
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA widespread bacterial phenazine forms S-conjugates with biogenic thiols and crosslinks proteins†
D.
Heine
a,
S.
Sundaram
a,
Matthias
Beudert
a,
K.
Martin
a and
C.
Hertweck
*ab
aLeibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Beutenbergstrasse 11a, 07745 Jena, Germany. E-mail: christian.hertweck@leibniz-hki.de
bFriedrich Schiller University, 07737 Jena, Germany
First published on 14th April 2016
Abstract
Phenazines are redox-active compounds produced by a range of bacteria, including many pathogens. Endowed with various biological activities, these ubiquitous N-heterocycles are well known for their ability to generate reactive oxygen species by redox cycling. Phenazines may lead to an irreversible depletion of glutathione, but a detailed mechanism has remained elusive. Furthermore, it is not understood why phenazines have so many protein targets and cause protein misfolding as well as their aggregation. Here we report the discovery of unprecedented conjugates (panphenazines A, B) of panthetheine and phenazine-1-carboxylic (PCA) acid from a Kitasatospora sp., which prompted us to investigate their biogenesis. We found that PCA reacts with diverse biogenic thiols under radical-forming conditions, which provides a plausible model for irreversible glutathione depletion. To evaluate the scope of the reaction in cells we designed biotin and rhodamine conjugates for protein labelling and examined their covalent fusion with model proteins (ketosynthase, carbonic anhydrase III, albumin). Our results reveal important, yet overlooked biological roles of phenazines and show for the first time their function in protein conjugation and crosslinking.
Introduction
From an evolutionary perspective, it is most remarkable how small biomolecules have been shaped into privileged structures that fulfil multiple biological functions. This is particularly true for the large family of phenazines, ancient N-heterocycles that are produced by a large variety of bacteria.1 Initially discovered as colourful bacterial pigments over 150 years ago, to date over one hundred natural phenazines have been characterised, most of which are endowed with highly diverse bioactivities.2 These findings inspired the chemical synthesis of thousands of phenazine derivatives and propelled the development of antibiotics and anticancer agents.1–3 However, phenazines are a remarkable example of antibiotic compounds that are actually crucial for microbial interaction and competitive fitness.4–6 They not only act as signalling molecules in microbial communication,7–9 mediating biofilm formation and morphogenesis of colonies,10 but also control gene expression11 and influence the intracellular redox state10 as well as exocellular electron transfer.12 Owing to their redox properties, phenazines play major roles in infection processes of pathogens such as Pseudomonas aeruginosa, especially in cystic fibrosis patients.13 In particular, pyocyanin (PYO, 1) and phenazine-1-carboxylic acid (PCA, 2), the key intermediate in phenazine biosynthesis,14 represent important virulence factors of P. aeruginosa that damage infected tissue (Fig. 1).15,16 The primary reason for the deleterious effects is phenazine-mediated redox cycling,1 which leads to high concentrations of reactive oxygen species (ROS). Naturally, ROS is effectively counteracted by reduced glutathione (GSH) as redox buffer, with formation of the oxidised disulfide form (GSSG).17,18 Despite an impressive body of data on phenazine functions, there are numerous open questions on their mode of action. Specifically, it was observed that phenazines may lead to a decrease in GSH levels by conjugation, but the reaction mechanism has remained elusive.19–22 Moreover, it was found that ROS formation alone is neither responsible for the observed cytotoxic effects nor for misfolding and aggregation of proteins in the model organism C. elegans when exposed to phenazines.23 Notably, addition of antioxidative agents failed to rescue the proteotoxic and protein aggregation phenotypes.23 In this context it is remarkable that phenazine-mediated toxicity may be attributed to autophagy, which can result from high titres of misfolded proteins.24 Yet, a link between non-functional proteins and phenazines has been missing. Furthermore, it is not yet sufficiently understood why phenazines have such broad activity profiles and highly diverse biological targets.2,25 Here, we report the discovery of unprecedented phenazine–pantetheine conjugates and the elucidation of their biogenesis, which revealed an overlooked mode of action of phenazines. Using two different synthetic probes we analysed the ability of phenazines to form thiol conjugates and crosslink proteins by a radical mechanism, which may clarify previously inexplicable disease symptoms and enigmatic phenazine functions.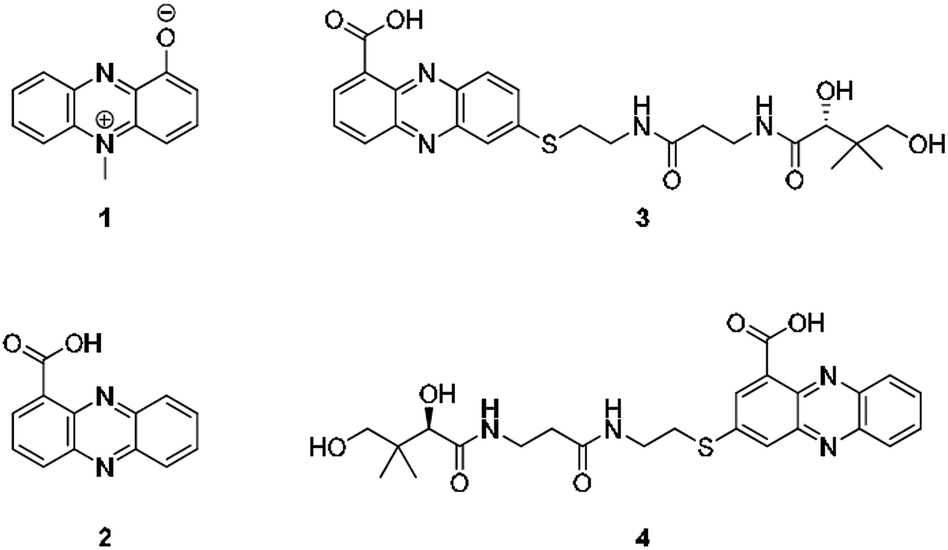 | ||
| Fig. 1 Structures of pyocyanin (1), PCA (2), and panphenazines A (3) and B (4) discovered in this study. | ||
Results and discussion
Discovery of panphenazines
Through genome mining26 of the rare actinomycete Kitasatospora sp. HKI 714 we found that this soil-derived actinobacterium has the potential to produce prenylated phenazines. The genomics-guided search led to the discovery of a series of new endophenazines along with acylated phenazine derivatives.27 As the strain produced large amounts of PCA (2), the universal precursor of phenazine derivatives, we expected to discover more congeners. In fact, metabolic profiling of concentrated culture extracts indicated the presence of unusual sulfur-containing phenazines. HRMS data (m/z = 501.1807 ([M + H]+ for 3), m/z = 501.1808 ([M + H]+ for 4)) and sulfur isotope pattern analysis suggested a molecular formula of C24H29O6N4S for both compounds. Because of the extremely low titres of the new compounds (≈5 μg L−1) the strain needed to be grown on a large scale (200 L). By semipreparative HPLC we eventually succeeded in isolating both compounds, named panphenazine A (3, 1.4 mg) and B (4, 0.9 mg), in sufficient amounts for NMR analyses. NMR data confirmed the phenazine core, and the downfield-shift (145.2 ppm) of the substituted quaternary C-7 of 3 suggested the presence of a thioether-bound side chain. The adjacent ethylene unit belongs to a cysteamine residue, and HMBC data pointed towards a peptide bond to the carboxy terminus of β-alanine. The remaining part of the molecule represents pantoic acid, which is connected to the β-alanine by another amide bond. The side chain is therefore identical to pantetheine, the acyl group carrier of coenzyme A or acyl carrier proteins during fatty acid and polyketide biosynthesis. The position of the side chain was confirmed by COSY and HMBC experiments. In an analogous way we elucidated the structure of compound 4, which differs from 3 only in the substitution site (C-3 in lieu of C-7) (Fig. S1 and Table S1†). Thus, panphenazines 3 and 4 are highly unusual pantetheine adducts of PCA (2).Biomimetic synthesis of PCA thiol conjugates
Considering the hundreds of known natural phenazines, reports on sulfur-containing derivatives are scarce. The few reported examples are two N-acetylcysteine conjugates, dermacozine J28 and SB 212305,29 and a partially characterised pyocyanin glutathione adduct.21,22 To date, practically nothing is known about their formation. Various enzymes have been described that mediate C–S bond formation in natural product biosynthetic pathways.30–36 However, inspection of the endophenazine (epa) biosynthesis gene cluster in the Kitasatospora sp. HKI 714 genome27 did not reveal any candidate genes that could be involved in the formation of the thioconjugates. Considering the substitution pattern, it was conceivable that 3 and 4 have resulted from a non-enzymatic reaction. Possible scenarios are (a) an oxidative aromatic coupling, (b) a Michael addition followed by reoxidation of the aromatic system, and (c) a coupling reaction involving radicals.To elucidate the mechanism of phenazine thioether formation, we performed a series of experiments using PCA (2) and N-acetylcysteamine as a simplified surrogate of pantetheine. Since addition of mild oxidizing agents or bases or incubation of PCA with disulfides (cysteine, pantethine) did not lead to S-conjugates, mechanisms involving oxidation or nucleophilic attack of a deprotonated thiol were ruled out. Instead, we found that thiol conjugation proceeded smoothly under the influence of sunlight. PCA has UV absorption maxima at 250 nm and 370 nm, which could serve for photo-excitation. Irradiation of the reaction mixture with an UV lamp (370 nm) yielded various regioisomeric conjugates, 5 (10%), 6 (8%), 7 (5%) and 8 (6%) (Fig. 2). HPLC-HRMS analysis indicated that, in addition to the monosubstituted conjugates, various other derivatives containing two or three N-acetyl cysteamine moieties were formed. Using these reaction conditions, we mimicked the biogenesis of the panphenazines from pantetheine and PCA. In contrast to the experiment with N-acetylcysteamine, irradiation of the solution at 370 nm yielded exclusively two regioisomers. HPLC-HRMS comparison unequivocally proved that the products are identical with the conjugates 3 and 4 isolated from Kitasatospora sp. HKI 714 (Fig. S2†). Furthermore, the photoreaction proved to be a viable route for coupling cysteine and glutathione with PCA, as evidenced by HRMS and MSn analyses (Fig. S3 and S4†).
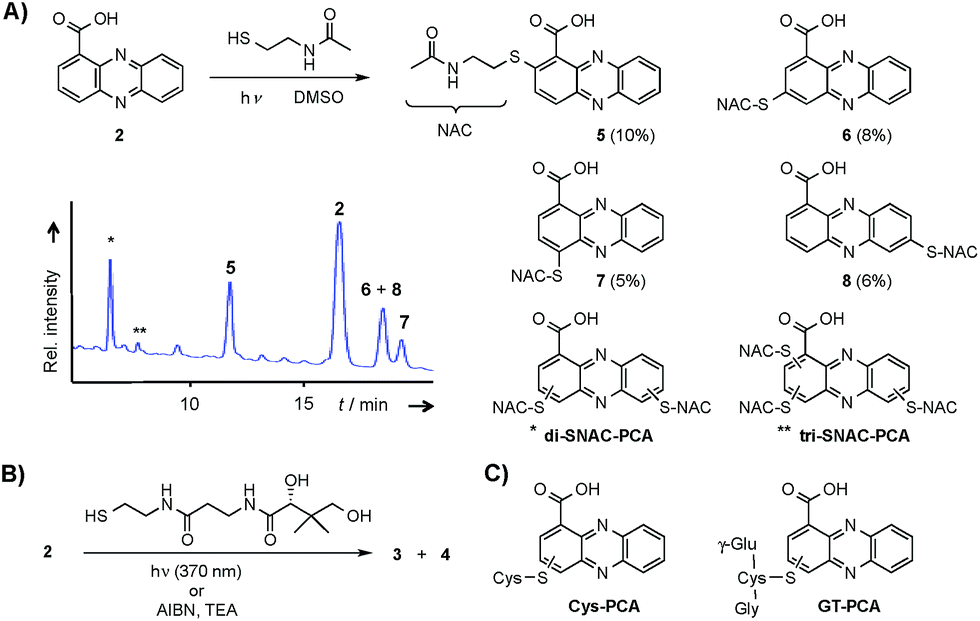 | ||
| Fig. 2 Formation of PCA-thiol conjugates. (A) Photo-induced conjugate formation of PCA and N-acetylcysteamine (NAC) at 370 nm (HPLC profile after 24 h); (B) biomimetic synthesis of 3 and 4; (C) general structure of cysteine–PCA and glutathione–PCA adducts resulting from irradiation. | ||
The successful photoreactions and the substitution pattern are highly suggestive for the involvement of resonance-stabilised radicals in thiol conjugation. To interrogate the reaction mechanism, we conducted the coupling reaction of PCA and N-acetylcysteamine in the dark, but added azobisisobutyronitrile (AIBN) as a radical initiator. HPLC-HRMS analyses clearly indicated the formation of the corresponding adducts. Likewise, 3 and 4 were formed after incubation of PCA with pantetheine and AIBN (Fig. S5†).
Mechanistic considerations
The radical mechanism is supported by two other observations: first, addition of an electron source such as triethylamine (TEA) substantially increases product formation (up to tenfold), which is in agreement with the reported mechanism of the formation of the phenazine radical anion.37 Second, addition of the radical scavenger t-butylhydroxytoluene (TBHT) hampered the photoreaction; only traces of conjugates could be detected. Thus, in addition to the observed substitution pattern of the thioconjugates, three lines of experimental evidence support the radical-based reaction mechanism (Fig. 3).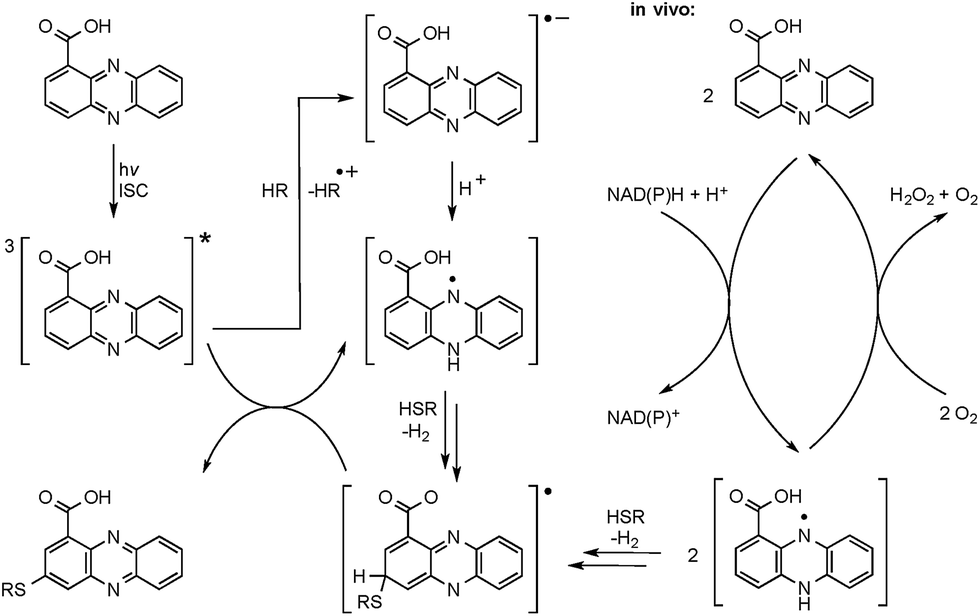 | ||
| Fig. 3 Proposed reaction mechanism for the radical-induced conjugate addition. Radical formation can proceed via a photochemical reaction (left) or by redox cycling inside the cell (right). Only one of the possible intermediates (resonance form) is shown, leading to the derivative substituted at position 7. ISC = intersystem crossing; HR, electron donor (triethylamine, thiol or other); HSR, thiol. | ||
However, a direct light-induced formation of the thiyl radical can be ruled out. The homolytic cleavage of the SH bond occurs at wavelengths below 300 nm,38 yet the PCA-thioconjugates are readily produced at 370 nm. Alternatively, it is more plausible that radical formation is initiated by photoexcitation of PCA into the triplet state. Transferring an electron from TEA (or an alternative electron source) leads to the phenazine radical anion (Fig. 3).37 Subsequent protonation generates a neutral radical, which can react with biogenic thiols (GSH, pantetheine, cysteamine, cysteine) to form a thiyl radical while regenerating PCA. By analogy to the aromatic substitution reaction with thiyl radicals,39 one may infer that the thiyl radicals readily attack the electron-rich phenazine core. After H abstraction and re-aromatization, the radical chain reaction continues, which may also lead to multiple substitutions as observed for N-acetylcysteamine. On the contrary, conjugate formation with pantetheine seems to appear more selectively, possibly due to an increased steric demand of the thiol. Even though various conjugates could be detected in the HPLC-HRMS trace, only two isomers (3 and 4) appear in higher amounts. It should be highlighted that light-independent avenues to the formation of phenazine radicals have been reported in the biological context. It is well known that PYO is chemically reduced through radical intermediates in the cell, and phenazine redox-cycling (intracellular as well as extracellular) may generate PCA radicals,1 which can react with thiols to form conjugates.
Phenazine protein adduct formation and crosslinking
Single-electron redox processes yield phenazine radicals in host cells; consequently, these reactive species may not only react with biogenic thiols, but also with SH residues of proteins, an important target of many small molecules.40,41 Such a process would be reminiscent of non-enzymatic reactions of sugars with proteins, yielding advanced glycation end-products (AGEs), which play important roles in aging and degenerative diseases.42 Considering the non-selective adduct formation of phenazines and the multitude of thiols in living cells, it is obvious that the corresponding conjugates would barely be detectable in vivo as individual entities. Thus we investigated whether PCA radicals form conjugates with individual proteins in vitro. For this, we synthesised a chemical probe (9) (Fig. S6†) in which the phenazine is fused to biotin via a tetraethylene glycol (TEG) linker (Fig. 4A).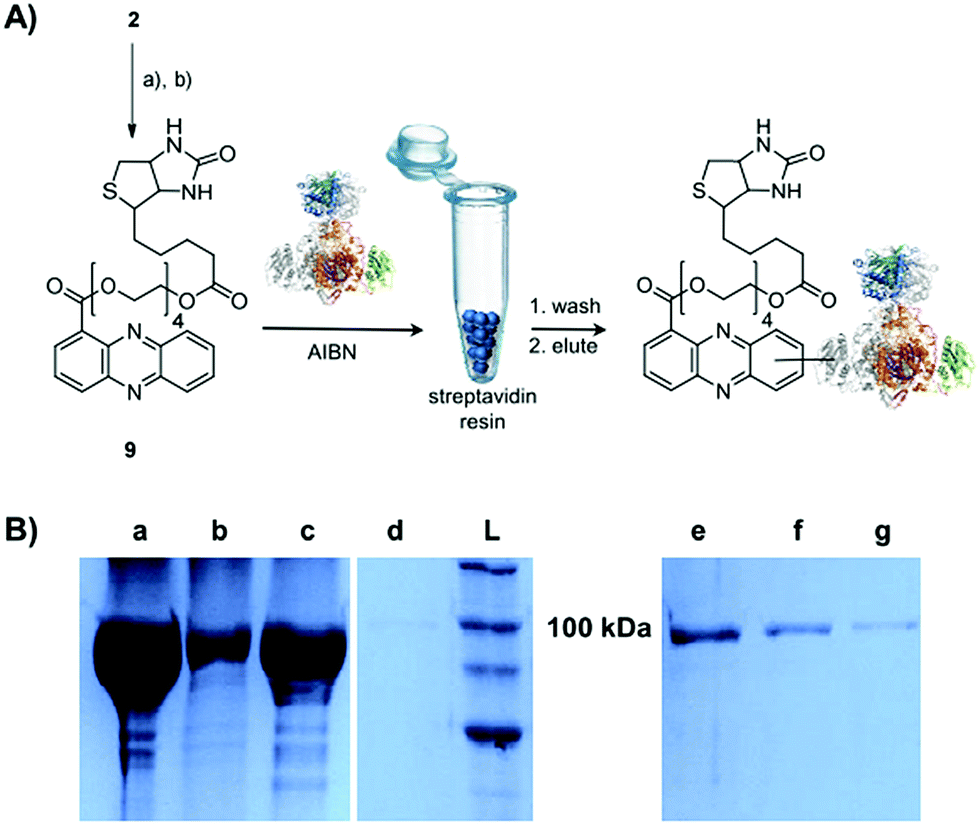 | ||
| Fig. 4 Evidence for radical-mediated protein conjugation by PCA. (A) Synthesis of biotin-based probe 9, thio-adduct formation with ketosynthase (KS-B) and purification with streptavidin resin; (a) tetraethylene glycol, EDC, DMAP, DCM, 86% (b) biotin, EDC, DMAP, DCM, 96%. (B) SDS-PAGE (10%) of KS-B after the incubation with the probe 9 for 24 h in the presence of AIBN. (a) Load, (b) flow-through, (c) first wash, (d) second wash, (e) first elution, (f) second elution, (g) third elution, (L) ladder. | ||
First, we incubated 9 and AIBN with a cysteine-rich didomain (ketosynthase-branching, KS-B, 14 cysteines) of a modular polyketide synthase (PKS) module.43–45 The reaction mixture was loaded onto a streptavidin column, and the biotinylated protein was eluted at 60 °C in buffer with 3 mM biotin after multiple washing steps. Subsequent sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis indicated that the protein was indeed linked to the biotin-labelled probe, unequivocally proving that the phenazine was bound to the PKS component (Fig. 4B). To gain insight into the fate of proteins in the presence of the phenazine and UV light, we also prepared a fluorescent probe (10). Although PCA shows fluorescence itself, the respective signal proved to be too weak for the detection of protein conjugates. Therefore we attached PCA to rhodamine B, a fluorophore with an exceptionally high fluorescence quantum yield. The KS-B didomain was subjected to the rhodamine-based probe 10 with irradiation at 370 nm, and the reaction mixture was analysed by SDS-PAGE on a 12% gel. Surprisingly, the fluorescent bands were substantially larger than expected. Instead of the predicted band at 100 kDa, we observed a fluorescent signal that corresponds to a molecular weight of over 200 kDa (Fig. S7†). One possible explanation for this finding is that the probe might have cross-linked two or more equivalents of the protein. When analysing the products on a lower concentrated gel (10%) for better separation, we detected several protein aggregates of various sizes (Fig. 5B).
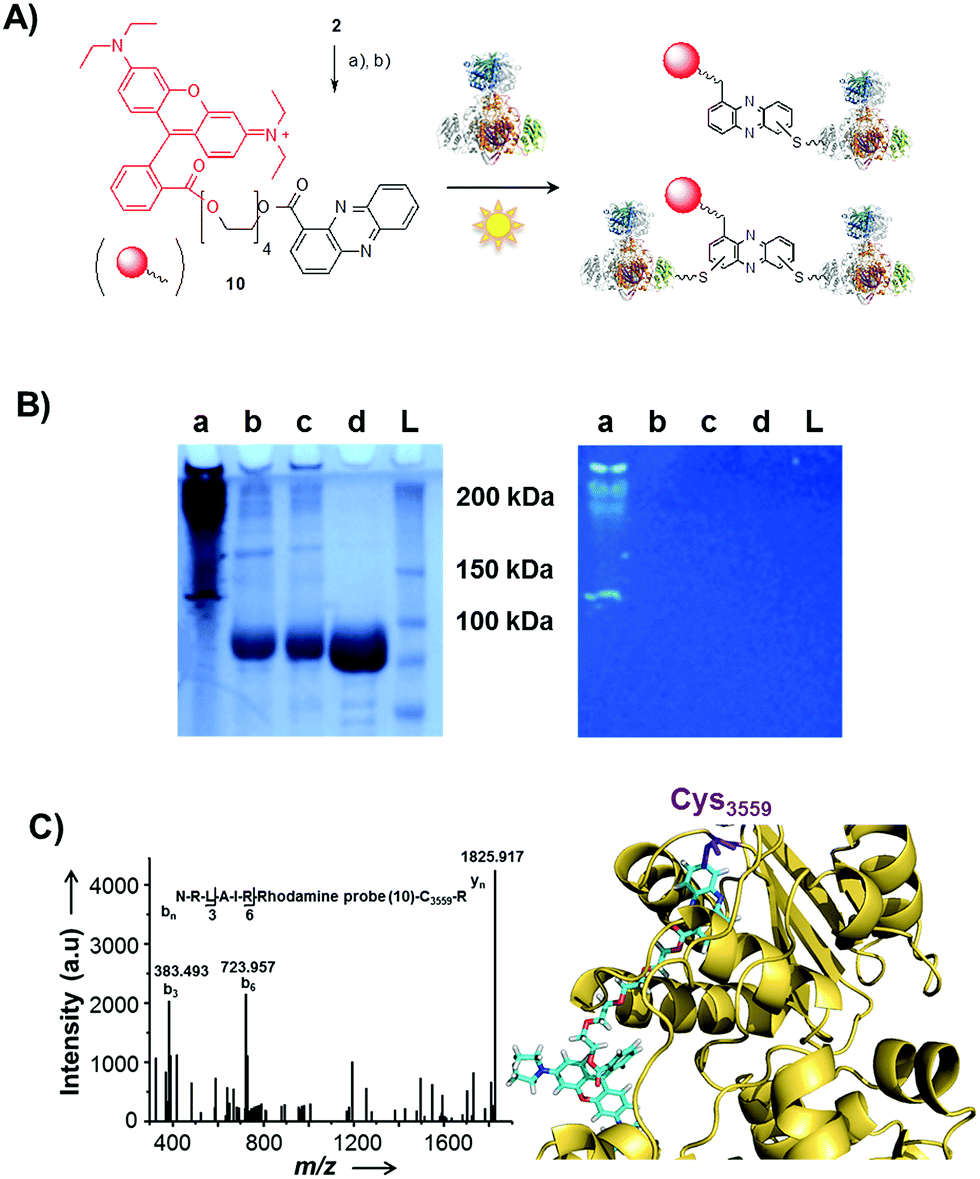 | ||
| Fig. 5 Photo-induced protein crosslinking by PCA. (A) Synthesis of PCA-rhodamine B conjugate 10 and thio-conjugate formation with KS-B, (a) tetraethylene glycol, EDC, DMAP, DCM, 86%, (b) rhodamine B, PyBOP, Hünigs base, DCM, 63%. (B) SDS-PAGE (10%) of KS-B after the incubation with probe 10 or rhodamine B in water (left picture: under white light; right picture at 370 nm); (contrast adjusted) (a) 10, 370 nm, 18 h, (b) rhodamine B, 370 nm, 18 h, (c) H2O, 370 nm, 18 h, (d) KS-B control; (L) ladder. (C) MALDI-MS/MS spectrum of the rhodamine-based probe (10) linked to a peptide fragment of KS-B and model of the rhodamine-based probe (10) linked to Cys3559 of KS-B (PDB code 4KC5). | ||
By tryptic digestion and MALDI analyses we corroborated that all protein complexes resulting from the incubation of KS-B with 9 and 10 are indeed derived from the KS (Fig. S8.1–S8.6†). In control experiments where the protein was irradiated in the presence of rhodamine B alone no aggregation could be observed (Fig. 5B). MS/MS analysis of the protein (lane a) revealed one of the peptides where 10 was bound to Cys3559 (Fig. 5C). A model was generated to show the proposed binding mode of 10 to this cysteine using Pymol (Fig. 5D). These findings unequivocally showed that the crosslinking activity correlates with the presence of the phenazine moiety and that multiple linkages are formed. To test the scope of the reaction we employed two proteins that are completely unrelated to a PKS, carbonic anhydrase III (5 cysteines) and albumin (35 cysteines). After incubation of carbonic anhydrase III with probe 10 we monitored the time course of its aggregation. Bands corresponding to the native protein slowly disappeared with concomitant formation of extensive protein aggregates bound to the fluorescent probe (Fig. S9†). Using the same conditions, albumin was functionalised with the rhodamine-based probe 10, yet formation of higher aggregates was negligible (Fig. S10†). Nonetheless, the monoadduct was ideally suited to confirm the binding of the probe to a cysteine side chain by MALDI analysis (Fig. S11 and S12†). Taken together, all selected proteins are derivatised with the phenazine probe, yet they differ in their tendency to form higher aggregates, which may be controlled by steric factors.
Conclusions
Considering the large number of studies on phenazines and their biological activities, it is surprising that their ability to capture biogenic thiols and crosslink proteins has so far been overlooked. The discovery of the panphenazines and the mechanistic investigations revealed that phenazine radicals form conjugates with a range of thiols. Thus, we provide a plausible model for irreversible GSH reduction in human endothelial cells.19 Furthermore, we report for the first time that phenazines form S-conjugates with proteins. Protein modification and aggregation mediated by small molecules are the focus of diverse research on biomaterials,46,47 and these processes are also relevant for protein aging and degenerative diseases.42 Our findings are significant because PCA is an important biosynthetic building block that is produced in large amounts by many pathogenic pseudomonads during infection processes.2,48 Since phenazine radicals are readily formed in cells by redox cycling, PCA and congeners may contribute to protein degeneration processes. Specifically, our findings may explain the broad activity profiles of phenazines including protein aggregation in C. elegans.23 Radical-based thiol conjugation and protein crosslinking by phenazines represent novel modes of action that are an important addition to the body of knowledge on this widespread group of virulence factors.6Experimental
General analytical procedures
All NMR experiments were performed on Bruker AVANCE II 300, AVANCE III 500 or a 600 MHz spectrometer, equipped with a Bruker Cryo Platform. The chemical shifts are reported in parts per million (ppm) relative to the solvent residual peak of chloroform-d1 (1H: 7.24 ppm, singlet; 13C: 77.00 ppm, triplet), methanol-d4 (1H: 3.30 ppm, quintet; 13C: 49.00 ppm, septet) or DMSO-D6 (1H: 2.50 ppm, quintet; 13C: 39.50 ppm, septet). HR ESI-MS was carried out on an Accela UPLC-system (Thermo Scientific) combined with an Exactive mass spectrometer (Thermo Scientific) equipped with an electrospray ion source. Solid phase extraction was carried out using Chromabond C18 ec Cartridges filled with 2000 mg of octadecyl-modified silica gel (Macherey-Nagel). HPLC-MS measurements were performed on a HPLC 1100 System connected to a 1100 Series LC/MSD Trap using an Zorbax Eclipse XDB-C8, 4.6 × 150 mm column, particle size 5 μm (Agilent Technologies). For thin layer chromatography TLC aluminium sheets silica gel 60 F254 (MERCK) were used. Open column chromatography was performed using silica gel 60; particle size 0.015–0.04 mm (Macherey-Nagel) and on Sephadex® LH-20 (Sigma-Aldrich). Semi preparative HPLC was performed on an 1260 Infinity System (Agilent Technologies) using the following columns: Zorbax Eclipse XDB-C8, 9.5 × 250 mm, particle size 5 μm (Agilent Technologies), Zorbax Eclipse XDB-C18, 9.5 × 250 mm, particle size 5 μm (Agilent Technologies) and EC 250/4 Nucleodur Sphinx RP, 10 × 250 mm, particle size 5 μm (Macherey-Nagel). All solvents for analytical and preparative HPLC were obtained commercially in gradient grade and were filtered prior to use. To avoid microbial growth 0.1% formic acid was added to the water, used for analytical and preparative HPLC. UV-spectra were obtained using a UV-spectrometer UV-1800 (Shimadzu). IR-spectra were recorded on an FT/IR-4100 ATR spectrometer (Jasco). For irradiation experiments an UV-lamp TL MINI 6 Watt 33-640 (Philips) and an UV-lamp F 6W T5 G5 129 3000K 280 lm (Sylvania) were used.Strain cultivation and isolation of panphenazines
The cultivation of Kitasatospora sp. HKI 714 and the fractionation of the crude extract were performed in exactly the same way as reported before.27 Compounds 1 and 2 were isolated by semipreparative HPLC (Zorbax Eclipse XDB-C18, 9.5 × 250 mm, 5 μm, flow rate 4 mL min−1, gradient starting from H2O/MeOH 90/10, in 2.5 min to 50/50, in 15.5 min to 46/54, in 7 min to 42/58, in 5 min to 0/100).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε): 236 (6.22), 274 (6.36), 381 (5.87), 427 (5.78) nm; HR ESI-MS: m/z = 501.1807 [M + H]+ (calculated 501.1809 for C24H29O6N4S).
ε): 261 (6.05), 372 (5.52), 408 (5.41) nm; HR ESI-MS: m/z = 501.1808 [M + H]+ (calculated 501.1809 for C24H29O6N4S).
ε): 236 (6.22), 274 (6.36), 381 (5.87), 427 (5.78) nm; HR ESI-MS: m/z = 501.1807 [M + H]+ (calculated 501.1809 for C24H29O6N4S).
ε): 261 (6.05), 372 (5.52), 408 (5.41) nm; HR ESI-MS: m/z = 501.1808 [M + H]+ (calculated 501.1809 for C24H29O6N4S).
General synthetic procedures
All reagents were obtained from commercial suppliers (Sigma Aldrich and Acros Organics) and were used without further purification. Carbonic anhydrase III (29.5 kDa) was obtained from GE Healthcare UK Limited and albumin (66 kDa) was obtained from Carl Roth GmbH. Chemical solvents were obtained at least in HPLC grade. Methanol and chloroform were distilled prior to use. Reactions were performed under inert atmosphere (Argon) using the Schlenk technique.Pantetheine
A solution of D-pantethine (200 mg, 0.36 mmol) and dithiothreitol (100 mg, 0.65 mmol) in a methanolic solution (4 mL; methanol:water; 1:1; v/v) was stirred for 2 days in an inert atmosphere (argon). The solvent was removed under reduced pressure and the product purified by column chromatography using silica gel (chloroform/methanol: 98/2: v/v) to give 195 mg of a colourless liquid. Yield 97%. 1H NMR (300 MHz, MeOD): δ 0.9 (6H, s), 2.4 (2H, t), 2.6 (2H, t), 3.3–3.6 (6H, m), 3.9 (1H, s) ppm. 13C NMR (75.5 MHz, MeOD): δ 20.9, 21.3, 24.5, 36.4, 40.4, 43.9, 70.3, 77.2, 79.5, 173.9, 176.1 ppm.
Synthesis of panphenazines (3 and 4)
Phenazine-1-carboxylic acid (2, 3 mg, 0.013 mmol) was dissolved in DMSO (300 μL) and pantetheine (4 mg, 0.013 mmol) was added. The stirred mixture was incubated at 370 nm for 24 h. The solvent was removed under reduced pressure and the crude product dissolved in MeOH. HPLC-HRMS proved the identity of one product with compound 4. Compound 3 was only produced in minor amounts under these conditions. The yield of 4 was quantified by integration of its peak area at a detector wavelength of 430 nm. Under these conditions yield was determined to be 2%. Repeating the reaction with the addition of TEA (1.9 μL, 0.014 mmol) increased the yield to 24%. In contrast, addition of TBHT instead of TEA hampered the reaction and no corresponding signal for 4 could be detected in the UV trace.Synthesis of phenazine N-acetylcysteamine conjugates (5–8)
Phenazine-1-carboxylic acid (2, 10 mg, 0.045 mmol) and N-acetylcysteamine (5 mg, 0.042 mmol) were dissolved in DMSO and incubated at 370 nm for 24 h. The solvent was removed under reduced pressure and the residue was subjected to semipreparative HPLC (Zorbax Eclipse XDB-C8, 9.5 × 250 mm, 5 μm, flow rate 4 mL min−1, gradient starting from H2O/ACN 90/10, in 3 min to 80/20, in 25 min to 60/40, in 2 min to 0/100). Yields of monosubstituted phenazines: 5 (10%), 6 (8%), 7 (5%), 8 (6%) (yellow solids).Synthesis of phenazine-biotine and -rhodamine probes
Tetraethyleneglycol phenazine-1-carboxylate (11)
Phenazine-1-carboxylic acid 2 (10 mg, 0.044 mmol) was dissolved in dichloromethane (5 mL) and cooled to 0 °C. Tetraethylene glycol (26 mg, 0.13 mmol, 3 eq.), DMAP (5 mg, 0.04 mmol) and EDC (8.4 mg, 0.05 mmol) were added. After 24 h, water was added and the aqueous phase extracted with dichloromethane (2 × 10 mL). The combined organic fractions were dried over sodium sulfate and concentrated under reduced pressure. Column chromatography over silica gel (chloroform/methanol: 98/2, v/v) gave the product (15.3 mg) as yellow oil. Yield: 86%. 1H NMR (600 MHz, MeOD): δ 3.47 (2H, t, 3J = 4.9 Hz), 3.54 (2H, m), 3.58 (2H, t, 3J = 4.9 Hz), 3.61 (2H, m), 3.66 (2H, m), 3.73 (2H, m), 3.93 (2H, t, 3J = 4.7 Hz), 4.65 (2H, t, 3J = 4.7 Hz), 7.96–7.99 (3H, m), 8.24–8.27 (1H, m), 8.31 (1H, dd, 3J = 6.8 Hz, 4J = 1.3 Hz), 8.33 (1H, m), 8.40 (1H, dd, 3J = 8.8 Hz, 4J = 1.3 Hz) ppm. 13C NMR (150.9 MHz, MeOD): δ 62.2, 66.0, 70.2, 71.4, 71.6, 71.7, 73.6, 79.5, 130.3, 130.9, 131.0, 132.7, 132.8, 132.9, 133.4, 134.0, 142.0, 143.8, 144.5, 144.9, 168.1 ppm; IR (film): 1119, 1214, 1237, 1274, 1350, 1421, 1523, 1727, 2874, 3018 cm−1, HRMS: (ESI+): m/z calculated for C21H25O6N2 [M + H]+ 401.1707, found: 401.1710.Biotin-labelled phenazine-1-carboxylic acid (9)
Tetraethylene-glycol phenazine-1-carboxylate (7 mg, 0.017 mmol) 11 and biotin (8.5 mg, 0.035 mmol, 2 eq.) were dissolved in dry dichloromethane (2 mL) and cooled to 0 °C. DMAP (2 mg, 0.0.17 mmol) and EDC (8 mg, 0.044 mmol, 2.5 eq.) were added. Water (5 mL) was added after 20 h and the aqueous phase extracted with dichloromethane (2 × 5 mL). The combined organic fractions were dried over sodium sulfate and concentrated under reduced pressure. Column chromatography over silica gel (chloroform/methanol: 95/5, v/v) gave 10.5 mg of a yellow solid. Yield: 96%. 1H NMR (500 MHz, CDCl3): δ 1.56–1.70 (6H, m), 2.28–2.32 (2H, m), 2.67–2.73 (1H, m), 2.81–2.88 (1H, m), 3.06–3.13 (1H, m), 3.57–3.65 (10H, m), 3.72 (2H, dd, 3J1 = 5.4 Hz, 3J2 = 4.1 Hz), 3.91 (2H, t, 3J1 = 4.8 Hz), 4.14–4.18 (2H, m), 4.23–4.28 (1H, m), 4.43–4.48 (1H, m), 4.65 (2H, t, 3J1 = 4.8 Hz), 7.82–7.86 (3H, m), 8.20–8.23 (1H, m), 8.23 (1H, dd, 3J1 = 7.1 Hz, 4J2 = 1.3 Hz), 8.27–8.30 (1H, m), 8.37 (1H, dd, 3J1 = 8.8 Hz, 4J2 = 1.3 Hz) ppm. 13C NMR (125 MHz, MeOD): δ ppm. 13C NMR (150.9 MHz, CDCl3): δ 24.6, 28.1, 28.2, 33.7, 40.4, 55.4, 60.1, 61.9, 63.3, 64.6, 69.0, 69.1, 70.4, 70.5, 70.6, 70.6, 129.1, 129.3, 130.3, 131.0, 131.2, 131.3, 132.1, 133.2, 141.0, 142.5, 143.1, 143.6, 166.4, 173.5, 174.1 ppm; IR (film): 747, 1038, 1119, 1179, 1236, 1264, 1455, 1522, 1700, 2867, 2925, 3236 cm−1, HRMS: (ESI+): m/z calculated for C31H39O8N4S [M + H]+ 627.2483, found: 627.2477.Rhodamine-labelled phenazine-1-carboxylic acid (10)
To a solution of rhodamine B (1.9 mg, 0.0043 mmol), tetraethyleneglycol phenazine-1-carboxylate 11 (1.7 mg, 0.0043 mmol) and PyBop (2.7 mg, 0.0052 mmol, 1.2 eq.) in anhydrous dichloromethane (1 mL) was added Hünigs base (2.9 μL, 4 eq.) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 24 h. The solution was concentrated under reduced pressure and the residue was subjected to column chromatography over silica gel (gradient chloroform/MeOH: 100/0–95/5, v/v). The crude product was subjected to semi-preparative HPLC using a Zorbax Eclipse XDB-C8 (acetonitrile–H2O, 0.1% formic acid; starting conditions: 20% acetonitrile, 40% in 4 min, 60% in 12.5 min, 100% in 0.5 min, 100% for 5 min, 20% in 1 min) to give 2.2 mg of a white solid. Yield: 63%. 1H NMR (CDCl3, 600 MHz) δ = 1.26–1.37 (12H, m), 3.45–3.49 (3H, m), 3.50–3.59 (9H, m), 3.61–3.64 (2H, m), 3.64–3.68 (1H, m), 3.69–3.72 (2H, m), 3.73–3.79 (1H, m), 3.90 (2H, t, 3J = 4.8 Hz), 4.12 (2H, t, 3J = 4.7 Hz), 4.63 (2H, t, 3J = 4.8 Hz), 6.78–6.81 (3H, m), 7.02 (1H, s), 7.04 (1H, s), 7.28 (1H, dd, 3J1 = 7.6 Hz, 4J2 = 0.7 Hz), 7.61–7.82 (3H, m), 7.85–7.91 (3H, m), 8.25 (1H, dd, 3J1 = 6.9 Hz, 4J2 = 1.1 Hz), 8.26 (1H, dd, 3J1 = 8.0 Hz, 4J2 = 1.0 Hz), 8.28–8.32 (2H, m), 8.28 (1H, d, 3J1 = 8.8 Hz) ppm. 13C NMR (CDCl3, 600 MHz) δ = 12.6, 12.6, 12.6, 13.7, 46.0, 46.0, 46.0, 47.4, 64.7, 64.8, 68.6, 69.2, 70.4, 70.5, 70.6, 70.7, 96.4, 113.6, 114.0, 119.0, 128.7, 129.0, 129.6, 129.8, 130.2, 130.3, 131.2, 131.3, 131.3, 131.4, 131.4, 131.7, 132.3, 132.8, 132.9, 133.0, 133.6, 141.1, 141.9, 142.5, 143.8, 153.8, 155.5, 157.8, 158.8, 165.0, 166.3 ppm. IR (film): 1077, 1126, 1183, 1342, 1410, 1462, 1590, 1651, 1722, 2353, 2860, 2923, 3318 cm−1, HRMS: (ESI+): m/z calculated for C49H53O8N4 [M]+ 825.3858, found: 825.3868.Phenazine-protein binding assays
The biotinylated probe 9 was directly mixed with the protein (KS-B – 104.7 kDa) in 20 mM Tris–HCl (pH 8.0). Final concentrations of the probe and protein were 0.5 mM and 20 μM respectively. The reaction mixture was incubated for 24 h under the following conditions: (a) at 254 nm (b) at 370 nm or (c) at 22 °C in the presence of 0.5 mM azobisisobutyronitrile (AIBN). For purification of the biotinylated KS-B, the reaction mixture was diluted twice with 50 mM Tris, 0.2 M NaCl, 0.1% SDS (pH 8.0) and incubated with 1 mL of streptavidin beads (Thermo Scientific) at 22 °C for 1 h with shaking at 400 rpm. The beads were washed twice with 50 mM Tris, 0.2 M NaCl, 2% SDS (pH 8.0) after incubation at 60 °C for 10 min. Elution of the bound protein was achieved by heating the sample thrice at 60 °C for 30 min in 50 mM Tris, 0.2 M NaCl, 2% SDS, 3 mM D-biotin (pH 8.0).The rhodamine-based probe 10 was incubated with the protein (KS-B, carbonic anhydrase III, albumin) in water or in DMSO. Final concentrations of the probe and the protein were 10 μM and 20 μM respectively. The reaction mixture was incubated for varying times between 15 min and 20 h under UV-light at 254 nm, at 370 nm, at 22 °C in the presence of 0.5 mM AIBN in the dark or at sunlight (specific times and conditions for each experiment are indicated in the respective figure legend). After the assay the protein samples were analysed by PAGE, on a 10% or 12% SDS-gel for KS-B. The structures of KS-B and albumin were visualised using Pymol. The probe 10 was generated as a fragment and added specifically to the Cys residue of the proteins by replacing the hydrogen atom in Pymol.
Acknowledgements
We thank H. Heinecke and A. Perner for NMR and HRMS measurements, M. Poetsch for MALDI experiments, K. Martin and K. Perlet for strain handling, and K.-D. Menzel and M. Steinacker for support in large-scale fermentation and downstream processing. We gratefully acknowledge the Studienstiftung des Deutschen Volkes (to D. H.) and the International Leibniz Research School for Microbial and Biomolecular Interactions (ILRS, to S. S.) for financial support.References
- S. Chincholkar and L. Thomashow, Microbial Phenazines, Springer, Berlin, 2013 Search PubMed.
- J. B. Laursen and J. Nielsen, Chem. Rev., 2004, 104, 1663–1686 CrossRef CAS PubMed.
- A. Cimmino, A. Evidente, V. Mathieu, A. Andolfi, F. Lefranc, A. Kornienko and R. Kiss, Nat. Prod. Rep., 2012, 29, 487–501 RSC.
- J. Davies and K. S. Ryan, ACS Chem. Biol., 2012, 7, 252–259 CrossRef CAS PubMed.
- A. Price-Whelan, L. E. P. Dietrich and D. K. Newman, Nat. Chem. Biol., 2006, 2, 71–78 CrossRef CAS PubMed.
- L. S. I. Pierson and E. A. Pierson, Appl. Microbiol. Biotechnol., 2010, 86, 1659–1670 CrossRef CAS PubMed.
- D. Romero, M. F. Traxler, D. Lopez and R. Kolter, Chem. Rev., 2011, 111, 5492–5505 CrossRef CAS PubMed.
- D. V. Mavrodi, J. A. Parejko, O. V. Mavrodi, Y. S. Kwak, D. M. Weller, W. Blankenfeldt and L. S. Thomashow, Environ. Microbiol., 2013, 15, 1462–2920 CrossRef PubMed.
- L. E. Dietrich, A. Price-Whelan, A. Petersen, M. Whiteley and D. K. Newman, Mol. Microbiol., 2006, 61, 1308–1321 CrossRef CAS PubMed.
- L. E. Dietrich, C. Okegbe, A. Price-Whelan, H. Sakhtah, R. C. Hunter and D. K. Newman, J. Bacteriol., 2013, 195, 1371–1380 CrossRef CAS PubMed.
- L. E. Dietrich, T. K. Teal, A. Price-Whelan and D. K. Newman, Science, 2008, 321, 1203–1206 CrossRef CAS PubMed.
- A. J. Stams, F. A. de Bok, C. M. Plugge, M. H. van Eekert, J. Dolfing and G. Schraa, Environ. Microbiol., 2006, 8, 371–382 CrossRef CAS PubMed.
- C. C. Caldwell, Y. Chen, H. S. Goetzmann, Y. Hao, M. T. Borchers, D. J. Hassett, L. R. Young, D. Mavrodi, L. Thomashow and G. W. Lau, Am. J. Pathol., 2009, 175, 2473–2488 CrossRef CAS PubMed.
- M. Mentel, E. G. Ahuja, D. V. Mavrodi, R. Breinbauer, L. S. Thomashow and W. Blankenfeldt, ChemBioChem, 2009, 10, 2295–2304 CrossRef CAS PubMed.
- R. C. Hunter, V. Klepac-Ceraj, M. M. Lorenzi, H. Grotzinger, T. R. Martin and D. K. Newman, Am. J. Respir. Cell Mol. Biol., 2012, 47, 738–745 CrossRef CAS PubMed.
- H. Ran, D. J. Hassett and G. W. Lau, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14315–14320 CrossRef CAS PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- Y. Q. O'Malley, K. J. Reszka, D. R. Spitz, G. M. Denning and B. E. Britigan, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2004, 287, L94–L103 CrossRef PubMed.
- M. Muller, Free Radical Biol. Med., 2002, 33, 1527–1533 CrossRef CAS PubMed.
- M. Muller, Free Radical Biol. Med., 2011, 50, 971–977 CrossRef CAS PubMed.
- R. Cheluvappa, R. Shimmon, M. Dawson, S. N. Hilmer and D. G. Le Couteur, Acta Biochim. Pol., 2008, 55, 571–580 CAS.
- M. Muller and N. D. Merrett, Chem. Biol. Interact., 2015, 232, 30–37 CrossRef CAS PubMed.
- A. Ray, C. Rentas, G. A. Caldwell and K. A. Caldwell, Neurosci. Lett., 2015, 584, 23–27 CrossRef CAS PubMed.
- A. J. McFarland, S. Anoopkumar-Dukie, A. V. Perkins, A. K. Davey and G. D. Grant, Arch. Toxicol., 2012, 86, 275–284 CrossRef CAS PubMed.
- B. Rada and T. L. Leto, Trends Microbiol., 2013, 21, 73–81 CrossRef CAS PubMed.
- J. M. Winter, S. Behnken and C. Hertweck, Curr. Opin. Chem. Biol., 2011, 15, 22–31 CrossRef CAS PubMed.
- D. Heine, K. Martin and C. Hertweck, J. Nat. Prod., 2014, 77, 1083–1087 CrossRef CAS PubMed.
- M. Wagner, W. M. Abdel-Mageed, R. Ebel, A. T. Bull, M. Goodfellow, H.-P. Fiedler and M. Jaspars, J. Nat. Prod., 2014, 77, 416–420 CrossRef CAS PubMed.
- M. L. Gilpin, M. Fulston, D. Payne, R. Cramp and I. Hood, J. Antibiot., 1995, 48, 1081–1085 CrossRef CAS PubMed.
- A. Braunshausen and F. P. Seebeck, J. Am. Chem. Soc., 2011, 133, 1757–1759 CrossRef CAS PubMed.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, T. Heinekamp, K. Willing, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 11092–11095 CrossRef CAS PubMed.
- D. H. Scharf, N. Remme, A. Habel, P. Chankhamjon, K. Scherlach, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 12322–12325 CrossRef CAS PubMed.
- S. Coyne, C. Chizzali, M. N. Khalil, A. Litomska, K. Richter, L. Beerhues and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 10564–10568 CrossRef CAS PubMed.
- E. Sasaki, X. Zhang, H. G. Sun, M. Y. Lu, T. L. Liu, A. Ou, J. Y. Li, Y. H. Chen, S. E. Ealick and H. W. Liu, Nature, 2014, 510, 427–431 CAS.
- K. V. Goncharenko, A. Vit, W. Blankenfeldt and F. P. Seebeck, Angew. Chem., Int. Ed., 2015, 54, 2821–2824 CrossRef CAS PubMed.
- B. Li, J. P. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk and S. K. Nair, Science, 2006, 311, 1464–1467 CrossRef CAS PubMed.
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916–11922 CrossRef CAS.
- G. A. Amaral, F. Ausfelder, J. G. Izquierdo, L. Rubio-Lago and L. Banares, J. Chem. Phys., 2007, 126, 024301 CrossRef CAS PubMed.
- S. Naumov and C. Schoneich, J. Phys. Chem. A, 2009, 113, 3560–3565 CrossRef CAS PubMed.
- M. Gersch, J. Kreuzer and S. A. Sieber, Nat. Prod. Rep., 2012, 29, 659–682 RSC.
- T. Böttcher, M. Pitscheider and S. A. Sieber, Angew. Chem., Int. Ed., 2010, 49, 2680–2698 CrossRef PubMed.
- P. Ulrich and A. Cerami, Recent Prog. Horm. Res., 2001, 56, 1–22 CrossRef CAS PubMed.
- T. Bretschneider, J. B. Heim, D. Heine, R. Winkler, B. Busch, B. Kusebauch, T. Stehle, G. Zocher and C. Hertweck, Nature, 2013, 502, 124–128 CrossRef CAS PubMed.
- D. Heine, T. Bretschneider, S. Sundaram and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 11645–11649 CrossRef CAS PubMed.
- D. Heine, S. Sundaram, T. Bretschneider and C. Hertweck, Chem. Commun., 2015, 51, 9872–9875 RSC.
- S. Adolph, V. Jung, J. Rattke and G. Pohnert, Angew. Chem., Int. Ed., 2005, 44, 2806–2808 CrossRef CAS PubMed.
- N. Reddy, R. Reddy and Q. Jiang, Trends Biotechnol., 2015, 33, 362–369 CrossRef CAS PubMed.
- G. V. Bloemberg and B. J. J. Lugtenberg, New Phytol., 2003, 157, 503–523 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00503a |
| This journal is © The Royal Society of Chemistry 2016 |