
N- and S-doped mesoporous carbon as metal-free cathode catalysts for direct biorenewable alcohol fuel cells†
Yang
Qiu‡
a,
Jiajie
Huo‡
a,
Fan
Jia
a,
Brent. H.
Shanks
a and
Wenzhen
Li
*ab
aChemical and Biological Engineering, Biorenewables Research Laboratory, Iowa State University, Ames, IA 50011, USA. E-mail: wzli@iastate.edu
bUS DOE Ames Lab, Ames, IA 50011, USA
First published on 6th November 2015
Abstract
Nitrogen and sulfur were simultaneously doped into the framework of mesoporous CMK-3 as metal-free catalysts for direct biorenewable alcohol fuel cells. Glucose, NH3, and thiophene were used as carbon, nitrogen and sulfur precursors, respectively, to prepare mesoporous N-S-CMK-3 with uniform mesopores and extra macropores, resulting in good O2 diffusion both in half cell and alcohol fuel cell investigations. Among all investigated CMK-3 based catalysts, N-S-CMK-3 prepared at 800 °C exhibited the highest ORR activity with the onset potential of 0.92 V vs. RHE, Tafel slope of 68 mV dec−1, and 3.96 electron transfer number per oxygen molecule in 0.1 M KOH. The alkaline membrane-based direct alcohol fuel cell (DAFC) with N-S-CMK-3 cathode displayed 88.2 mW cm−2 peak power density without obvious O2 diffusion issue, reaching 84% initial performance of that with a Pt/C cathode. The high catalyst durability and fuel-crossover tolerance led to stable performance of the N-S-CMK-3 cathode DAFC with 90.6 mW cm−2 peak power density after 2 h operation, while the Pt/C cathode-based DAFC lost 36.9% of its peak power density. The high ORR activity of N-S-CMK-3 can be attributed to the synergistic effect between graphitic-N and S (C–S–C structure), suggesting great potential to use N-S-CMK-3 as an alternative to noble metal catalysts in the fuel cell cathode.
1 Introduction
With increasing energy demands, declining petroleum reserves and increasing environmental pressure, having clean, sustainable, reliable and technical viable energy resources is one of the most significant challenges facing human society, industry and the economy.1,2 Sustainable energy conversion and storage technologies, such as fuel cells, metal-air batteries, etc., attract enormous attention and have been intensively studied and developed given their potential for high energy-conversion efficiency and environmental advantages.3–6 However, the sluggish oxygen reduction reaction (ORR), one of the half electrochemical reactions occurring at the cathode side, results in significant overpotential even at open circuit voltage operation (>250 mV), and largely limits the fuel cell's working efficiency and output power density.6,7 The noble metal-based catalysts, e.g. Pt, have been found to be the best ORR catalysts at low temperatures, but they suffer from several serious issues including their limited reserves on the earth, high cost and instability under the fuel cell operation environment.4,7 For direct alcohol fuel cells, noble metal-based ORR catalysts further have an alcohol poisoning issue, which is originated from the crossover of alcohol fuel from the anode to cathode.6,8 With the alcohol crossover, electrochemical oxidation of alcohol competes with the ORR at the cathode and results in a mixing potential, thus reducing the fuel cell operating voltage and efficiency. Therefore, there is a clear and urgent need to seek for alternative noble metal-free or even metal-free cathode catalysts with high ORR activity, low economic cost, robust stability and high tolerance towards alcohol fuels.One strategy is to lower the noble metal content in the ORR catalysts. Adzic and co-workers reported the underpotential deposition method to fabricate a Pt monolayer on Au and Co.9,10 MPt (M = Fe, Co etc.) alloys and core–shell (noble metal core) catalysts were also investigated to reduce the noble metal content.8,11 Another strategy is to replace Pt by transition metal macrocycle catalysts prepared through pyrolysis processes. Although these catalysts demonstrated competitive ORR activity relative to Pt, they suffer significant activity loss due to instability of the transition metal in the acid electrolyte.12 Recently, nitrogen doped carbon has been discovered as a metal-free ORR catalyst in a high pH medium. For example, metal-free nitrogen-doped carbon nanotubes were shown to have remarkable ORR activity and durability.13 It has been reported that the carbon materials could become non-electron-neutral by incorporating N atoms into the graphitic framework. The electronic property tuning effect of N atoms can significantly change the charge density and spin density of carbon atoms; thus benefiting the adsorption of oxygen and subsequent reduction reaction on carbon.13,14 Besides nitrogen, other elements, such as S, B and P, etc., can also be incorporated into the carbon framework as a dopant or co-dopant with N to further enhance the ORR activity based on the synergistic effects between the two dopants.15–19 Unfortunately, some undesired structures/phases (e.g. boron nitride, BN) could be formed under high temperature annealing conditions, and can significantly lower the ORR activity of the catalysts.14,20 Thus, rational choice of dual-dopants and its corresponding precursors play an important role in obtaining high ORR activity. However, few reports explain the criterion for choosing heteroatom precursors or investigate the precursor effect on ORR activity over heteroatom-doped nanocarbons.
Among nanocarbon materials, heteroatom-doped carbon nanotubes and graphene, one or two dimensional carbon networks with outstanding electric conductivity, have demonstrated excellent ORR performance, and have emerged as promising ORR catalyst candidates.21–23 However, their relatively low electrochemical surface area (ECSA) and randomly formed oxygen-inaccessible microspores may lead to internal diffusion issues in practical single fuel cell cathode applications.6,24 In comparison, mesoporous carbon nanostructures are more advantageous to serve as the cathode catalyst in a real fuel cell setting, because they feature not only high conductivity, but also well-ordered porous structure and tunable uniform mesopores (e.g. 4–20 nm) with a high ECSA (>1000 m2 g−1).25–27 Heteroatom (e.g. N) containing hydrocarbons are potentially attractive to serve as the carbon precursor; but some of them are very expensive with price even comparable to noble metals.28–30 Thus, carbon precursor cost should also be taken into consideration for the catalyst preparation and utilization in fuel cells.
Heteroatom-doped carbons are rarely directly employed as cathode catalysts in H2/O2 fuel cell, which is probably due to the membrane-electrode-assembly (MEA) fabrication issues,7,31 underperforming the electricity generation of H2/O2 fuel cells. For low temperature fuel cells directly fed with biomass-derived alcohols, such as ethanol and polyols, they are also attractive because they do not need a fuel reforming system and rigorous MEA fabrication process.32,33 In addition, the direct biorenewable alcohol fuel cells can simultaneously convert chemical energy into electrical energy and co-produce desirable biobased chemicals with the advantages of high efficiency, quiet operation and low CO2 emission.5,8 Therefore, they are poised to take a significant role in our future energy landscape. US DOE has identified the top ten important chemicals, including ethanol, polyols (glycerol, sorbitol, etc.),34 which can be economically derived from biomass, and can serve as building blocks for chemicals, fuels and energy production in the future.
Herein, we report a nitrogen and sulfur dual-doped, metal-free mesoporous carbon (N-S-CMK-3) with outstanding electrocatalytic activity for ORR and its utilization as a cathodic catalyst in a direct biorenewable alcohol fuel cell, which exhibits remarkable performance and operation stability as compared with a noble metal Pt/C cathode fuel cell. A ubiquitous environment-friendly renewable biomass compound, glucose, serves as the carbon sources. Highly active N and S are introduced from ammonia (NH3) and thiophene precursors, which can efficiently prevent N-S-CMK-3 from having clogged inside pores and channels thereby obtaining a highly organized mesoporous structure with high surface area. Simultaneously generating macropores (pore diameter > 200 nm) together with mesopores improves the adsorption of O2 and increases the electrocatalytic activity for ORR. After ruling out the transition metal (Fe) effect, high ORR activity and fuel cell performance can be largely ascribed to the synergistic effect of N and S atoms with specific structures (e.g. graphitic-N and C–S–C). The high ORR activity of N-S-CMK-3 with robust stability and excellent tolerance of alcohol crossover in the direct alcohol alkaline fuel cell is superior to that of noble metals and holds promise of serving as cathode catalyst for other energy conversion and storage devices, such as the H2/O2 fuel cells and metal–air batteries.
2 Experimental
2.1 Material preparation
The synthesis of CMK-3 uses SBA-15 as the template. For the synthesis of SBA-15,35 Pluoronic P123 (5.0 g) was dissolved in a hydrochloric solution (190 mL, 1.6 M) with 350 rpm stirring at 35 °C. After the dissolution of P123, tetraethyl orthosilicate (11.0 g) was added into the solution and stirred at 35 °C for another 24 h. The solution was then moved into a polytetrafluoroethylene bottle for an additional 24 h treatment at 100 °C. After that, the solid was filtered and washed with deionized water (18.2 MΩ) and dried in an oven overnight. A final calcination in air at 550 °C for 6 h was used to remove the surfactant. Then, SBA15 (2.8 g) was mixed with deionized water (15 g), glucose (3.5 g), and sulfuric acid (0.39 g). The mixture was treated at 100 °C for 6 hours, and then at 160 °C for another 6 hours. Deionized water (15 g), glucose (2.24 g), and sulfuric acid (0.25 g) was again added, mixed, and treated under the same condition described above. The sample was then heated at 900 °C and kept for 10 h under flowing nitrogen. The sample was treated with 10% hydrofluoric acid in ethanol solution (50% by volume) to remove silica. After filtration and drying, the base CMK-3 was obtained.The nitrogen and sulfur dual-doped N-S-CMK-3 catalysts were synthesized under high temperature annealing. 0.5 g thiophene and 0.5 g iron chloride (FeCl3) were dissolved into 15 mL of ethanol with magnetic stirring, followed by directly adding 0.5 g CMK-3, and the mixture was left under magnetic stirring at ambient temperature for 12 h. The pre-treated CMK-3 was obtained by filtrating and washing with deionized water for 5 times to remove iron chloride residue followed by drying at 80 °C for 12 h. After completely removing the liquid solution, the pre-treated CMK-3 was annealed in ambient pressure NH3 gas with a 60 mL min−1 flow rate. The temperature was kept at 700, 800 and 900 °C for 2 h, respectively, after a heating rate of 5 °C min−1. After cooling down to room temperature, the sample was subjected to a 6.0 M hydrochloric acid treatment to further remove the Fe residue, and the final N-S-CMK-3 was obtained by washing with deionized water to a pH of 7 and drying at 60 °C for 12 h. Single-doped N-CMK-3 and S-CMK-3 were synthesized by using similar method discussed above without thiophene or NH3 dopants, respectively.
2.2 Material characterization
Composition was measured using an inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer). The sample was prepared by dissolving 10 mg of catalyst in 4 mL of fresh aqua regia, followed by diluting to 250 mL with deionized water (18.2 MΩ). The XRD patterns were obtained from a Rigaku Ultima IV X-ray diffraction (XRD) systems with Cu Kα radiation (λ = 1.5406 Å), with a tube current of 44 mA and a tube voltage of 40 kV. JEOL 2100 200 kV scanning and transmission electron microscope (STEM) and corresponding element energy-dispersive X-ray spectrometer (EDS) were utilized to determine the catalyst structure and element concentration. JEOL 5800LV and FEI Quanta 250 field-emission scanning electron microscope (SEM) were used for macro morphology determination. X-ray photoelectron spectroscopy (XPS) with Mg K alpha X-ray (1253.6 eV) (Kratos Amicus/ESCA 3400) was also used to determine the heteroatom concentrations and its corresponding status through different binding energy peak. Surface area and porosity characterization were performed via nitrogen physisorption in a Micromeritics ASAP 2020 operated at −196 °C. The specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) equation and the pore volume was determined at a relative pressure of 0.973. A DXR Raman microscope (Thermo Scientific, Waltham, MA, USA) was used for spectra acquisition with 780 nm excitation at 14 mW laser power, 10× objective, and 50 μm slit aperture (resolution is 4–8 cm−1). The exposure time is 30 s.2.3 Electrocatalytic characterization
The electrochemical tests were carried out both in the half-cell and fuel cell reactors. The half-cell test was conducted in a three-electrode setup consisting of a rotating disk electrode (working electrode), a coiled platinum counter electrode, which was isolated by a fritted glass tube from the main test electrolyte, and a Hg/HgO reference electrode with 0.1 M KOH filling solution. Potential was applied by using a multi-channel potentiostat (Biological). To fabricate the working electrode, a 2.0 mg mL−1 solution of catalyst ink was prepared by dispersing N-S-CMK-3 in mixture of iso-propanol and 5 wt% Nafion solution (one-propanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nafion solution = 9:1 v/v), followed by ultrasonication until no aggregation was visible. Then, 30 μL of ink was dropped onto the surface of a glassy carbon electrode with a catalyst loading of 0.306 mg cmgeo−2 (Pt loading of 61.2 μgPt cmgeo−2), and a uniform catalyst film was obtained after 30 min ambient temperature drying in air. In order to compare the ORR performance of N-S-CMK-3 to other control samples, the electrodes of 20 wt% Pt/C, CMK-3 and mono-element doped CMK-3 were fabricated by using the same method discussed above. The ORR performance of the catalyst was investigated by cyclic voltammetry (CV) test, which was performed at 50 mV s−1 scan rate after purging the electrolyte with N2 and O2 for 30 min respectively, followed by the linear sweep voltammetry (LSV) toward ORR activity evaluation in O2-saturated electrolyte with 10 mV s−1 scan rate at different rotating rate from 2000 rpm to 400 rpm.
:Nafion solution = 9:1 v/v), followed by ultrasonication until no aggregation was visible. Then, 30 μL of ink was dropped onto the surface of a glassy carbon electrode with a catalyst loading of 0.306 mg cmgeo−2 (Pt loading of 61.2 μgPt cmgeo−2), and a uniform catalyst film was obtained after 30 min ambient temperature drying in air. In order to compare the ORR performance of N-S-CMK-3 to other control samples, the electrodes of 20 wt% Pt/C, CMK-3 and mono-element doped CMK-3 were fabricated by using the same method discussed above. The ORR performance of the catalyst was investigated by cyclic voltammetry (CV) test, which was performed at 50 mV s−1 scan rate after purging the electrolyte with N2 and O2 for 30 min respectively, followed by the linear sweep voltammetry (LSV) toward ORR activity evaluation in O2-saturated electrolyte with 10 mV s−1 scan rate at different rotating rate from 2000 rpm to 400 rpm.
Rotating ring-disk electrode (RRDE) was employed to measure the concentration of HO2− intermediate and electron transfer number per oxygen (n) with scanning rate of 10 mV s−1 at rotating rate of 1600 rpm. The HO2− intermediated, generated on the surface of disk electrode (0.19625 cm2 geometric surface area), can be immediately oxidized on Pt-ring electrode surface at 0.8 V vs. RHE applied potential. The electron transfer number n and HO2− intermediate production percentage (HO2−%) were determined as follows:28
 | (1) |
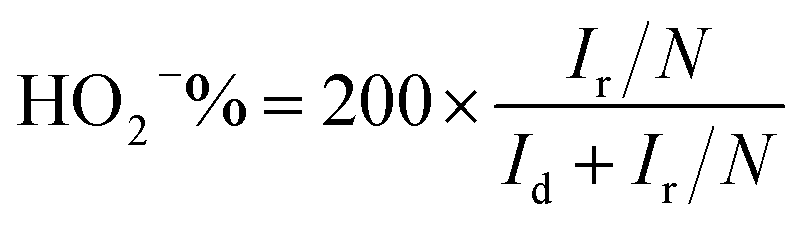 | (2) |
All experimental results for the ORR activity in this paper were reported as current density versus applied potential (vs. RHE), and the current densities were normalized using geometric surface area of a glassy carbon electrode with 5 mm diameter. The Hg/HgO reference electrode was calibrated with respect to the reversible hydrogen electrode (RHE) before use. The calibration was conducted in standard three-electrode system with Pt wire as the working and counter electrodes, and the Hg/HgO electrode as the reference electrode. The 0.1 M KOH electrolyte was purged and saturated with high purity H2 for 30 min before calibration, and LSV test was then run at a scan rate of 0.5 mV s−1, and the potential range of −0.93 V to −0.85 V vs. Hg/HgO, where the current crossed zero is taken to be the thermodynamic potential for the hydrogen electrode reactions. The result is E(RHE) = E(Hg/HgO) + 0.888 V in 0.1 M KOH electrolyte.
The fuel cell tests were performed on a Scribner Fuel Cell System 850e (Scribner Associates, USA), and the fuel cell fixture with an active area of 5 cm2 was purchased from Fuel Cell Technology Inc. The cathode electrode composed of 70 wt% N-S-CMK-3 catalyst and 30 wt% AS-4 ionomer was mixed and directly sprayed onto an A-201 anion exchange membrane to obtain a catalyst loading of 2.0 mg cm−2, while commercial Pt/C blended with 5 wt% PTFE solution (Pt/C:PTFE = 9:1 w/w) was sprayed onto a PTFE-untreated carbon cloth liquid diffusion layer with a 1.0 mgpt cm−2 catalyst loading. The membrane electrode assembly (MEA) was constructed by pressing the anode carbon cloth with anion exchange membrane, which has been sprayed with cathode catalyst. 2.0 M KOH with 1.0 M fuel (glycerol, ethanol or sorbitol) was pumped into the anode compartment at 1.0 mL min−1, while high purity O2 (99.999%) regulated at 400 mL min−1 was fed to cathode side with 30 psi backpressure.48 The cell temperature was kept at 50 °C, and the relative humidity for both anode and cathode is 100%. The MEA was activated at 0.1 V cell voltage until the current density got stable. The I–V polarization curve was obtained by applying a constant voltage and collecting corresponding current density.
The overall electron transfer numbers per oxygen molecule involved in the typical ORR process were calculated from the slopes of the Koutecky–Levich plots using the following equations:14,30
 | (3) |
| B = 0.2nFCo(Do)2/3ν−1/6 | (4) |
| Jk = nFkCo | (5) |
485C mol−1), Do is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm s−1), v is the kinetic viscosity (0.01 cm2 s−1), and Co is the concentration of O2 (1.2 × 10−3 mol L−1). The constant 0.2 is adopted when the rotating speed is in rpm, and k is the electron transfer rate constant.
3 Results and discussion
3.1 Material synthesis and characterization
The well-ordered mesoporous carbons, N-S-CMK-3, were synthesized via carbonization and subsequent N and S doping processes, as shown in Scheme 1. Glucose served as the carbon source to mix with the mesoporous SBA-15 silica template,35 prior to calcination. Hydrofluoric acid (HF) was used to etch away the SBA15 template to obtain pure CMK-3 mesoporous carbons. Then, a mixture of CMK-3 and thiophene as well as iron(III) chloride (FeCl3) was annealed at 800 °C under anhydrous ammonia (NH3) gas purging, to facilitate heteroatom N and S doping into the graphitic framework of CMK-3. The N-S-CMK-3 mesoporous carbons were finally exposed to hydrochloric acid (HCl) washing and overnight vacuum drying (for synthesis details, see Experimental section). | ||
| Scheme 1 Schematic of the synthesis of metal-free mesoporous N-S-CMK-3 catalysts. | ||
In this work, glucose was chosen as the carbon precursor for mesoporous carbon synthesis owning to its unbranched carbon structure that facilitates formation of a stable and well-ordered mesoporous structure with narrow pore size distribution through carbonization.27 Glucose can be ubiquitously obtained in large quantities at low price, while some previously reported heteroatom-containing carbon precursors (e.g. Ferrocene, Vitamin B12, PDI) have 10 fold higher price.28–30 Together with its additional advantages of non-toxicity and non-volatility, glucose is a promising green carbon precursor for mesoporous carbon synthesis. We chose ammonia and thiophene as the N and S dopant precursors based on consideration of their small molecular weight, simple molecular structure and high N/C and S/C ratios (1.0 and 0.5, respectively). These precursors can not only largely prevent pore clogging by CN and CS species in the doping process, but also facilitate formation of high N and S content mesoporous carbon with very large surface area (>1000 m2 g−1), since ammonia etching can assist in the formation of extra macropores (pore diameter > 200 nm).36 Single and multi-doped CMK-3 carbons were prepared for comparison through the same processes as described above, and a commercial Pt/C, the best-known ORR catalyst, was also studied as the baseline performance in this work.
The as-synthesized mesoporous N-S-CMK-3 catalysts were first characterized using scanning electron microscopy (SEM), as shown in Fig. 1a. The morphology of the N-S-CMK-3 was irregular with visible macropores on the surface, which had no distinguishable morphology change compared with that of CMK-3 (Fig. S1†). The underlying highly organized hierarchical porous structures were observed by transmission electron microscopy (TEM) characterization (Fig. 1b). The TEM images revealed an average carbon rod diameter of 6 nm, which is consistent with the pore size of the SBA-15 template (inset in Fig. S3a†), indicating successful carbonization of glucose within the silica template. However, it is interesting to note that the carbon structure varied when the heteroatom doping temperature increased from 700 °C to 900 °C (Fig. S2†). At a doping temperature of 700 °C, apparent clogging originated from the thiophene derived CS species during the doping process became more serious and the carbon sheets were randomly formed, which could undesirably decrease the surface area and pore volume of carbon catalysts, leading to a ORR performance drop.37 The doping temperature of 900 °C appeared to facilitate the decomposition of the mesoporous carbon and a resulting porous structure was dominantly observed instead of the organized hierarchical porous structures (Fig. S2†), because of the overreaction of ammonia with carbon during the heteroatom doping process. Ammonia etching of the carbon may follow the reactions shown below:36
| 2NH3 → N2+3H2 | (6) |
| C+2H2 → CH4 | (7) |
 | ||
| Fig. 1 (a) SEM image (b) TEM image and (c) Nitrogen adsorption–desorption isotherms and pore size distribution (Inset) of N-S-CMK-3 800 °C; (d) Small-angle XRD patterns of N-S-CMK-3 800 °C and CMK-3; (e) Raman spectra of N-S-CMK-3 800 °C, N-SCMK-3, S-CMK-3 and CMK-3. | ||
The ammonia etching can be regulated by tuning the doping temperature. A moderate etching rate at around 800 °C (doping temperature) favors the formation of macropores and not only diminishes the CS species clogging effect, but also significantly increases the surface area of the resulting N-S-CMK-3 mesoporous carbon. To better investigate the temperature effect on catalysts structure, the BET surface areas of the N-S-CMK-3 catalysts were measured to be 784 m2 g−1 with (N-S-CMK-3 700 °C), 1023 m2 g−1 (N-S-CMK-3 800 °C) and 921 m2 g−1 (N-S-CMK-3 900 °C), all of which were lower than the 1126 m2 g−1 of CMK-3, but much higher than previously reported carbon nanotube and graphene ORR catalysts, such as mesoporous graphene and carbon hybrids11,17 (the BET surface area and total pore volume of all the as-synthesized CMK-3 based catalysts are shown in Table S1†). Therefore, 800 °C was determined to be the optimal doping temperature to prepare well-ordered mesoporous N-S-CMK-3. The surface texture of the N-S-CMK-3 catalysts was investigated by N2 adsorption–desorption isotherms (type IV), and the typical hysteresis loops was observed to reveal the mesoporous nature of the well-ordered N-S-CMK-3 catalysts. The pore diameter of N-S-CMK-3 was centered at 3.7 nm according to the Barrett–Joyer–Halenda (BJH) model (inset in Fig. 1c),38 which is close to the dominant pore diameter of 3.8 nm for pure CMK-3 (inset in Fig. S3b†); whereas larger mesopores with pore size ranging from 4.8 nm to 15.0 nm and macropores (>200 nm) were only obtained in N-S-CMK-3 after heteroatom doping. This wide pore size distribution further confirms the ammonia etching reaction with the N-S-CMK-3 mesoporous carbon. Zhang et al. reported that the combination of macropores and large mesopores could enhance the rate of desolvation and adsorption of O2 and increase the electrocatalytic activity for ORR.39 Consistent with that report, the N-S-CMK-3 (800 °C) with the unique mesoporous and extra macroporous structures exhibited the highest ORR performance among all investigated CMK-3 based catalysts as shown in Section 3.2 “Electrocatalytic analysis in half cell”.
The well-ordered mesoporous carbon structure was also verified by small-angle XRD measurement, as shown in Fig. 1d. Three well-resolved diffraction peaks were observed in the small-angle XRD pattern of N-S-CMK-3, which can be assigned to (100), (110), and (200) reflections of 2D-hexagonal arrangements, and was very similar to that of the pristine CMK-3. Furthermore, two broad diffraction peaks centered at 25° and 44°, corresponding to the graphite (002) and (101) facets,30 were observed in both the N-S-CMK-3 and CMK-3 wide-angle XRD patterns (Fig. S4†), indicating no significant structure change during the N-, S-doping process, and these are in good agreement with the results observed from TEM images. More structural information for N-S-CMK-3 was obtained from Raman spectroscopy (Fig. 1e). Two typical bands observed at 1340 cm−1 and 1580 cm−1 can be assigned to the D- and G-bands, corresponding to the disordered carbon structure and interplane sp2-hybridized graphitic carbon sheet, respectively.30,40 The ID/IG ratio of N-S-CMK-3 is similar to that of CMK-3 and lower than that of N-CMK-3 and S-CMK-3, suggesting a more graphitic structure with relatively low defect sites for the N-S-CMK-3 sample.
To determine heteroatoms content in as-synthesized N-S-CMK-3, catalysts were characterized by energy-dispersive X-ray spectroscopy (EDS). The EDS spectra reveal the presence of N, S and O atoms in the graphitic framework, while no Fe signal was observed (Fig. S5a†), indicating the Fe was removed by hydrochloric acid (HCl) washing and the high ORR activity could be only ascribed to the N-S-CMK-3 catalyst. Moreover, the EDS elemental mapping in conjunction with the TEM images illustrates a homogeneous distribution of N and S in the N-S-CMK-3 catalyst (Fig. S5b–d†). X-ray photoelectron spectroscopy (XPS) was further used to characterize N, S and O valence states. From Fig. 2a, the content of the elements were determined by XPS to be 3.84 atom% of N, 0.83 atom% of S and 1.14 atom% of O, and negligible 0.12 atom% of Fe in N-S-CMK-3 for the 800 °C heteroatom doping temperature. Although the small amounts of Fe were quantified, no Fe signal was observed using the high resolution spectrum of XPS (Fig. S6a†), suggesting the removal of Fe through HCl washing. The Fe result from XPS could be due to the signal to noise resolution limitation of XPS. The high-resolution S2p spectrum was found to be fitted into two peaks centered at 163.5 eV and 165.0 eV, representing to S2p 3/2 and S2p 1/2; whereas SOx groups, which have been reported inactive for ORR, was not found (Fig. 2b).41,42 Similarly, the high-resolution N1s spectra showed three nitrogen forms existed, which can be typically illustrated as pyridinic-N (398.3 eV), pyrrolic-N (399.9 eV), graphitic-N (401.2 eV) and N+–O− (403.6 eV) structures, as shown in Fig. 2c.19,28,43 The C1s high-resolution spectrum was deconvoluted into single peaks corresponding to 284.5 eV (C1) for sp2-hybridised carbon atoms bonded to neighboring carbon and hydrogen atoms, 285.5 eV (C2) for C–N–C and C–S–C, and 288.0 eV (C3) for different oxygen-containing groups (e.g. COOH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–OH),28 further confirming that N and S were successfully doped into the CMK-3 carbon framework (Fig. S6b†). Inductively coupled plasma-mass spectrometry (ICP-MS) was also used to further test the content of Fe residue in N-S-CMK-3 catalysts. The Fe concentration was detected as only 0.09 wt%, which is close to the ICP-MS detection limit, further indicating successful Fe elimination for the N-S-CMK-3 mesoporous carbon.
O, and C–OH),28 further confirming that N and S were successfully doped into the CMK-3 carbon framework (Fig. S6b†). Inductively coupled plasma-mass spectrometry (ICP-MS) was also used to further test the content of Fe residue in N-S-CMK-3 catalysts. The Fe concentration was detected as only 0.09 wt%, which is close to the ICP-MS detection limit, further indicating successful Fe elimination for the N-S-CMK-3 mesoporous carbon.
 | ||
| Fig. 2 (a) XPS survey spectrum; high-resolution spectra of (b) S2p and (c) N1s of N-S-CMK-3 800 °C. | ||
3.2 Electrocatalytic analysis in half cell
Before electrocatalytic ORR activity investigation, the Hg/HgO reference electrode was calibrated with respect to the reversible hydrogen electrode (RHE), and the result is E(RHE) = E(Hg/HgO) + 0.888 V in 0.1 M KOH electrolyte, as shown in Fig. S7.†All applied potential reported in this paper was versus the reversible hydrogen electrode (RHE).
The ORR electrocatalytic activity of N-S-CMK-3 was first investigated by cyclic voltammetry (CV) tests, as shown in Fig. S8.† Featureless voltammetric currents without an obvious redox peak were obtained when the 0.1 M KOH electrolyte was saturated with N2 (short dash curve), as a result of the well-known typical capacitance effect on porous carbon materials. In contrast, when an O2-saturated electrolyte was used, a well-defined cathodic ORR peak, centered at 0.66 V with 5.45 mA cm−2 current density, was observed over as-synthesized N-S-CMK-3, demonstrating obvious ORR electrocatalytic activity. To obtain deeper insight of ORR steps on the N-S-CMK-3 catalysts, linear sweep voltammetry (LSV) tests were carried out at different rotating rate ranging from 2000 rpm to 400 rpm in 0.1 M KOH electrolyte saturated with O2 (Fig. 3a). The voltammetric profiles demonstrate the current density increased as the applied potential became more negative, and the CMK-3 based catalysts were first investigated with different annealing temperature.
 | ||
| Fig. 3 (a) Polarization curves of LSV tests at different rotating speeds for N-S-CMK-3 800 °C conducted in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 (inset shows the corresponding K-L plots); (b) Comparative polarization (RDE) curves for N-S-CMK-3 catalysts with different doping temperatures, and (c) comparative polarization curves for different catalysts with the same loading measured at 1600 rpm in O2-saturated 0.1 M KOH solution; (d) Comparative polarization curves (RRDE) for different catalysts with the same loading measured at 1600 rpm in O2-saturated 0.1 M KOH solution; (e) HO2− yields (top) during the ORR and the corresponding electron transfer number (n, bottom) of the prepared catalysts measured by RRDE. | ||
As shown in Fig. 3b, N-S-CKM-3 800 °C exhibited the best ORR performance with the highest limiting current density of 5.50 mA cm−2 at 0.5 V and the most positive onset potential of 0.92 V among three N-S-CMK-3 catalysts annealed by different temperature. The possible reason is that the N-S-CMK-3800 °C maintained the largest surface area (1023 m2 g−1) after heteroatom doping, while 700 °C and 900 °C led to surface area reduction to 784 m2 g−1 and 921 m2 g−1, respectively (Table S1†). In addition, an apparent clogging with the thiophene derived CS species occurred at 700 °C doping process, which could undesirably decrease the surface area and pore volume of carbon catalysts, and lead a O2 diffusion issue for ORR; while 900 °C doping temperature can cause overreaction of ammonia with mesoporous carbon and result in decomposition of porous structure, which was predominantly observed instead of the organized hierarchical porous structures in SEM images (Fig. S2†). Thus, the highly ordered mesoporous structure with extra macropores was formed by moderate etching reaction of NH3 at 800 °C doping temperature, which provided the largest surface area and benefited the best ORR performance among these three N-S-CMK-3 catalysts.
Comparative studies of solely doped N-CMK-3, S-CMK-3, and pristine CMK-3 as well as with Pt/C were also conducted using the same LSV method. As shown in Fig. 3c, with N or S solely doped into CMK-3 carbon frameworks, the onset potential (potential at which J = 0.1 mA cm−2) increased from 0.79 V (CMK-3) to 0.83 V (S-CMK-3) and 0.87 V (N-CMK-3), respectively, and their corresponding ORR current density at 0.5 V raised from 1.98 (CMK-3) to 2.88 (S-CMK-3) and 3.30 (N-CMK-3) mA cm−2, respectively. Evidently, the ORR activity of CMK-3 was improved with involvement of heteroatoms, which could be attributed to the positive change in the charge density or/and spin density of neighbor carbon atoms (serving as ORR active sites), facilitating 4e− transfer ORR. Furthermore, by simultaneously incorporating N and S, the ORR activity of N-S-CMK-3 was further enhanced, and exhibited an ORR current density of 5.50 mA cm−2 at 0.5 V and an onset potential of 0.92 V with Tafel slope of 68 mV dec−1, which were comparable to the best-known ORR catalyst, Pt/C (Fig. S9†). In addition, only a 33 mV half-wave potential difference (E1/2) was observed between the N-S-CMK-3 and Pt/C, indicating promising potential of N-S-CMK-3 to be an alternative to Pt/C as ORR catalysts.
The ORR catalytic activity of various catalysts was further compared in Table S2,† including transition-metal based carbon materials (e.g. Fe–N/C, Mn3O4/porous glassy carbon) and metal free carbon materials (e.g. N–S mesoporous graphene, N–S doped graphene/CNT).14,21,43–46 Chen et al. reported the N and S doped graphene-CNT with an onset potential of 0.85 V and electron transfer number of 3.8,21 which are lower than that of N-S-CMK-3 reported in this work, probably due to the uniform mesoporous structure with high surface area (1023 m2 g−1). Liu et al. synthesized the heteroatom doped carbon materials (FeSO4-PEI) exhibited a decent Tafel slope,45 but its onset potential, 0.79 V, with electron transfer number of 3.8 affects its feasibility as a highly active ORR catalyst. Obviously, among the catalysts summarized in Table S2,† N-S-CMK-3 provided more positive onset potential, higher ORR limiting current density, and sharper Tafel plot, all of which further confirmed its intrinsic high catalytic activity towards ORR. It should be noted that the onset potential and Tafel slope exhibited by ORR catalysts are very sensitive to its loading, and the reported catalyst loading is significantly varied from 0.0001 to 0.4 mg cm−2, as summarized in Table S2† (or to even higher value). Thus, the electron transfer number (n), obtained from RDE and RRDE, and mass-transport limiting current were investigated to further evaluate the intrinsic ORR kinetics of different ORR catalysts.
The electron transfer number (n) at a given applied potential was calculated by Koutecky–Levich (K–L) equation (for K–L equation details see Experimental section). The K–L plots (J−1vs. ω1/2) of the N-S-CMK-3 800 °C catalyst exhibited a high linearity, and the slopes within investigated applied potentials (0.2 to 0.5 V) were approximately constant (inset in Fig. 3a), implying a close n value for ORR at such potential range. Consequently, the n value of N-S-CMK-3800 °C was determined to be 4.0 at such applied potential range, which is same as the n value of Pt/C (Fig. S10†), suggesting a complete ORR process with 4e− transfer reduction to H2O. In addition, the n value of solely doped N-CMK-3, S-CMK-3 and blank CMK-3 were also calculated by using K-L equation. As shown in Fig. S10,† the n value of CMK-3, S-CMK-3 and N-CMK-3 were calculated to be 2.2, 2.8 and 3.4 at 0.5 V vs. RHE, respectively, which are in good accordance to the trend of their corresponding limiting current density as discussed above. The kinetic limiting current density (Jk) at 0.5 V was also determined by K-L equation and was displayed in Fig. S10.† Although both N-CMK-3 and S-CMK-3 showed moderate performance for ORR, only N-S-CMK-3 exhibited an elegant ORR activity with complete 4e− pathway (n = 4) as comparable to Pt/C. Remarkably, the Jk of N-S-CMK-3 (32.6 mA cm−2) slightly surpassed that of Pt/C (25.6 mA cm−2) at 0.5 V and was much higher than that of N-CMK-3, S-CMK-3 and CMK-3 samples.
To further investigate the ORR activity of N-S-CMK-3 and its corresponding reaction mechanism, RRDE tests were conducted to directly measure the concentration of HO2− intermediate and determine the electron transfer number (n) of catalysts. The generated HO2− intermediate on the surface of disk electrode can be immediately oxidized on the surface of Pt ring electrode at 0.8 V vs. RHE applied potential, thus RRDE is reliable to in situ determine n toward the N-S-CMK-3 catalysts. As shown in Fig. 3d, N-S-CMK-3 showed the lowest ring current density ascribing to the HO2− oxidation, and the HO2− yield on disk electrode was below 1.5% in the potential range from 0.25 V to 0.6 V, while the average HO2− yield of other CMK-3 based catalysts were 92% (CMK-3), 62% (S-CMK-3), 27% (N-CMK3), respectively (Fig. 3e). The large amount of HO2− intermediate production could decrease the n and drastically reduce the ORR performance toward catalysts. Consequently, the average n value of N-S-CMK-3 at investigated potential was determined to be 3.96 without apparent change in such potential range (Fig. 3e). This is much higher than those over other investigated catalysts (e.g. n = 3.45 for N-CMK-3, n = 2.76 for S-CMK-3, and n = 2.15 for CMK-3), which is in good accordance with the results obtained from the K-L equation calculation in RDE tests. The performance of non-doped and mono-doped CMK-3 were far from satisfactory. However, with both N and S heteroatoms doping into carbon structure of mesoporous CMK-3, the catalyst ORR activity was significantly improved. This could be ascribed to the additional S atoms doping into N-CMK-3, further leading the positive charge and asymmetric spin density of neighboring carbon atoms, which are serving as the ORR active sites (the details were discussed in Section 3.4 “Insights into the synergistic effect of N and S on ORR”).14,47 The synergistic effect between N and S heteroatoms on neighbor carbons could facilitate the complete oxygen reduction reaction with 4e− transfer, and decrease the HO2− intermediate yield. On the other hand, the high surface area (1023 m2 g−1) of N-S-CMK-3 with a uniform structure combining mesopores and macropores could provide more active sites, which is beneficial to the utilization of O2 fuel, leading to a higher ORR activity. However, when only S was doped, the clogging issue could severely decrease the surface area of S-CMK-3. Together with these possible reasons, the N-S-CMK-3 exhibited a higher ORR limiting current density, and showed the best ORR activity among all investigated CMK-3 catalysts in this work, which is competitive to the that for Pt/C, suggesting its great potential to serve as a Pt/C alternative for ORR at the cathode side of fuel cells, Li-ion battery, and other energy storage and conversion devices.
Since the N-S-CMK-3 will be used as the cathode catalyst in direct alcohol fuel cell, the catalyst durability and alcohol fuel-crossover influence on ORR should be considered. The durability of the catalysts was first tested by cycling the catalyst between 0.1 V and 1.2 V at 10 mV s−1 scan rate under oxygen purging. After 3000 potential cycles, the N-S-CMK-3 exhibits a robust stability with only 46 mV of half-wave potential shift (Fig. 4a), which is superior to 52 mV for the commercial Pt/C catalyst (Fig. S11†). The stability of N-S-CMK-3 was further measured at a constant voltage of 0.5 V for 45000 s (12.5 hours). The results indicate slowly decreasing ORR performance with N-S-CMK-3 as 85.9% of the current was maintained after 45000 s. Conversely, Pt/C exhibits more serious performance degradation as only 76.1% of the current remained after an identical stability test (Fig. S12†). For fuel-crossover tolerance, glycerol, the coproduct (about 10 wt%) from biodiesel manufacturing, was added into the half cell reactor with the N-S-CMK-3 catalysts after 1800 s (chronoamperometry test at 0.6 V); comparative study of Pt/C catalysts was also carried out using the same method. Interestingly, when glycerol was added into electrolyte, the current density of N-S-CMK-3 had nearly no change, strongly indicating its high catalytic activity for ORR while being nearly inert towards glycerol oxidation (Fig. 4b). Conversely, Pt/C exhibits an instantaneous current density spike due to its high activity toward glycerol oxidation, and thus its vulnerability to glycerol crossover.
 | ||
| Fig. 4 (a) Polarization curves of N-S-CMK-3 800 °C before and after 3000 potential cycles in O2-saturated 0.1 M KOH solution; (b) glycerol crossover tolerance test of N-S-CMK-3 800 °C and commercial Pt/C conducted by chronoamperometric response at 0.6 V vs. RHE. | ||
3.3 Electrocatalytic analysis in single fuel cell
Single fuel cell performance is considered the ultimate catalyst evaluation method from both engineering and application points of view. The N-S-CMK-3 was fabricated into an alkaline membrane fuel cell, and its electrocatalytic activity toward ORR and single glycerol fuel cell performance tested. The steady-state polarization curves with different cathodic catalysts in the glycerol alkaline fuel cell are shown in Fig. 5a. It is observed that the open circuit potential of the fuel cell with N-S-CMK-3, CMK-3 and Pt/C were 0.86 V, 0.70 V and 0.85 V, respectively. Due to the outstanding ORR electrocatalytic activity and apparent O2 diffusion advantage, the glycerol alkaline membrane fuel cell with N-S-CMK-3 cathode yields a high peak power density of 88.2 mW cm−2, which is much higher than that with the CMK-3 cathode, 42.4 mV cm−2, and 84% of the fuel cell performance when compared with the best-known ORR catalyst, Pt/C, at cathode (105 mW cm−2 peak power density). It is worth to mention that the Fe loading would be extremely low (2.4 μg cm−2 cathode) as converted from 0.12 wt% of Fe determined by XPS. Based on the I–V curve analysis, the fuel cell with N-S-CMK-3 cathode displays Pt-like behavior both in kinetic dominated regime (current density < 50 mA cm−2) and ohmic resistance controlled regime (current density > 50 mA cm−2),48,49 implying a considerably high ORR activity and less performance loss owing to the resistance to ionic current.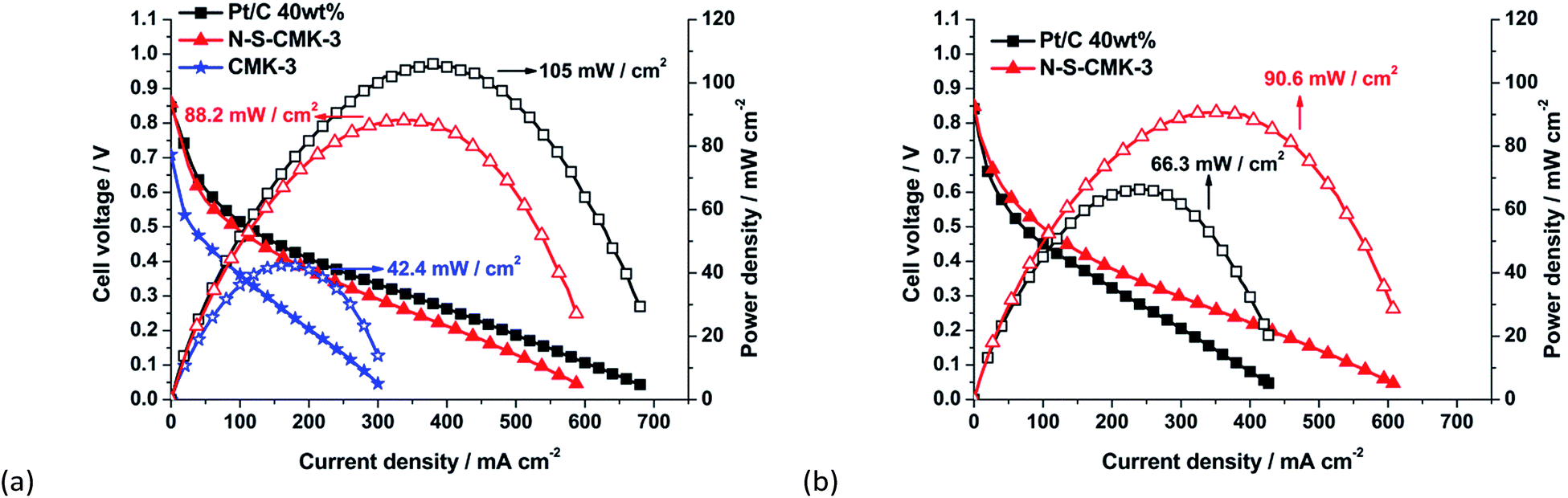 | ||
| Fig. 5 Polarization and power density curves before (a) and after (b) 2 h continuous current scanning of direct glycerol fuel cell with the N-S-CMK-3 cathode catalysts (loading 2.0 mg cm−2) and Pt/C 40 wt% anode catalysts (loading 1.0 mgpt cm−2) at 50 °C. | ||
We also tested the fuel cell performance using other bioalcohol fuels, ethanol and sorbitol. Ethanol is the first generation of biofuel and have been widely used as gasoline additive,34 while sorbitol is an important sugar alcohol that can be produced from cellulose hydrolysis and potentially used as precursors to generate the biorenewable chemicals.34,50,51 The N-S-CMK-3 cathode alkaline fuel cell reached 80% of that with the Pt/C cathode alkaline fuel cell using sorbitol, and even higher than that of Pt/C when ethanol is used as the fuel (Fig. S13†). It should be noted that the catalyst layer for N-S-CMK-3 cathode is thicker than that for Pt/C catalyst, which could affect the O2 diffusion toward ORR on fuel cell cathode side, leading to a slightly lower peak power density for N-S-CMK-3 cathode fuel cell relative to the Pt/C cathode one. To further study the O2 diffusion effect, different backpressures (30 psi and ambient pressure) were used. From Fig. S14,† the peak power density of N-S-CMK-3 cathode glycerol fuel cell was obtained as 82.7 mW cm−2, which has only 6.2% performance dropping in contrast to the 88.2 mW cm−2 achieved by using 30 psi backpressure. Thus, the O2 diffusion issue is negligible when the backpressure changes from 30 psi to ambient pressure. This good O2 diffusion property can likely be attributed to its uniform mesoporous structure with a wide range of mesopores and macropores as well as the high surface area.
The durability of catalyst also serves as an important criterion to determine the suitability of N-S-CMK-3 in the cathodic side for ORR. As shown in Fig. 5b, the glycerol fuel cell shows 90.6 mW cm−2 of peak power density after the 2 h scan current test, which is even slightly higher than the 88.2 mW cm−2 during initial performance and indicates robust stability and high selectivity to ORR in cathode of glycerol fuel cell. In contrast, the performance of the Pt/C cathode glycerol fuel cell was decreased dramatically from 105 mW cm−2 to 66.3 mW cm−2 (36.9% performance dropping) after 2 h, consistent with the ORR performance drop in the half cell fuel-crossover test, because glycerol could be readily oxidized on the Pt/C catalysts surface thereby decreasing the fuel cell performance after the fuel-crossover. Together with the excellent ORR performance in the glycerol alkaline fuel cell, good O2 diffusion and expected durability, the as-synthesized N–S-CMK-3 could effectively serve as the cathodic catalyst in alkaline fuel cell.
3.4 Insights into the synergistic effect of N and S on ORR
To acquire the insight into synergistic effect of N and S heteroatoms on ORR activity, all of the CMK-3-based catalysts were characterized by XPS. The N/C and S/C atomic ratios were both significantly decreased from 6.40 atom% to 1.27 atom% and 1.11 atom% to 0.20 atom%, respectively, when the doping temperature was increased from 700 °C to 900 °C (Fig. 6a and Table S1†), which is similar to previous reports. The broad N1s peak was then deconvoluted into four peaks, assigned to pyridinic-N, pyrrolic-N, graphitic-N and N+–O− (inactive to ORR) structures, respectively (Fig. S15† and 2c).19,43 Interestingly, when combined with the ORR performance and electron transfer number, n, for the N-S-CMK-3 catalysts (Fig. 3b), Fig. 6 reveals a correlation between the graphitic-N species and the ORR activity. For example, the N-S-CMK-3 (800 °C) with a higher proportion of graphitic-N centers favors higher ORR activity with complete 4e− transferred reaction, whereas those containing a lower portion of graphitic-N sites exhibit lower ORR activity for both the 4e− and 2e− reactions. In addition, although the graphitic-N content has been found to be close for N-CMK-3 and N-S-CMK-3 (Fig. 6b and S16†), the N-CMK-3 without S doping exhibits moderate ORR activity with n of 3.4 at 0.5 V, which is much lower than that of N and S dual-doped N-S-CMK-3. Thus, it could be hypothesized that the doped graphitic-N and S atoms can increase both the catalyst activity toward ORR and the synergistic effect between N and S, thus greatly enhancing the ORR activity when combined in the N-S-CMK-3 catalysts.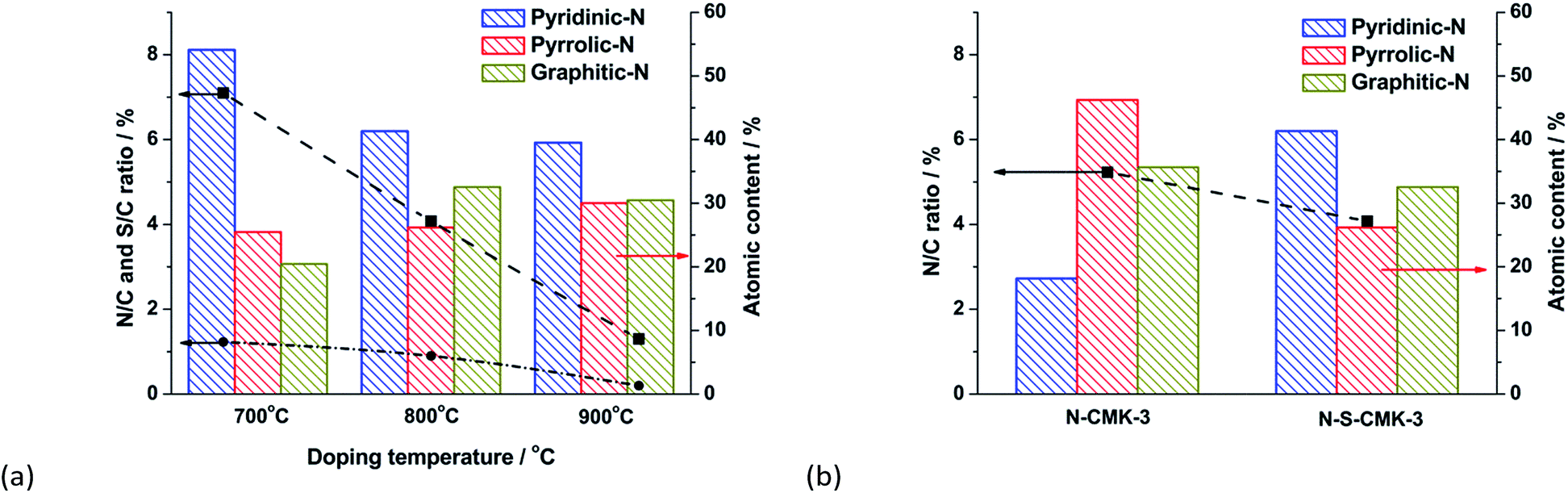 | ||
| Fig. 6 (a) Histograms of different nitrogen-based functional groups content (right y axis) and N/C, S/C atomic ratios (left y axis) characterized on the N-S-CMK-3, versus doping temperature; (b) Histograms of different nitrogen-based functional groups content (right y axis) and N/C atomic ratios (left y axis) characterized on the N-CMK-3 and N-S-CMK-3, respectively. | ||
The ORR activity of these catalytic materials has been found to be determined by atomic charge densities and spin densities, with the latter playing a more important role than the former.52 Previous work has been reported that the carbon atoms with (1) positive spin density and charge density larger than 0.15, or (2) relatively small negative spin density of ≥ −0.01 and large charge density of ≥ 0.2, were most likely to serve as ORR active sites.14,52 For solely doped N–C, the incorporated graphitic-N atom can significantly change the charge density of neighbor carbon atoms due to the larger electronegativity of N (3.04) than that of carbon (2.55). The neighbor carbon atoms near graphitic-N atom in N–C structure show the non-uniform charge density and spin density value, while the carbon atom spin density is zero and charge density is small and uniformly distributed in pure carbon materials, such as graphene.14,53 Thus, some neighbor carbon atoms become catalytic active sites favoring 4e− transfer ORR. On the other hand, for S–C, the small electronegativity difference between S (2.58) and C (2.55) leads a negligible charge transfer. But big sulfur atom incorporated into carbon networks can induce the strain and stress of carbon rings, resulting in a change in charge density of neighbor carbon atoms. In addition, the mismatch of outermost orbitals of sulfur and carbon features a highly asymmetric spin density. Hence, the carbon atoms near the S atom possess a positive spin density, and serve as ORR active sites. Meanwhile, the substituting S atoms in S–C structure also have a positive spin density, thus making them possible to be catalytic active sites toward ORR.14
For N- and S-dual doped carbon materials, the atomic charge density and spin density of carbon atoms were simultaneously affected by incorporated graphitic-N and S atoms, and more active sites were obtained. Therefore, the ORR performance of N-S-CMK-3 mesoporous carbon was significantly improved when both N and S were incorporated into carbon networks. The previous DFT calculation results as well as the experimental results in this work can well explain the synergistic effect of N and S on neighbor carbon atoms, and further highlight the remarkable ORR activity of N-S-CMK-3 with the highest electron transfer number, n = 3.96, as compared to that on solely doped and pristine CMK-3 catalysts.14,41,52
4 Conclusions
In summary, we have demonstrated the successful synthesis of novel N- and S-dual-doped N-S-CMK-3 mesoporous carbons using biorenewable glucose as the carbon source and ammonia and thiophene as highly efficient N and S dopants. The high surface area (1023 m2 g−1), derived from a moderate NH3 etching effect, and the synergistic effect of graphitic-N and S atoms resulted in remarkable electrocatalytic ORR activity. N-S-CMK-3 annealed at 800 °C exhibits the highest ORR activity (close to that of Pt/C) with the onset potential of 0.92 V vs. RHE, Tafel slope of 68 mV dec−1, and 3.96 electron transfer number per oxygen molecule in 0.1 M KOH. By directly using N-S-CMK-3 as the cathode catalyst in single direct glycerol fuel cell, it exhibited 88.2 mW cm−2 peak power density without obvious O2 diffusion limitation, which represents 84% of the initial performance of the one with a Pt/C cathode. More interestingly, the peak power density of the fuel cell with N-S-CMK-3 can maintain a peak power density at 90.6 mW cm−2 after two hours operation, while the one with Pt/C cathode lost its peak power density to 66.3 mW cm−2. The robust stability and alleviation of fuel-crossover issue (resistance to alcohol oxidation) demonstrated in both half cell and single fuel cell tests strongly suggest the N-S-CMK-3 could be a promising substitute to replace noble metal and serve as cathodic catalyst for alcohol alkaline fuel cells, H2/O2 fuel cells and likely other energy conversion and storage devices.Acknowledgements
We acknowledge financial support from NSF-CBET 1501113 and ISU and Ames Lab Startup Funds, and are grateful to Dr Dapeng Jing and James Anderegg for XPS characterization on CMK-3 materials and Dr Le Xin for fruitful discussion. W. Li thanks his Richard Seagrave Professorship support.Notes and references
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS PubMed.
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981 CrossRef CAS PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- E. H. Yu, X. Wang, U. Krewer, L. Li and K. Scott, Energy Environ. Sci., 2012, 5, 5668 CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187 CrossRef CAS.
- W. Z. Li, W. J. Zhou, H. Q. Li, Z. H. Zhou, B. Zhou, G. Q. Sun and Q. Xin, Electrochim. Acta, 2004, 49, 1045 CrossRef CAS.
- S. R. Brankovic, J. X. Wang and R. R. Adzic, Surf. Sci., 2001, 474, L173 CrossRef CAS.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249 CrossRef CAS.
- S. J. Guo, S. Zhang and S. H. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed.
- S. Y. Wang, E. Iyyamperumal, A. Roy, Y. H. Xue, D. S. Yu and L. M. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756 CrossRef CAS PubMed.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. F. Li, G. Y. Fang, H. G. Nie, Z. Liu, X. M. Zhou, X. Chen and S. M. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- H. L. Jiang, Y. H. Zhu, Q. Feng, Y. H. Su, X. L. Yang and C. Z. Li, Chem.–Eur. J., 2014, 20, 3106 CrossRef CAS PubMed.
- L. J. Yang, S. J. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Z. Wang, Q. Wu, J. Ma, Y. W. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed.
- D. C. Higgins, M. A. Hoque, F. Hassan, J. Y. Choi, B. Kim and Z. W. Chen, ACS Catal., 2014, 4, 2734 CrossRef CAS.
- Y. X. Zhang, J. Zhang and D. S. Su, ChemSusChem, 2014, 7, 1240 CrossRef CAS PubMed.
- W. W. Gao, X. Feng, T. Y. Zhang, H Huang, J. Li and W. B. Song, ACS Appl. Mater. Interfaces, 2014, 6, 19109 CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS PubMed.
- J. X. Xu, Y. Zhao, C. Shen and L. H. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 12594 CAS.
- R. L. Liu, D. Q. Wu, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 CAS.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Marchionni, F. Vizza, L. Q. Wang and P. K. Shen, Fuel Cells, 2010, 10, 582 CrossRef CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
- P. Chen, L. K. Wang, G. Wang, M. R. Gao, J. Ge, W. J. Yuan, Y. H. Shen, A. J. Xie and S. H. Yu, Energy Environ. Sci., 2014, 7, 4095 CAS.
- H. Wang, X. J. Bo, Y. F. Zhang and L. P. Guo, Electrochim. Acta, 2013, 108, 404 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- Z. Y. Zhang, G. M. Veith, G. M. Brown, P. F. Fulvio, P. C. Hillesheim, S. Dai and S. H. Overbury, Chem. Commun., 2014, 50, 1469 RSC.
- A. H. Lu and F. Schuth, Adv. Mater., 2006, 18, 1793 CrossRef CAS.
- W. Kicinski, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1 CrossRef CAS.
- D. Y. Zhang, Y. Hao, L. W. Zheng, Y. Ma, H. X. Feng and H. M. Luo, J. Mater. Chem. A, 2013, 1, 7584 CAS.
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027 CrossRef CAS PubMed.
- T. Y. Ma, J. R. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54(15), 4646–4650 CrossRef CAS PubMed.
- J. J. Shi, X. J. Zhou, P. Xu, J. L. Qiao, Z. W. Chen and Y. Y. Liu, Electrochim. Acta, 2014, 145, 259–269 CrossRef CAS.
- Y. Gorlin, C. J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2(12), 2687–2694 CrossRef CAS.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 Search PubMed.
- Z. C. Wang, L. Xin, X. S. Zhao, Y. Qiu, Z. Y. Zhang, O. A. Baturina and W. Z. Li, Renewable Energy, 2014, 62, 556 CrossRef CAS.
- Q. G. He, Q. Li, S. Khene, X. M. Ren, F. E. Lopez-Suarez, D. Lozano-Castello, A. Bueno-Lopez and G. Wu, J. Phys. Chem. C, 2013, 117, 8697 CAS.
- L. Xin, Z. Y. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Z. Li, ChemSusChem, 2013, 6, 674 CrossRef CAS PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS PubMed.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- S. Y. Wang, L. P. Zhang, Z. H. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed.
- Y. Qiu, L. Xin and W. Z. Li, Langmuir, 2014, 30, 7893–7901 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06039g |
| ‡ Yang Qiu and Jiajie Huo contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |