, H. K. Chan, J. Driscoll and R. D. Sacks, Lab Chip, 2005, 5, 1123–1131 RSC.
- P. R. Lewis, R. P. Manginell, D. R. Adkins, R. J. Kottenstette, D. R. Wheeler, S. S. Sokolowski, D. E. Trudell, J. E. Byrnes, M. Okandan and J. M. Bauer, IEEE Sens. J., 2006, 6, 784–795 CrossRef.
- S. K. Kim, H. Chang and E. T. Zellers, Anal. Chem., 2011, 83, 7198–7206 CrossRef CAS PubMed.
- R. P. Manginell, J. M. Bauer, M. W. Moorman, L. J. Sanchez, J. M. Anderson, J. J. Whiting, D. A. Porter, D. Copic and K. E. Achyuthan, Sensors, 2011, 11, 6517–6532 CrossRef CAS PubMed.
- W. R. Collin, G. Serrano, L. K. Wright, H. Chang, N. Nuñovero and E. T. Zellers, Anal. Chem., 2013, 86, 655–663 CrossRef PubMed.
- R.-S. Jian, Y.-S. Huang, S.-L. Lai, L.-Y. Sung and C.-J. Lu, Microchem. J., 2013, 108, 161–167 CrossRef CAS.
- L. K. Wright and E. T. Zellers, Analyst, 2013, 138, 6860 RSC.
- Y. Qin and Y. Gianchandani, J. Microelectromech. Syst., 2014, 23, 980–990 CrossRef.
- A. Garg, M. Akbar, E. Vejerano, S. Narayanan, L. Nazhandali, L. Marr and M. Agah, Sens. Actuators, B, 2014, 212, 145–154 CrossRef.
- H. Zhu, R. Nidetz, M. Zhou, J. Lee, S. Buggaveeti, K. Kurabayashi and X. Fan, Lab Chip, 2015, 15, 3021–3029 RSC.
- C. Jin, P. Kurzawski, A. Hierlemann and E. T. Zellers, Anal. Chem., 2008, 80, 227–236 CrossRef CAS PubMed.
- T. Górecki, J. Harynuk and O. Panić, J. Sep. Sci., 2004, 27, 359–379 CrossRef.
- J. Dallüge, J. Beens and U. A. T. Brinkman, J. Chromatogr., A, 2003, 1000, 69–108 CrossRef.
- J. B. Phillips, R. B. Gaines, J. Blomberg, F. W. van der Wielen, J. M. Dimandja, V. Green, J. Granger, D. Patterson, L. Racovalis and H. J. de Geus, J. High Resolut. Chromatogr., 1999, 22, 3–10 CrossRef CAS.
- M. Libardoni, C. Fix, J. H. Waite and R. Sacks, Anal. Methods, 2010, 2, 936–943 RSC.
- S.-J. Kim, S. M. Reidy, B. P. Block, K. D. Wise, E. T. Zellers and K. Kurabayashi, Lab Chip, 2010, 10, 1647–1654 RSC.
- S.-J. Kim, G. Serrano, K. D. Wise, K. Kurabayashi and E. T. Zellers, Anal. Chem., 2011, 83, 5556–5562 CrossRef CAS PubMed.
- G. Serrano, D. Paul, S.-J. Kim, K. Kurabayashi and E. T. Zellers, Anal. Chem., 2012, 84, 6973–6980 CrossRef CAS PubMed.
- W. R. Collin, A. Bondy, D. Paul, K. Kurabayashi and E. T. Zellers, Anal. Chem., 2015, 87, 1630–1637 CrossRef CAS PubMed.
- J. J. Whiting, C. S. Fix, J. M. Anderson, A. W. Staton, R. P. Manginell, D. R. Wheeler, E. B. Myers, M. L. Roukes and R. Simonson, Proc. Solid-State Sensors, Actuators and Microsystems Conference, 2009, 1666–1669, Jun. 21–25, Denver, CO Search PubMed.
- B.-X. Chen, T.-Y. Hung, R.-S. Jian and C.-J. Lu, Lab Chip, 2013, 13, 1333 RSC.
- J. Liu, J.-H. Seo, Y. Li, D. Chen, K. Kurabayashi and X. Fan, Lab Chip, 2013, 13, 818–825 RSC.
- K. Scholten, X. Fan and E. T. Zellers, Lab Chip, 2014, 14, 3873–3880 RSC.
- S. I. Shopova, I. M. White, Y. Sun, H. Zhu, X. Fan, G. Frye-Mason, A. Thompson and S.-J. Ja, Anal. Chem., 2008, 80, 2232–2238 CrossRef CAS PubMed.
- M. Agah, J. A. Potkay, G. Lambertus, R. Sacks and K. D. Wise, J. Microelectromech. Syst., 2005, 14, 1039–1050 CrossRef.
- G. Lambertus, A. Elstro, K. Sensenig, J. Potkay, M. Agah, S. Scheuering, K. Wise, F. Dorman and R. Sacks, Anal. Chem., 2004, 76, 2629–2637 CrossRef CAS PubMed.
- S. Reidy, G. Lambertus, J. Reece and R. Sacks, Anal. Chem., 2006, 78, 2623–2630 CrossRef CAS PubMed.
- K. Scholten, X. Fan and E. T. Zellers, Appl. Phys. Lett., 2011, 99(14), 141108 CrossRef PubMed.
- A. M. Kummer, A. Hierlemann and H. Baltes, Anal. Chem., 2004, 76, 2470–2477 CrossRef CAS PubMed.
- J. Beens, H. Boelens, R. Tijssen and J. Blomberg, J. High Resolut. Chromatogr., 1998, 21, 47–54 CrossRef CAS.
- P. A. Martos, A. Saraullo and J. Pawliszyn, Anal. Chem., 1997, 69, 402–408 CrossRef CAS PubMed.
- A. Hierlemann, A. J. Ricco, K. Bodenhöfer, A. Dominik and W. Göpel, Anal. Chem., 2000, 72, 3696–3708 CrossRef CAS PubMed.
- G. Baysinger, in CRC Handbook of Chemistry and Physics, National Institute of Standards and Technology, 2014 Search PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, http://webbook.nist.gov/chemistry/, accessed June 15, 2015 Search PubMed.
- K. W. Scholten, W. R. Collin, X. Fan and E. T. Zellers, Nanoscale, 2015, 7, 9282–9289 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |