
Naked-eye nanobiosensor for therapeutic drug monitoring of methotrexate
H.
Yockell-Lelièvre
a,
N.
Bukar
a,
J. L.
Toulouse
a,
J. N.
Pelletier
abc and
J.-F.
Masson
*ad
aDépartement de chimie, Université de Montréal, CP 6128 Succ. Centre-Ville, Montreal, QC, Canada H3C 3J7. E-mail: jf.masson@umontreal.ca; Tel: +1-514-343-7342
bPROTEO,
cCentre for green chemistry and catalysis/Centre de chimie verte et de catalyse (CGCC/CCVC),
dCentre for self-assembled chemical structures (CSACS),
First published on 29th July 2015
Abstract
Sensing of methotrexate at clinically-relevant concentrations was achieved with a plasmon-coupling assay. In this assay, free methotrexate and folic acid Au nanoparticles competed for human dihydrofolate reductase (hDHFR)-functionalized Au nanoparticles (Au NP). The hDHFR-functionalized Au NPs were immobilized on a small glass sensor inserted in a portable 4-channel LSPR reader. This allowed rapid (minutes) and sensitive (nanomolar range) measurement of methotrexate concentration by means of total internal reflection plasmonic spectroscopy. The large bathochromic shifts of the plasmon-coupling assay led to striking colour changes visible to the naked eye for methotrexate at clinically-relevant concentrations. The results demonstrate the potential for therapeutic drug monitoring of a widely used chemotherapy agent, as assessed with the naked eye.
Introduction
Therapeutic drug monitoring (TDM) is essential to ensure safety and efficacy over the course of treatment of many diseases.1 Currently, the majority of TDM is performed in centralized laboratories, using relatively lengthy and complex protocols often requiring specialized, dedicated scientific equipment. Separation techniques coupled to mass spectrometric detection or immunoassays are the current workhorses of clinical biochemistry laboratories for TDM.1 As a result of its high cost, this infrastructure is generally restricted to major healthcare centres, which limits the accessibility of TDM. In addition, certain TDM protocols – such as the standard assay for quantification of methotrexate (MTX) – rely on expensive, proprietary multi-well sample plates, producing a dilemma whereby partially-filled plates are used at increased cost-per-sample, or additional turn-around time is required to wait for samples to fill the plate. Such factors contribute to the underutilisation of TDM. Thus, new TDM methods are required to address these challenges.2Colorimetric test strips are amongst the simplest point-of-care test technologies that have had a tremendous impact on the healthcare of patients worldwide. For example, lateral flow immunoassays on test-strips have led to a series of important applications, the best known of which is the pregnancy test.3–5 These sensors are often based on naked-eye detection, as a simple and effective means to read an analytical test. Often, the colorimetric readout is enabled by the use of gold nanoparticles (Au NPs), which present a strong extinction band in the visual range due to their localized surface plasmon resonance (LSPR) properties.6,7 Au NPs are also involved in many solution-based colorimetric tests because of their ability to drastically change colour upon aggregation.8,9 The striking colour changes can also result from the plasmon coupling that accompanies dimerization of Au NP.10,11 Thus, analyte-controlled plasmon coupling can serve as an effective detection mechanism by measuring the shift in wavelength of the absorption band, or, in the best of cases where a large shift occurs, by directly observing colour changes. The design of dimerization or aggregation assays sensitive for therapeutic drugs could provide simple and effective analytical tools for TDM to routinely ascertain that drug concentrations are within the range for efficient treatment and prevent potential toxicity.
Methotrexate (MTX) is an antimetabolite drug commonly used for the chemotherapeutic treatment of various cancers.12,13 It is an unreactive analogue of folate, thereby inhibiting the dihydrofolate reductase (hDHFR) enzyme necessary for synthesis of DNA precursors and thus cell replication. Adequate dosage of MTX for a cancer patient is of the utmost importance: while being mainly cytotoxic toward cancer cells, MTX also acts upon some healthy tissues, leading to toxicity issues. However, insufficient MTX levels are ineffective in halting cancer progression.14 Since MTX absorption and clearance in an individual are influenced by many factors, including age, gender, metabolism, genetics, disease state and renal function, TDM of MTX is highly valuable for optimising the course of treatment. TDM of MTX allows for monitoring drug metabolism and patient compliance (for home-administered treatments), and to adjust treatment schedule in accordance to individualized factors.
Currently, TDM of MTX concentration in a patient's bloodstream is performed with a dedicated analyser or, less commonly, by HPLC-MS.15 Accessibility using either approach is undermined by high instrument cost and requirement for highly-trained personnel. Designing a more affordable assay for MTX that could be used by frontline healthcare workers could improve patient follow-up during chemotherapy.
Other strategies that have been proposed for MTX sensing include methods based on electrochemistry,16–21 nanoparticle-based optical detection22–24 or bioluminescence.25 We recently introduced a colorimetric test for MTX based on a competitive assay involving folic acid-derived Au NP and human dihydrofolate reductase (hDHFR).23 MTX competed with the folic acid-Au NPs for binding to hDHFR, leading to a high LSPR response (shift of wavelength, or color) at low concentrations of MTX, and the absence of LSPR response at high concentrations of MTX. Due to the relatively small absolute wavelength shift of this LSPR test, a UV-Vis spectrophotometer was required to read-out the assay. To address this shortcoming, we recently introduced a multi-channel and portable SPR instrument to monitor MTX in clinical samples using a similar Au NP competition assay.24 For that assay, hDHFR was bound to a gold film deposited onto the surface of a dove prism. The Debye length of the surface-immobilized sample was adjusted to facilitate the interaction of hDHFR and the folic acid-Au NP.26 The SPR assay was sensitive to clinical concentrations of MTX and correlated well with established techniques.
Depositing Au NPs functionalized with a molecular receptor on a solid support leads to colorimetric sensors. While the previous MTX sensor relied on SPR detection for quantifying MTX, here we present a competitive assay for TDM of MTX in the form of a Au NP sensor that provides colorimetric visualisation. In this assay, a solution of MTX and folic acid-Au NPs enter competition for another set of Au NPs functionalized with hDHFR on the sensor surface (Fig. 1). The experimental and theoretical parameters influencing the performance of the competitive LSPR assay are also reported in this manuscript. MTX can be indirectly measured by colorimetry using this assay, or by the naked eye.
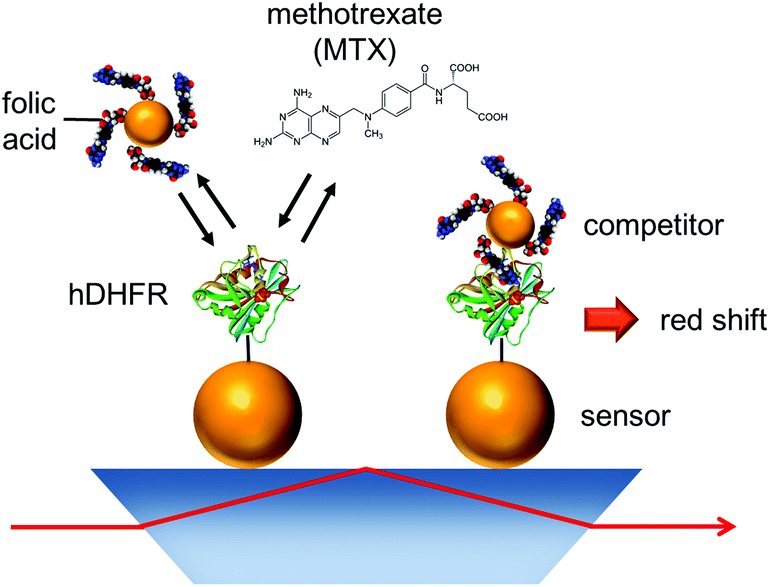 | ||
| Fig. 1 Schematic representation of competitive assay to monitor MTX. | ||
Experimental
Gold nanoparticle synthesis
Ultrapure water (18.2 MΩ cm) was used for all synthesis. All glassware was washed with aqua regia and rinsed with ultrapure water (Caution! Aqua regia is highly corrosive). All chemicals were purchased from Sigma Aldrich. Au NPs were characterized with UV-vis absorption spectroscopy.27Au NPs of 12 nm diameter were synthesized by the microwave-assisted citrate reduction. Sodium citrate dihydrate (0.3 g) and hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O; 40 mg) were dissolved in 1 L of water at room temperature. The solution was then heated in a 1000 W commercial microwave oven at full power for 5 min. After cooling, the suspension was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM for 20 min in order to increase its concentration by about two orders of magnitude.
000 RPM for 20 min in order to increase its concentration by about two orders of magnitude.
Another set of Au NPs with a diameter of 60 nm was synthesized using a seeded growth method.28 The concentrated 12 nm Au NPs solution (1 mL) was mixed with 3 mL of a 200 mM solution of hydroxylamine in 400 mL of water, and 5 mL of a 50 mM solution of HAuCl4·3H2O were added dropwise. After 5 min of stirring, the suspension was concentrated by centrifugation at 15000 RPM for 2 min. A TEM image of the nanospheres is provided in Fig. 2.
 | ||
| Fig. 2 (Top) Calculated electric field surrounding a 60 nm spherical (left) and a 80 nm raspberry-shaped (right) Au NP in water, excited by polarized light at their respective maximum plasmon resonance wavelength. (Bottom) TEM images of the nanospheres (left) and nanoraspberries (right). The scale bar of TEM images is 50 nm. | ||
Nanoraspberries of 80 nm diameter were synthesized using a protocol elaborated by Xie et al.29 To 2.5 mL of a 50 mM solution of HAuCl4·3H2O diluted in 500 mL of water, 25 mL of a 0.1 M solution of HEPES (previously adjusted to pH 7.4) were added and stirred for 1 h at room temperature. The suspension was then concentrated by centrifugation at 15000 RPM for 1 min. To obtain 25 nm Au NPs, the previously synthesized 80 nm raspberry-shaped Au NPs (non-concentrated) were heated for 5 min at full power in a 1000 W commercial microwave oven. The heating induced Ostwald ripening of the particles, producing spherical, 25 nm Au NPs. The suspension was then concentrated by centrifugation at 15000 RPM for 2 min. A TEM image of the nanoraspberries was acquired to confirm the shape and size (Fig. 2).
Preparation of the colorimetric LSPR sensors
Surface capping of the 60 nm spherical and 80 nm raspberry-shaped AuNPs with thiol-terminated polystyrene (PS, Mn = 8000 g mol−1) was conducted via ligand exchange in acetone.30 The samples were purified by repeated rinsing cycles of suspension–centrifugation at 15000 RPM in acetone and dried, then dissolved in chloroform to produce a highly concentrated suspension (∼1013 NP mL−1). A small excess of unbound PS was added to the suspensions to ensure proper spacing between the Au NPs. This excess was necessary to avoid plasmon coupling of neighbouring Au NPs in the sensing layer in absence of the competition assay.
Dove prisms (20 × 12 × 3 mm) were cleaned in hot piranha solution for 90 min (Caution! Piranha solution is highly corrosive). The LSPR sensors were fabricated by spreading and drying a small droplet of the suspension of PS-capped Au NPs onto the surface of a dove prism using a glass pipette. The PS was removed by etching with an oxygen plasma for 2 hours. The surface of the sensors was functionalized overnight with 2 mg mL−1 3-MPA-LHDLHD-OH in DMF.31 The peptide monolayer has been observed to reduce nonspecific adsorption of biomolecules.31–34 The prisms were then cleaned with ethanol and water. The carboxyl groups on the surface were activated for 20 min with an aqueous solution of N-ethyl-N′-(3-dimethylaminopropyl)-carbodiimide (EDC; 39 mg mL−1) and 5 mM N-hydroxysuccinimide (NHS; 14 mg mL−1) and washed with water and PBS pH 4.5. They were then reacted overnight in an aqueous solution of Nα′Nα-bis(carboxymethyl)-L-lysine (225 mg/30 mL water).31 The remaining activated carboxyl groups were deactivated for 5 min with 1 M ethanolamine at pH 8.5. After washing the prisms with water, they were placed in a solution of copper(II) sulfate pentahydrate (1 g per 30 mL) for 2 hours. Cu2+ is chelated by the surface-bound, modified Lys. The prisms were then washed with water and with ethanol, and dried under nitrogen flow. They were stored in the dark until use.
At the time of use, hexa-histidine-tagged human dihydrofolate reductase (hDHFR) was added to the peptide-modified prisms, such that hDHFR bound to the Cu2+ by virtue of its histidine tag. His-tagged hDHFR was heterologously expressed in Escherichia coli, purified and characterized as previously described.24 It was stably stored at −80 °C, and aliquots were thawed on ice for use.
Surface functionalization of the competitor Au nanoparticles
The competitor Au NPs were synthesized by reaction of 50 mL of a 100 μM solution of folic acid in ethanol and 3 mL of concentrated Au NPs (12, 25 or 60 nm) suspension added dropwise under constant stirring. After one hour, the suspension was centrifuged at 15000 RPM for 1 minute and re-suspended in clean ethanol under sonication, and re-centrifuged. This rinsing cycle was repeated three times, and the sample was dried under a flow of nitrogen. The competitor Au NPs were stored in the dark at 4 °C until use. Prior to use, they were re-suspended in PBS, by dipping the eppendorf tube for 5 s in a sonic bath.
LSPR measurements
Colorimetric LSPR-based experiments were performed on a portable 4-channel SPR instrument based on a dove prism.24 The prepared dove prism sensor was inserted in the instrument. A 4-channel PDMS fluidic cell was then mounted in the instrument.24 For all measurements, the solutions were injected in the fluidic cell and reacted under static flow conditions. The instrument allowed the analysis of one sample in triplicate, and a reference. PBS was injected in the reference channel for all steps. The sensor was first conditioned with PBS for 1 min, and 300 μL of a 40 μg mL−1 solution of hDHFR was immobilized on the surface of the colorimetric sensor for 10 min. After a rinsing step with PBS (1 min), solutions containing different concentrations of MTX and a constant concentration of competitor Au NPs were injected in the cell for 10 min followed by a rinsing with PBS. Spectra were collected from 450 to 750 nm (Ocean Optics USB4000 with a grating centred at 600 nm). Data was acquired and processed in real-time with Labview software.Results and discussion
Design and properties of the colorimetric LSPR sensor
The LSPR sensor was based on a total internal reflection instrument analogous to a 4-channel SPR instrument recently developed in our group.24 In this LSPR sensor, the spectrophotometer covered the visible range of 450 to 750 nm, instead of the typical range for SPR of 550 to 850 nm. As a result, the plasmon resonance at 520 nm that is typical of spherical Au NPs could be observed with the LSPR sensor. Another difference consisted in the substrate, consisting to an array of Au NPs coated the surface of the dove prism. In this configuration of LSPR, the evanescent field of the light undergoing total internal reflection excited the plasmon resonance of the Au NPs immobilized to glass surface. This configuration is advantageous for biosensing, as the light never travels in the often highly absorptive or turbid biological samples. In addition, the sequence of chemical reactions is much simpler in a flow cell in comparison to working in solution; fewer centrifugation steps are required and they can be performed on smaller volumes in the fluidic cell. Thus, we selected the 4-channel LSPR sensor for all experiments in this study.The fabrication of the LSPR sensor relied on the formation of a monolayer of Au NPs on the surface of a dove prism. To this effect, the Au NPs were functionalized with a monolayer of polystyrene (Mn = 8000 g mol−1). Excess PS was added in order to increase the spacing between Au NPs on the surface in order to avoid surface coupling between neighbouring Au NPs. When no PS excess was present, the colour on the surface of the prism was blue indicating strong plasmon coupling, while the prisms prepared in presence of an excess PS led to a red coloration characteristic of isolated Au NP (Fig. 3). Plasmon coupling should be exclusive to the competition assay to achieve maximum sensitivity and large colour changes. To verify this hypothesis, the LSPR wavelength on the surface of the dove prism was compared with the resonance wavelength in solution. The plasmon resonance of spherical Au NPs of 60 nm diameter was 550 nm for an aqueous suspension, compared to 558 nm for identical Au NPs immobilized on the dove prism also exposed to water. Similarly, the measured plasmon resonance of 80 nm raspberry-shaped Au NPs was 590 nm both in solution and deposited on the substrate, indicating the absence of plasmon coupling. Thus, the immobilisation of the Au NPs on the dove prism did not significantly induce plasmon coupling.
 | ||
| Fig. 3 Photograph of the dove prisms coated with PS-capped Au NPs, after plasma etching. No PS excess was added to the prism on the left, causing visible plasmon coupling between Au NPs in the surface layer. | ||
The optical properties of Au NPs depend on their size and shape.6,7 The selection of the size and shape of the Au NPs on the surface of the LSPR sensor is thus a determinant of the optical properties of the sensor. A strong and sharp plasmonic band generally leads to superior plasmonic sensing due to better spectral resolution. For spherical Au NPs, the absorbance cross-section is proportional to the particle volume. However, Au NPs with diameters larger than 60 nm show an increased scattering contribution to their extinction coefficient, causing a broadening of their plasmonic band.35 Hence, Au NPs with sizes around 60 nm were selected to optimise the signal of the LSPR sensor.
Both spherical and branched Au NPs (with shapes like stars or raspberries) have previously demonstrated good performances as LSPR sensors36–39 and both were considered for this assay. When compared to spherical particles within the same size range, branched Au NPs present surface asperities that have both red-shifting and tip-confining effects on their plasmon resonance. While the varying length of the surface features can, in some cases, considerably broaden their plasmon band,29 the fairly regular, short protuberances of the raspberries synthesized here do not significantly induce plasmon band broadening and they remain interesting LSPR probe candidates. The sensitivity of LSPR sensors depends on the field penetration depth of the plasmon resonance into the dielectric layer. Theoretical simulations provide an in-depth look at the field distribution for particles of different shapes and sizes. The electric field distribution around nanoparticles (Comsol Multiphysics 4.2) was thus estimated for a 60 nm Au sphere and a 80 nm Au raspberry (Fig. 2). While the enhanced electrical field surrounding the surface protuberances of the raspberry spreads out for about 20 nm, the field surrounding the sphere reaches over 40 nm. In classical direct detection assays, Au NPs with very short field penetration depth are usually very sensitive. Thus, raspberry-shaped Au NPs are predicted to be better suited for assays involving the direct detection than spheres. However, a longer field depth is more appropriate in the case of a competitive assay involving a second Au NP in a dimerization assay. The entire study was then conducted using 60 nm spherical Au NPs as sensors.
Influence of the competitor Au NP size
The distance between the surface-bound Au NPs on the LSPR and the competitor Au NPs suspended in solution greatly affects plasmon coupling and thus, the change in colour that can be observed. According to the “plasmon ruler principle”, the magnitude of the plasmon shift between a pair of spherical metallic particles varies as a function of the third power of the ratio of their mean diameter on the center-to-center distance.40 In the current case of our MTX assay, the interparticle gap was dictated by the size of the molecules bound to the surface of the Au NPs on the LSPR sensor and on the competitor Au NPs. In this MTX assay, the gap is a function of the size of the peptide monolayer and the size of the hDHFR-folate complex, which is estimated as approximately 4 nm. Thus, the binding shift monitored on the sensor-bound Au NPs and the competitor Au NPs should increase with larger competitor Au NP. Hence, a larger size for the competitor is desirable in order to produce a noticeable colour change on the sensor.To this effect, Au NPs of 12, 25 and 60 nm diameter were tested as competitor Au NPs. The concentrations of the different sized competitors were set to give an absorbance of 5 at their maximum plasmon resonance wavelength (the absorbance was measured at 1 for a 5-fold diluted sample). This corresponded to approximately 20, 2.5 and 0.16 nM for 12, 25 and 60 nm Au NPs, respectively (the concentrations reported here are for NPs). These concentrations were selected to maximize the shift in the absence of MTX, hence maximizing the range of the MTX assay and the magnitude of colour change observed in the assay. In the absence of MTX, the plasmon coupling resulted in shifts of 27 nm for Au NPs of 25 nm diameter, which was nearly twice that observed with 12 nm NPs (shift = 15 nm). However, the plasmon coupling shift was much lower for Au NPs of 60 nm diameter (shift = 3 nm).
Increasing the particle diameter from 12 to 25 nm resulted in larger shifts as expected, but the unexpected decrease in shift upon using 60 nm Au NP appears to result from poor colloidal stability, as noted by a strong colour change of the suspension. The folic acid competitor Au NPs must retain colloidal stability in the assay buffer.26 Here, the Au NPs are stabilised in PBS due to the surface charges induced by the presence of negatively-charged folate ions bound on their surface. This charge prevented aggregation of Au NPs caused by NP size-dependant van der Waals attractive forces. We recently reported that the Debye length of folate-capped Au NPs should be less than 1 nm to achieve optimal interaction with the sensor surface-bound hDHFR,26 and thus the solution conditions used here were identical to the ones reported in the previous communication. The Debye length was consistent with the size of the folate ion (0.8 nm).26 While 25 nm Au NP are well stabilized in PBS with a ∼1 nm Debye length, the same parameters will cause 60 nm Au NPs to aggregate over the course of a few minutes, thus thwarting the performance of the assay. The 25 nm Au NPs were therefore selected for the MTX assay, in order to obtain the largest shift and thus, colour change.
The 60 nm Au NPs on the LSPR sensor are sufficiently large to host a few 25 nm competitor Au NPs. It is important to understand the influence of multiple Au NP binding events on individual sensor-bound Au NPs. As large scale aggregation should be observed for Au NP suspensions, and since TEM cannot be performed on the LSPR substrates, simulations were performed with Comsol Multiphysics 4.2 to predict the shift expected when a 60 nm Au sphere is in close proximity to one, two or three 25 nm Au NPs, all distanced by a gap ranging from 2 to 6 nm (Fig. 4). The calculated values of the shifts at the 4 nm gap corresponding to the anticipated distance of Au NP in the MTX assay were 19, 25 and 33 nm, respectively for one, two and three 25 nm Au NPs coupled to the 60 nm Au NP on the LSPR sensor. The experimentally observed plasmon coupling of the Au NP competitor with the LSPR sensor led to a 27 nm shift, in good agreement with the simulations for two Au NP competitors binding to a single 60 nm Au NP on the LSPR sensor.
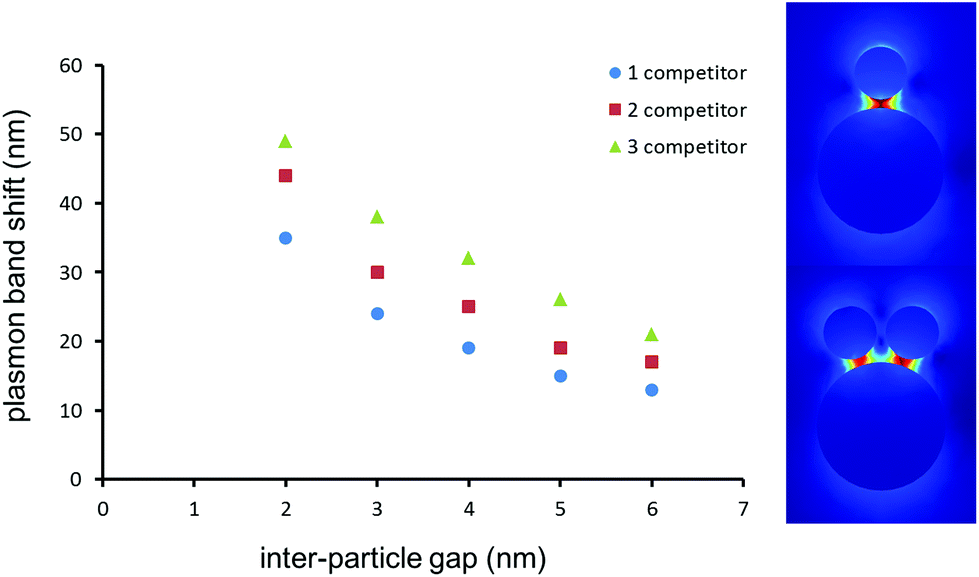 | ||
| Fig. 4 (Left) Calculated plasmon shift for a 60 nm-diameter spherical Au NPs placed near one, two or three 25 nm competitor Au NPs at the distances specified. (Right) Calculated electric field surrounding a dimer and a trimer spaced by a 4 nm gap. | ||
Performance of plasmonic sensors for MTX sensing
The MTX sensor was calibrated for a range of concentrations from 1 to 1000 nM. This range includes key maximal safety thresholds for MTX in clinical samples, which are in the range of 1000 nM 48 h post-administration, and 100 nM 72 h post-administration.41 The LSPR sensors were calibrated with a constant concentration of 2 nM of 25 nm competitor Au NPs. The calibration curve was established with the shift measured 10 min after the injection of MTX and competitor Au NPs. The LSPR sensor responded linearly for the injection of different concentrations of MTX between 1 and 1000 nM in PBS (Fig. 5). The LOD was measured at 5 nM for this MTX sensor. The LOD with the colorimetric assay compares advantageously with other detection techniques in SPR (28 nM),24 LSPR (155 nM),23 FPIA (30 nM),15 LC-MS (25 nM)15 and other commercial immunoassays such as Ark MTX (LOD = 20 nM). The coefficient of variation was between 15 and 20% for triplicate analysis.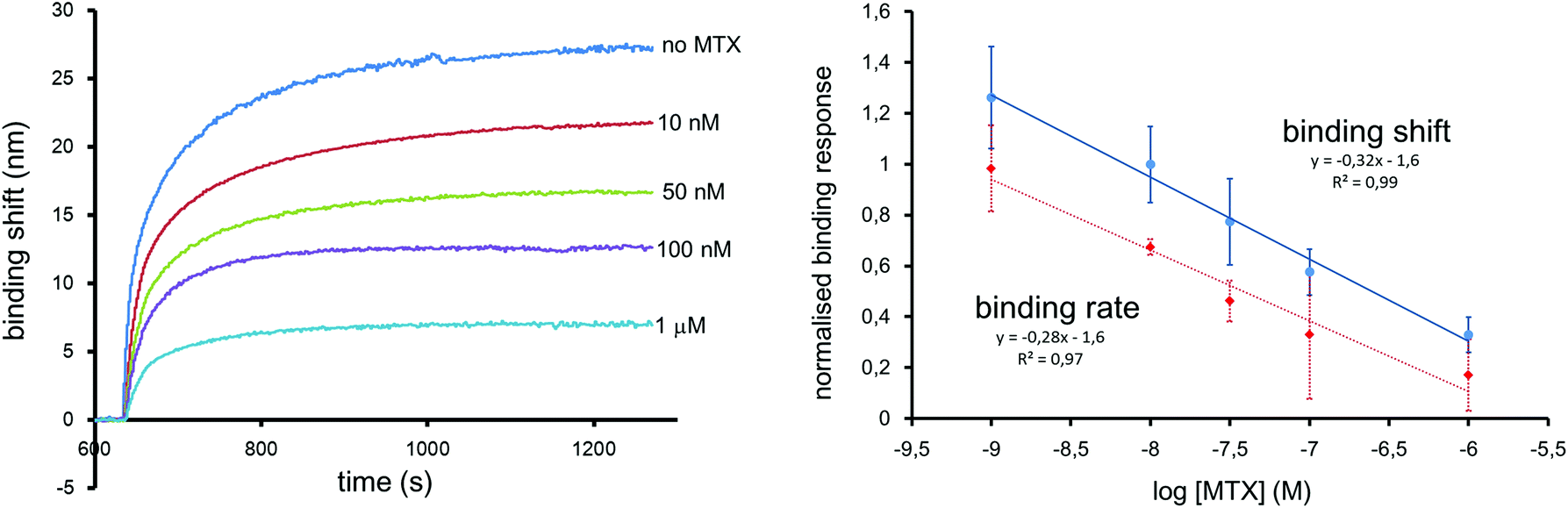 | ||
| Fig. 5 (Left) Response over time of the colorimetric sensor for MTX measured using 60 nm Au spheres as sensors and 25 nm spheres as competitor. A large wavelength shift was observed for 0 nM MTX, while a good discrimination between 100 nM (maximal safety level) and 1 μM was also observed. (Right) Calibration curves extracted from the measured binding shifts and binding rates. Error bars represent the standard deviation of a triplicate measurement. The calibration using the binding shift was offset for clarity. | ||
The analysis time was shortened by determining the binding rate during the first seconds following sample injection, as previously demonstrated.24 Normalized calibration curves were obtained from the binding shifts after 10 minutes and from the binding rate calculated from the slope of the first 12 seconds after injection (Fig. 5). Both calibration methods extracted from the same data set are in good agreement, demonstrating that quantification is possible in 12 seconds. The results can be confirmed from equilibrium binding data.
Colorimetric sensing with naked-eye detection was also achieved with the SPR sensor. The large 25 nm shift for the lowest MTX concentrations resulted in observable colour changes. Photographs of dove prism sensors exposed to 0, 100 and 1000 nM MTX standards illustrate the resulting colour changes (Fig. 6). The colour difference is sufficient to distinguish the MTX concentration following MTX administration (above 1000 nM), near the 72 h post-administration safety threshold of 100 nM, and where patients that have completely metabolised MTX. These colour changes can be easily distinguished by the naked eye. Once the samples sensors were dried after being withdrawn from the instrument, the plasmon shift was permanent and the colour remained intact for at least 12 months. Traceability, archiving and verification are key issues in clinical measurements. Long-term colour stability is an additional advantage of this colorimetric MTX sensor for traceability and verification of TDM results.
 | ||
| Fig. 6 Photograph of the actual colorimetric test-strips for MTX. A blank solution (which would be corresponding to the complete metabolism of MTX) showed a bluish-purple color “S”, 100 nM led to an intense pink coloration of the “S” and 1 μM resulted in a light blush pink “S”, similar to the initial coloration of the strip. Semi-quantitative interpretation of the results was possible by naked-eye. | ||
Conclusions
We demonstrated that the colorimetric LSPR sensor for MTX based on the formation of a dimer of Au NPs was rapid (12 seconds) and sensitive to concentrations in the clinical range. The fabrication method used for the colorimetric sensor was simple and inexpensive and it is envisioned that it could be easily transposed to larger production scale. Both the sensor and the competitor used for the analysis can be prepared in advance and stored for long periods of time, so only 2 injection steps of 10 minutes each were needed for each sample. Naked-eye detection was demonstrated and could provide semi-quantitative information in a clinical situation where a rapid assessment of MTX levels are required in the course of the treatment of patients.Acknowledgements
Financial support was provided by the Institut Mérieux, the Université de Montréal, the National Science and Engineering Research Council (NSERC) of Canada, the Canadian foundation for innovation (CFI), and the Centre for self-assembled chemical structures (CSACS).Notes and references
- A. Dasgupta, Therapeutic drug monitoring, Elsevier, Oxford, UK, 2012 Search PubMed.
- J.-F. Masson and J. N. Pelletier, Nanomedicine, 2015, 10, 521–524 CrossRef CAS PubMed.
- D. H. Choi, S. K. Lee, Y. K. Oh, B. W. Bae, S. D. Lee, S. Kim, Y.-B. Shin and M.-G. Kim, Biosens. Bioelectron., 2010, 25, 1999–2002 CrossRef CAS PubMed.
- X. Mao, Y. Ma, A. Zhang, L. Zhang, L. Zeng and G. Liu, Anal. Chem., 2009, 81, 1660–1668 CrossRef CAS PubMed.
- R. Wong and H. Tse, Lateral-flow immunoassay, Springer, 2009 Search PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- D. Vilela, M. C. González and A. Escarpa, Anal. Chim. Acta, 2012, 751, 24–43 CrossRef CAS PubMed.
- W. Zhao, W. Chiuman, M. A. Brook and Y. Li, ChemBioChem, 2007, 8, 727–731 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- J. Walling, Invest. New Drugs, 2006, 24, 37–77 CrossRef PubMed.
- D. Aumente, D. S. Buelga, J. C. Lukas, P. Gomez, A. Torres and M. J. Garcia, Clin. Pharmacokinet., 2006, 45, 1227–1238 CrossRef CAS PubMed.
- R. Bouquie, G. Deslandes, B. Nieto Bernaldez, C. Renaud, E. Dailly and P. Jolliet, Anal. Methods, 2014, 6, 178–186 RSC.
- Y. Guo, Y. Chen, Q. Zhao, S. Shuang and C. Dong, Electroanalysis, 2011, 23, 2400–2407 CrossRef CAS.
- G. G. Oliveira, B. C. Janegitz, V. Zucolotto and O. Fatibello-Filho, Cent. Eur. J. Chem., 2013, 11, 1837–1843 CrossRef CAS.
- R. Selesovska, L. Janikova-Bandzuchova and J. Chylkova, Electroanalysis, 2015, 27, 42–51 CrossRef CAS.
- Y. Wei, L. Luo, Y. Ding, X. Si and Y. Ning, Bioelectrochemistry, 2014, 98, 70–75 CrossRef CAS.
- Z. Zhu, F. Wang, F. Wang and L. Xi, J. Electroanal. Chem., 2013, 708, 13–19 CrossRef CAS.
- Z. Zhu, H. Wu, S. Wu, Z. Huang, Y. Zhu and L. Xi, J. Chromatogr., A, 2013, 1283, 62–67 CrossRef CAS PubMed.
- Z. Chen, S. Qian, X. Chen, W. Gao and Y. Lin, Analyst, 2012, 137, 4356–4361 RSC.
- S. S. Zhao, M. A. Bichelberger, D. Y. Colin, R. Robitaille, J. N. Pelletier and J.-F. Masson, Analyst, 2012, 137, 4742–4750 RSC.
- S. S. Zhao, N. Bukar, J. L. Toulouse, D. Pelechacz, R. Robitaille, J. N. Pelletier and J.-F. Masson, Biosens. Bioelectron., 2015, 64, 664–670 CrossRef CAS.
- R. Griss, A. Schena, L. Reymond, L. Patiny, D. Werner, C. E. Tinberg, D. Baker and K. Johnsson, Nat. Chem. Biol., 2014, 10, 598–603 CrossRef CAS PubMed.
- N. Bukar, S. S. Zhao, D. M. Charbonneau, J. N. Pelletier and J.-F. Masson, Chem. Commun., 2014, 50, 4947–4950 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- H. Yockell-Lelièvre, J. Desbiens and A. M. Ritcey, Langmuir, 2007, 23, 2843–2850 CrossRef PubMed.
- O. R. Bolduc, P. Lambert-Lanteigne, D. Y. Colin, S. S. Zhao, C. Proulx, D. Boeglin, W. D. Lubell, J. N. Pelletier, J. Fethiere, H. Ong and J.-F. Masson, Analyst, 2011, 136, 3142–3148 RSC.
- O. R. Bolduc, C. M. Clouthier, J. N. Pelletier and J.-F. Masson, Anal. Chem., 2009, 81, 6779–6788 CrossRef CAS PubMed.
- O. R. Bolduc and J.-F. Masson, Langmuir, 2008, 24, 12085–12091 CrossRef CAS PubMed.
- O. R. Bolduc, J. N. Pelletier and J.-F. Masson, Anal. Chem., 2010, 82, 3699–3706 CrossRef CAS PubMed.
- J. H. Hodak, A. Henglein and G. V. Hartland, J. Phys. Chem. B, 2000, 104, 9954–9965 CrossRef CAS.
- N. Cennamo, G. Agostino, A. Donà, G. Dacarro, P. Pallavicini, M. Pesavento and L. Zeni, Sensors, 2013, 13, 14676–14686 CrossRef PubMed.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415–2419 RSC.
- T. K. Sau, A. L. Rogach, F. Jäckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS PubMed.
- B. Sepúlveda, P. C. Angelomé, L. M. Lechuga and L. M. Liz-Marzán, Nano Today, 2009, 4, 244–251 CrossRef.
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- B. C. Widemann and P. C. Adamson, Oncologist, 2006, 11, 694–703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |