
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantum dot induced cellular perturbations involving varying toxicity pathways†
Abdullah
Al-Ali‡
a,
Neenu
Singh‡
a,
Bella
Manshian
b,
Tom
Wilkinson
a,
John
Wills
a,
Gareth J. S.
Jenkins
a and
Shareen H.
Doak
*a
aInstitute of Life Science, College of Medicine, Swansea University, Singleton Park, Swansea, SA2 8PP, Wales, UK. E-mail: s.h.doak@swansea.ac.uk; Fax: +44 (0) 1792 602147; Tel: +44 (0) 1792 295388
bBiomedical NMR unit-MoSAIC, Department of Medicine, KU Leuven, B-3000 Leuven, Belgium
First published on 6th January 2015
Abstract
The unique optical and electronic properties of quantum dots (QD) have led to rapid progress in their development and application, particularly in innovative therapeutic and diagnostic products. Along with the great pace at which QD are being developed, research is being focussed on fabricating less toxic QD with novel surface functionalities. The present study was therefore focused on assessing the impact of varying QD surface chemistry on cellular uptake and a range of indicators for cell perturbation following exposure. The study demonstrated that despite a low intrinsic cytotoxicity of three QD with different surface functional groups, they were all capable of inducing an acute inflammatory response and alterations in transcriptional gene activity, without affecting cell cycle regulation. Further, this investigation demonstrated that although the QD were capable of inducing an inflammatory and oxidative stress response, there was clearly variation in the degree of molecular change according to surface chemistry, which correlated with the degree of cellular uptake. These findings therefore highlight the potential for chronic inflammatory responses following exposure to QD, but in addition, they also demonstrate the importance of studying a wide range of toxicity pathways to generate a comprehensive picture of biological response to nanomaterials.
Introduction
Quantum Dots (QD) are semiconductor engineered nanocrystals that have unique optical and electrical properties. Consequently, they are promising novel opportunities for sensing clinically relevant molecules and biomarkers to support molecular disease imaging and therapeutic intervention.1,2 Indeed, QD have been demonstrated to improve imaging and sensing of infectious disease such as respiratory syncytial virus (RSV) and detection of E.coli at levels as low as 104 bacteria ml−1 of sample.3–6 QD have also been used in biological applications including the labelling of cells and organelles, tracing movement of cells in tissue, and tracking macromolecules in cells.7–10The most common colloidal QD are a combination of the periodic table elements from groups II and IV (e.g. cadmium selenide (CdSe), cadmium sulphide (CdS), and zinc oxide (ZnO)). Consequently, although QDs provide a unique advantage in non-invasive imaging technologies, their transition metal-based core is believed to be highly toxic.11 Particularly in the internal cellular environment, heavy metal ions may be released from QD as a result of photolytic and oxidative conditions that can effectively degrade QD, promoting toxicity. To make QD more biocompatible, their heavy metal core can be capped with a zinc sulphide (ZnS) shell and often a further surface coating can be applied to suit specific applications (e.g. functional groups with specific charges or bio-molecular targets). However, despite the improved QD designs, there remains evidence that these materials can still be toxic and QD have been associated with cytotoxicity, pulmonary inflammation, reactive oxygen species (ROS) induction and transcription disturbances involving stress defence and DNA repair genes.12–14
QD-associated toxic effects may be variable depending on the exposure route; possibilities of which include inhalation/ingestion (due to environmental and occupational exposure), or intravenous/intraperitoneal (for specific biomedical applications). However, a primary cell type of importance regardless of exposure route are macrophages, as they play a central role in the clearance of foreign materials (xenobiotics, pharmaceuticals or nanoparticles) in major organs and tissues such as the lungs, liver and blood. These cells are programmed to engulf invading pathogens or particulate material and subsequently elicit an inflammatory response to help eradicate the foreign substances; but this in turn may impinge on normal cellular pathways leading to toxicity.15 Furthermore, the impact of macrophage exposure to a nano-entity is an important consideration given that investigations utilising this cell-type (e.g. monocytic THP-1 cells) have been shown to be a promising model to assess the ability of QDs with different functionalities to act as suitable labels for biological imaging.16
Although QD are promising major advances in biomedical applications, with promises of wide reaching applications in biological and biomedical fields, their fabrication with a diverse range of surface functionalities may govern QD-cell surface interactions and potential adverse biological effects.17 There are limited studies that assess correlations between QD that have hydrophilic versus hydrophobic surface functionalities, with cellular uptake and multiple toxicity end-points/pathways in macrophages. Thus, it is imperative that a more detailed understanding of QD-macrophage cell interactions that affect biological functions at cellular and molecular levels are developed. The present study therefore investigates the impact of several QDs with varying surface chemistry (representing hydrophilic and hydrophobic nanomaterials, and differential surface charge) on differentiated THP-1 macrophage cells, through the assessment of their capacity to alter cell-cycle progression, induce cytotoxicity or inflammatory responses and cause gene expression profile alterations.
Results
Physico-chemical characterisation
This investigation focused on three QD: CdSe/ZnS hexadecylamine (HDA) coated QD (hydrophobic, HDA is neutrally charged), carboxylated CdSe/ZnS QD (hydrophilic, carboxyl functional groups are negatively charged) and amino polyethylene glycol (PEG) coated CdSe/ZnS QD (hydrophilic with a positively charged functional group). A range of physico-chemical properties were characterised for all three CdSe/ZnS QD under study. Transmission Electron Microscopy (TEM) was used to determine size, morphology and crystal structure of the QD. All QD were largely spherical and were ∼4–5 nm in size (Fig. 1). Additionally, energy-dispersive X-ray spectroscopy (EDX) analysis was utilised to assess purity and it demonstrated that the only elements present in the QD included: Se, Cd, S and Zn (not shown).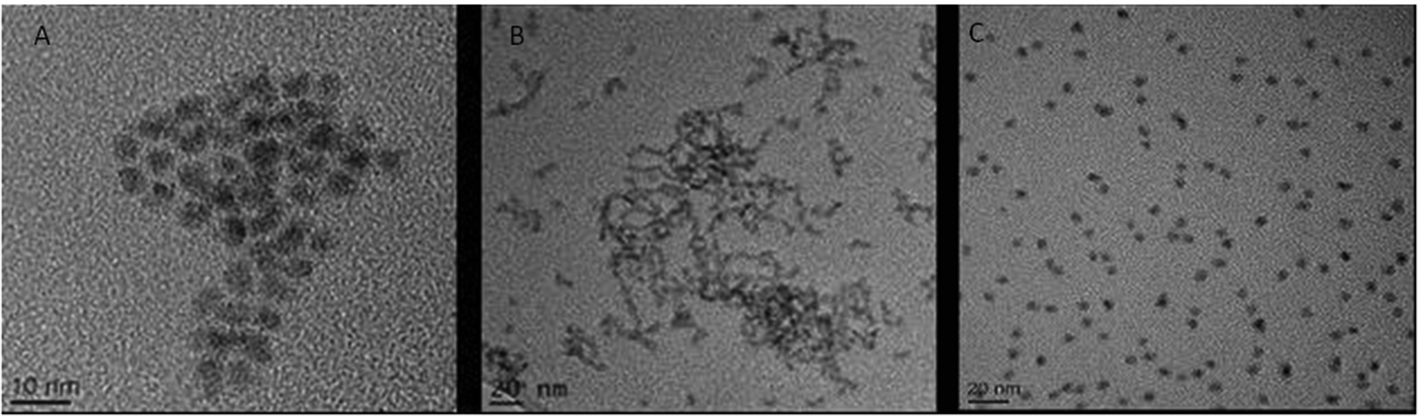 | ||
| Fig. 1 TEM images of the three CdSe/ZnS QD. (A) CdSe QD capped with PEG. (B) HDA capped CdSe/ZnS QD. (C) Carboxylated CdSe/ZnS QD. | ||
Dynamic light scattering (DLS) was used to measure size distribution of the CdSe/ZnS QD with the different surface coatings (carboxylated, PEG and HDA) dispersed in medium with varying quantities of FBS and the data is detailed in Table 1. The QD size distribution was obtained following subtraction of the size profile generated by media alone. Interestingly, the quantity of serum in the media had a big impact on the level of agglomeration of the QDs (Table 1). The QD capped with HDA and dispersed in 10% FBS containing DMEM culture media demonstrated a size distribution ranging from 4 nm to 82 nm (Table 1) with the greatest (peak) particle size at 6 nm, indicating the majority of the particles were mono-dispersed. In contrast, when dispersed in 2% FBS containing DMEM media the NPs demonstrated a wider size distribution and greater peak size (Table 1), indicating more agglomeration. This pattern was also true for the other QD, where both carboxylated- and PEG-QD were largely mono-dispersed in full serum complement media. However, in general, the carboxylated QD demonstrated the smallest size range indicating that they agglomerated to a lesser extent than the PEG- or HDA-QD (Table 1).
| HDA-QDs | Carboxylated-QDs | PEG-QDs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Serum content of media | Size range (nm) | Peak size (nm) | Zeta potential (mV) | Size range (nm) | Peak size (nm) | Zeta potential (mV) | Size range (nm) | Peak size (nm) | Zeta potential (mV) |
| 10% FBS | 4–82 | 6 | −8.4 | 3–124 | 6–8 | −10.45 | 3–569 | 5–8 | 2.7 |
| 2% FBS | 3–355 | 150 | −12.85 | 3–68 | 10 | −13.35 | 4–2566 | 5–8 | 2.6 |
| 0% FBS | 188–2056 | 600 | −6.025 | 197–1477 | 500–600 | −28.1 | 95–923 | 100–200 | −1.6 |
The surface coating on a nanoparticle influences their subsequent charge, which has an impact on their behaviour in media with respect to agglomeration, or in cells where the charge may be an important factor influencing cellular uptake. Thus, a zeta sizer was utilised to determine the surface charge of the QD under investigation. The carboxylated CdSe/ZnS QDs had the highest negative charge in water, while the PEG-QD had a very slight positive charge and the HDA were close to neutral (Table 1). However, in reality all the NPs examined demonstrated a largely neutral charge as they were between +30 mV/−30 mV. Only charges outside of this range are considered positively or negatively charged respectively.18
THP-1 cellular uptake of QD
The Image Stream imaging flow cytometer was used to measure differentiated THP-1 cellular uptake of the test QD in either 10% or 2% FBS containing media. When THP-1 cells were assessed for QD uptake after 24 h exposure to carboxylated QD, not only was significant internalisation observed, but these QD also demonstrated the highest fluorescent intensity levels when compared to the other assessed QD (Fig. 2). The data demonstrated a highly significant fluorescent intensity level of approximately 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 664 RFU when THP-1 cells were treated with the QD in 10% FBS containing media. Whereas a lower but still highly significant level of uptake (approximately 32063 RFU) was observed when the cells were treated with the QD in 2% FBS containing media (Fig. 2A).
664 RFU when THP-1 cells were treated with the QD in 10% FBS containing media. Whereas a lower but still highly significant level of uptake (approximately 32063 RFU) was observed when the cells were treated with the QD in 2% FBS containing media (Fig. 2A).
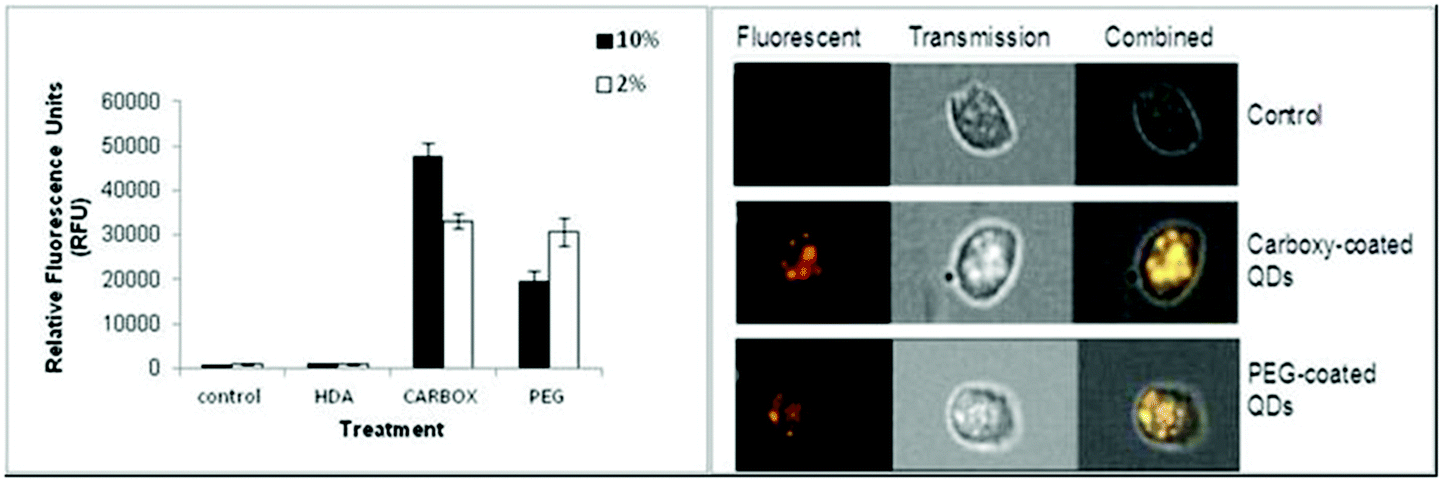 | ||
| Fig. 2 Image Stream of analysis of HDA-QD, carboxylated-QD or PEG-QD uptake into differentiated THP-1 cells (A). RFU reflects the level of QD uptake into THP-1 cells exposed to QD in the presence of 10% or 2% containing media. (B) A snapshot from the Image Stream software illustrating the increasing fluorescence signal intensity in cells exposed to the carboxylated QD or PEG-QD as compared to untreated control cells. (***) Indicates high statistical significance p ≤ 0.001. | ||
The Image Stream analysis of differentiated THP-1 cells exposed to PEG QD also demonstrated cellular uptake but at a lower level when compared with the carboxylated QD (Fig. 2). A highly significant increase of fluorescence intensity was seen in both 10% and 2% FBS containing RPMI-1640 culture media. However the 10% FBS containing RPMI culture media had a slightly lower fluorescent intensity level than when compared with the 2% FBS containing media. In contrast, the HDA coated QD did not appear to be internalised by the THP-1 cells in either 10% or 2% FBS containing media. The QD exposure in 10% FBS containing RPMI-1640 media reached an average of 919 RFU. Similarly, the exposure in 2% FBS containing culture media demonstrated an average of 844 RFU, indicating undetectable cellular uptake, as the control background RFU was an average of 890 RFU.
Therefore, the study demonstrated significant cellular uptake of carboxylated and PEG QD by the monocytic THP-1 cells with the order of uptake as follows: Carboxylated- > PEG- > HDA-QD.
Cytotoxicity induced by QD on THP-1 cells
To determine whether cellular uptake of the three test QDs, as analysed by Image Stream analysis resulted in THP-1 cell toxicity, cell viability assessment was performed under varying experimental conditions (10% and 2% FBS) and time points (24 and 72 h exposures). Following 24 h exposure the carboxylated-, PEG- and HDA-QD demonstrated no significant cytotoxicity regardless of the serum content of the media. A slight dose-dependent increase in cell viability was observed with the HDA-QD, but was not a significant trend (Fig. 3E).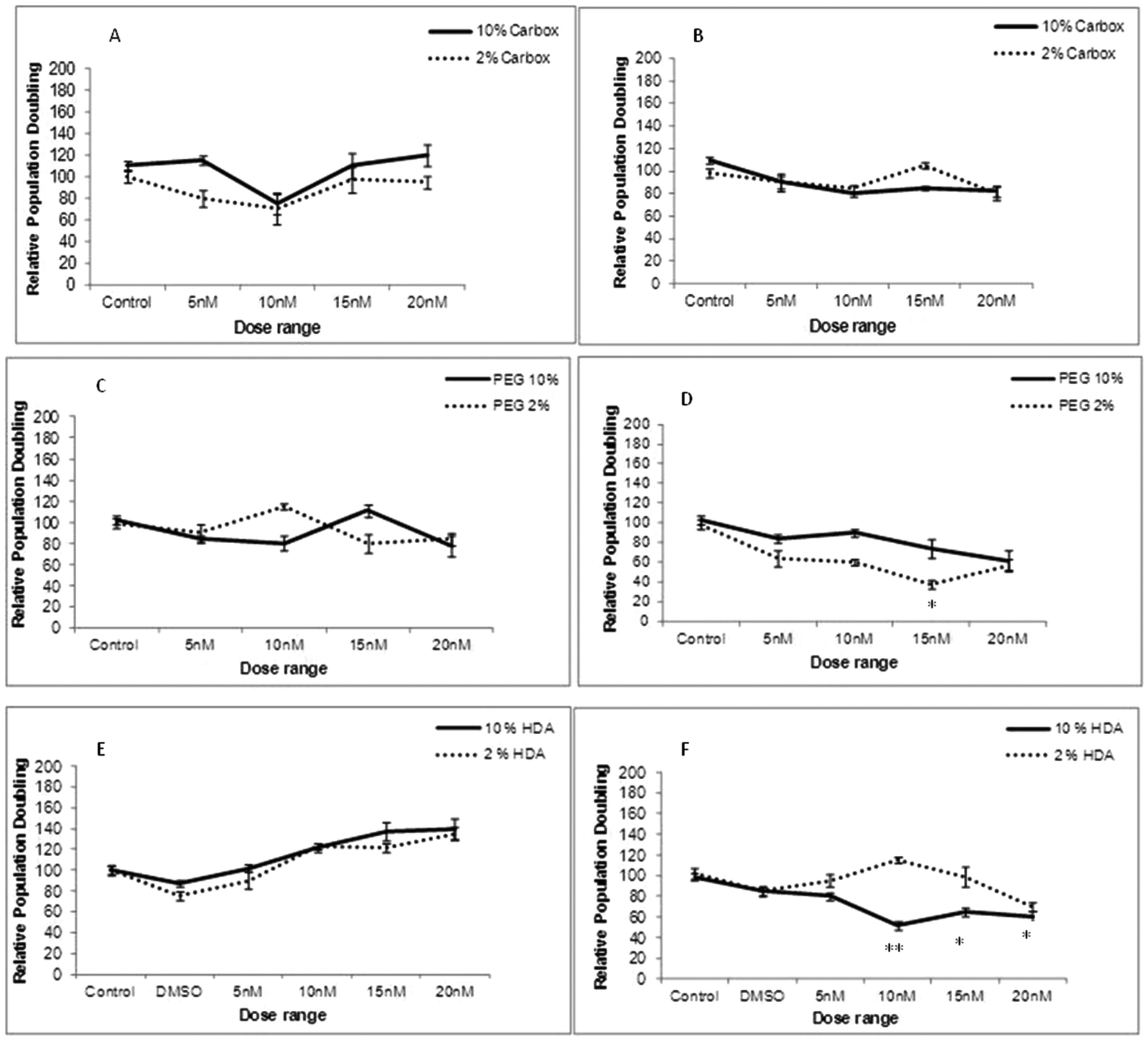 | ||
| Fig. 3 Cytotoxicity evaluation of THP-1 cells treated with carboxylated, PEG or HDA CdSe/ZnS QD for 24 h (A, C and E respectively) or 72 h (B, D and F) in the presence of 10% and 2% FBS containing RPMI-1640 media. *p < 0.05 and **p < 0.01 compared to control cells. | ||
However, when exposure time was increased to 72 h some cytotoxicity became apparent. HDA-QD in 10% FBS media induced a dose-dependent decrease in cell viability, with significant cytotoxicity achieved from 10 nM. In 2% FBS containing media, exposure to the QD again resulted in a dose-dependent decrease in cell viability, but not to the same extent as in 10% serum and a significant decrease was not achieved over the dose range applied (Fig. 3F). The PEG-QD also resulted in a dose-dependent decrease in viability following 72 h exposures. However, in 2% FBS this only reached significance at 15 nM and did not reach significance in 10% FBS containing media (Fig. 3D). In contrast, the carboxylated-QD did not induce significant cytotoxicity over the dose range applied, regardless of the serum content of the media (Fig. 3A, B).
In summary, the study showed that QD cytotoxicity was only observed after long-term exposure of 72 h and not at 24 h. Additionally, there was no direct correlation between the level of cellular uptake and cytotoxicity, suggesting that the intrinsic cytotoxicity of carboxylated-QD, which showed the highest level of uptake, is low.
Cell cycle analysis
THP-1 cells were examined for any changes or disruptions that may occur to their cell cycle dynamics following exposure to the three test QD. However, the cells were only exposed to the top dose (20 nM) as no change in cell cycle dynamics were observed (Table 2).| Cycle phase | Control % | Carboxylated % | PEG % | HDA % |
|---|---|---|---|---|
| G0/G1 | 47 | 39.8 | 46.5 | 48.4 |
| S | 9.9 | 12.7 | 10.4 | 5.6 |
| G2/m | 34.2 | 36.8 | 34.4 | 33.9 |
Of particular interest was that alongside the cell cycle histograms, data could also be generated to quantify the QD cellular uptake in the THP-1 cells with each treatment. There was noticeable cellular uptake when THP-1 cells were exposed to carboxylated- and PEG-QD, while there was no noticeable cellular uptake with the HDA-QD. These findings corresponded to the Image Stream analysis (data not shown). Thus, despite cellular uptake of the carboxylated- and PEG- QD, these materials had no impact on the cell cycle dynamics, demonstrating that the accumulation of QDs in THP-1 cells do not disrupt cell cycle regulation.
Cytokine and chemokine induction
IL-1β cytokine and IL-8 chemokine protein levels were assessed by ELISAs following exposure of the QD to differentiated THP-1 cells over a range of time points. With regard to IL-1β, there was no significant up-regulated activity observed with all three QD samples over all time points tested, regardless of the presence of 2% or 10% serum containing media (data not illustrated). In contrast, all treatments demonstrated significant up-regulated levels of the IL-8 chemokine following 2 and 4 h exposure times (Fig. 4). The extent of IL-8 up-regulation was very similar for all three QD. When comparing treatments between 10% FBS and 2% FBS containing culture media, 2% FBS treatments generally demonstrated lower (but still significantly increased) IL-8 levels as compared to the 10% FBS treatments. This response appeared to be maintained at 4 h, but by 8 h the IL-8 expression levels were substantially reduced to almost background levels with all test QD, indicating that the THP-1 cells appeared to induce a fast response to the internalised particles that returned to normal by 8 h post exposure (Fig. 4C).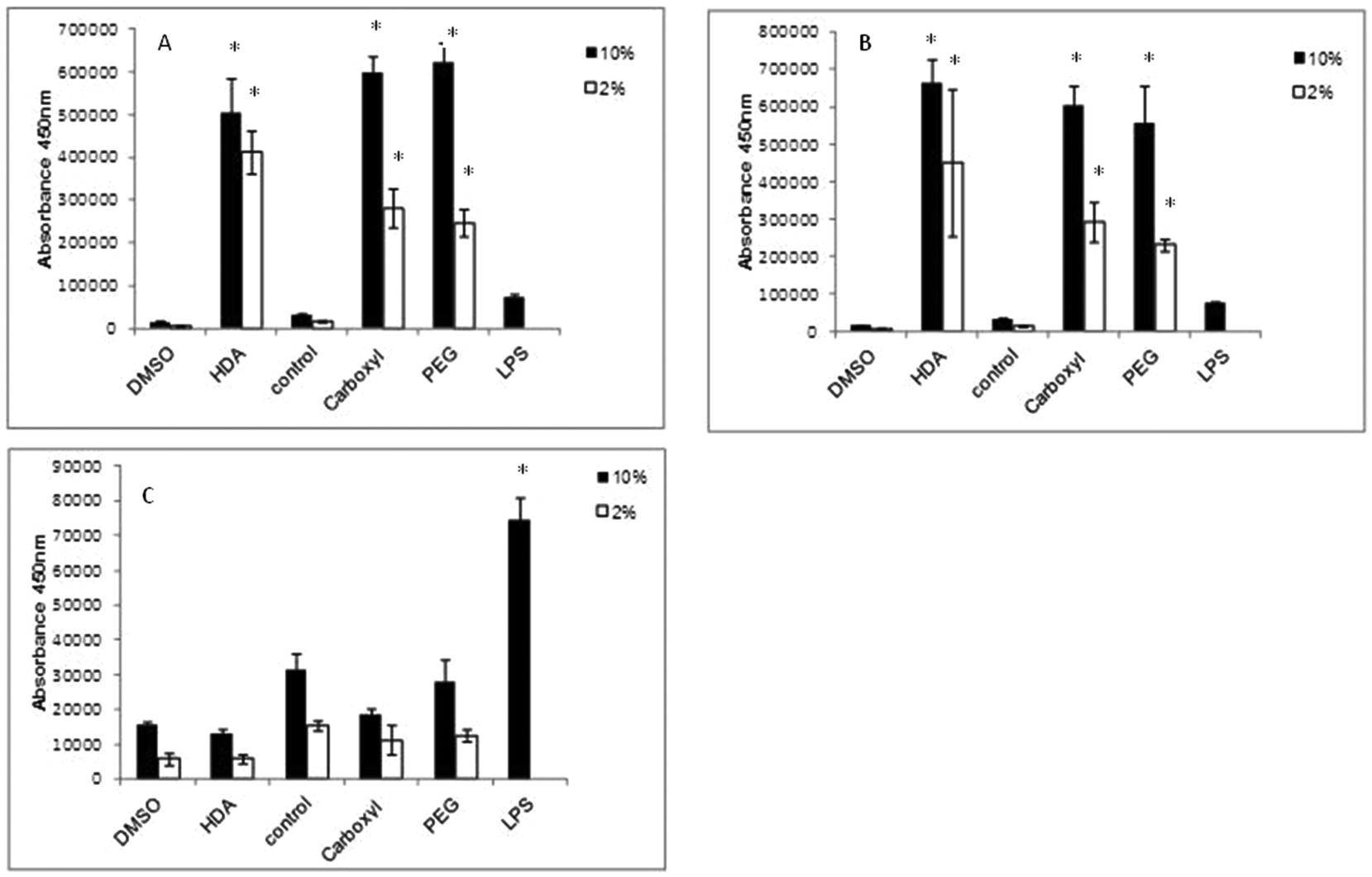 | ||
| Fig. 4 IL-8 ELISA conducted on differentiated THP-1 cells exposed to QD. All three CdSe/ZnS QD exposed to THP-1 cells for (A) 2 h, (B) 4 h and (C) 8 h. HDA CdSe/ZnS QD are displayed separately on the left of the graphs because HDA QD samples were suspended in DMSO and therefore needed to be compared to the DMSO control, while carboxylated and PEG-QD were dissolved in H2O (control). (*) Indicates statistical significance p < 0.05. | ||
In this study, LPS was used as the positive control, but it was found to only give high IL-8 expression levels at 8 h, with little IL-8 up-regulation at the 2 and 4 h time points. This was expected as LPS requires 8 h to induce an inflammatory response. But what is interesting is the scale of the QD-induced IL-8 response at the earlier time points as compared to LPS after 8 h. The QD were clearly capable of inducing a strong IL-8 response, suggesting that they are rapidly internalised by the macrophages, followed by a swift response to their uptake.
Gene expression analysis
Gene expression arrays were performed on differentiated THP-1 cells exposed to all three test QD to assess the subsequent oxidative stress pathway expression profiles induced in response. The expression array utilised for this purpose was the RT2 Profiler™ Human Oxidative Stress and Antioxidant Defense PCR Array (PAHS-065A; SABioscience Qiagen Company/UK). A total of 84 genes involved in oxidative stress were examined using this array. A total of 84 genes involved in oxidative stress were analyzed for a relative fold change in gene expression following differentiated THP-1 cell exposure to the 3 different CdSe/ZnS-QD. The details of the up- and down-regulation (fold change) of genes in QD exposed cells relative to untreated control THP-1 cells are shown in Fig. S1.†When differentiated THP-1 cells were exposed to HDA-QD, 12 genes in total demonstrated substantial alterations in their transcriptional levels as compared to the control. Of these 12 genes, 5 were up regulated and 7 were down-regulated out of the 84 genes analysed in total (Table 3). In regard to the up-regulated genes there were four (MPV17, PTGS2, and TTN) that were two-fold up-regulated whereas NOS2 and TXNRD1 were greater than three-fold over-expressed. The down-regulated genes included GPX4, NUDT1, PREX1 and SIRT2 which were all just over two-fold suppressed, while APOE was nearly three-fold down-regulated as compared with the control. Interestingly, both PNKP and PRDX2 were substantially (six-fold) down-regulated.
| Symbol | Description | Fold change |
|---|---|---|
| Exposure to HDA CdSe/ZnS QD | ||
| MPV17 | Mitochondrial inner membrane protein | 2.141 |
| NOS2 | Nitric oxide synthase 2, inducible | 3.4064 |
| PTG52 | Prostaglandin-endoperoxide synthase 2 | 2.0969 |
| TTN | Titin | 2.0255 |
| TXNRD1 | Thioredoxin reductase 1 | 3.2677 |
| APOE | Apolipoprotein E | −3.3213 |
| GPX4 | Glutathione peroxidise 4 (phospholipid hydroperoxidase) | −2.1611 |
| NUDT1 | Nudix (Nucleoside diphosphate linked moiety X)- type motif 1 | −2.3979 |
| PNKP | Polynucleotide kinase 3′-phosphatase | −6.2843 |
| PRDX2 | Peroxiredoxin 2 | −6.2409 |
| PREX1 | Phosphatidylinositol-3,4,5,-triphosphate-dependent | −2.5171 |
| Rac exchange factor 1 | ||
| SIRT2 | Sirtuin 2 | −2.5522 |
| Exposure to Carboxylated CdSe/ZnS QD | ||
| BNIP3 | BCL2/adenovirus E1B 19 kDa interacting protein 3 | 3.1613 |
| GLRX2 | Glutaredoxin 2 | 2.2509 |
| GPX7 | Glutathione peroxidise 7 | 2.2046 |
| MPV17 | Mitochondrial inner membrane protein | 2.1592 |
| MSRA | Methionine sulfoxide reductase A | 2.4462 |
| NOS2 | Nitricoxide synthase 2, inducible | 2.3465 |
| PRDX1 | Peroxiredoxin 1 | 2.2666 |
| PRDX4 | Peroxiredoxin 4 | 2.2982 |
| PRNP | Prion protein | 2.8889 |
| SOD1 | Superoxide dismutase 1, soluble | 2.1295 |
| TXNDC2 | Thioredoxin domain containing 2 (spermatozoa) | 2.5856 |
| TXNRD1 | Thioredoxin reductase 1 | 4.259 |
| APOE | Apolipoprotein E | −3.4093 |
| GPX4 | Glutathione peroxidise 4 (phospholipid hydroperoxidase) | −2.265 |
| NUDT1 | Nudix (Nucleoside diphosphate linked moiety X)- type motif 1 | −2.0272 |
| PNKP | Polynucleotide kinase 3′-phosphatase | −3.8624 |
| PRDX2 | Peroxiredoxin 2 | −2.6564 |
| PREX1 | Phosphatidylinositol-3,4,5,-triphosphate-dependent | −4.256 |
| Rac exchange factor 1 | ||
| STK25 | Serine/threonine kinase 25 | −2.0698 |
| Exposure to PEG CdSe/ZnS QD | ||
| BNIP3 | BCL2/adenovirus E1B 19 kDa interacting protein 3 | 2.3164 |
| MPV17 | Mitochondrial inner membrane protein | 2.6793 |
| MSRA | Methionine sulfoxide reductase A | 2.6242 |
| TXNRD1 | Thioredoxin reductase 1 | 3.2307 |
| APOE | Apolipoprotein E | −14.4014 |
| CAT | Catalase | −2.9042 |
| CYBA | Cytochrome b-245, alpha polypeptide | −4.1935 |
| DUSP1 | Dual specificity phophatase 1 | −2.0394 |
| GPX4 | Glutathione peroxidise 4 (phospholipid hydroperoxidase) | −5.7284 |
| GTF21 | General transcription factor IIi | 2.2786 |
| NCF1 | Neutrophil cytosolic factor 1 | −2.9244 |
| OXSR1 | Oxidative stress responsive 1 | −3.6506 |
With regard to carboxylated-QD, 19 genes in total demonstrated substantial alterations in their transcriptional levels as compared to the control following exposure. Of these 19 genes, 12 were up regulated and 7 were down regulated as detailed in (Table 3). The up-regulated genes largely demonstrated 2-fold increases in expression (GLRX2, GPX7, MPV17, MSRA, NOS2, PRDX1, PRDX4, SOD1 and TXNDC2), but BNIP3 and PRNP were slightly higher with a 3-fold up-regulation. Interestingly, TXNRD1 was over-expressed to the highest extent, reaching a 4-fold up-regulation. In regards to down-regulated genes, GPX4, NUDT1, PRDX2 and STK25 only demonstrated approximately 2-fold decreases in expression, while APOE, PNKP and PREX1 were further suppressed by 3–4 fold when compared with the controls.
Finally, exposure to the PEG-QD altered the expression of 14 genes. Of these, 4 were up-regulated and 10 were down-regulated out of the 84 genes analysed in total (Table 3). All up-regulated genes (BNIP3, MPV17, and MSRA) demonstrated an approximate two-fold increase in expression with the exception of TXNRD1, which presented with a slightly higher 3-fold increase when compared with the control. The down-regulated genes largely presented an approximate 2–3-fold down-regulation (CAT, DUPS1, GTF2I, NCF1, OXSR1), but both CYBA and GPX4 were further suppressed to levels 4–6 fold lower than the control. Interestingly, APOE demonstrated a dramatic down-regulation of 14-fold lower transcriptional levels than the control.
Intriguingly, when the oxidative stress response gene expression profiles induced by exposure to the three test QD were compared, several similarities were observed with some common genes altered by more than one QD sample treatment (Table 4). For example MPV17 and TXRND1 were up-regulated to almost exactly the same extent in THP-1 cells by all three QD assessed; whereas APOE, GPX4 and PRDX2 were the commonly down-regulated genes. APOE and GPX4 were most substantially down regulated in the PEG-QD treated cells, while the HDA-QD were responsible for the greatest suppression of PRDX2.
| Gene | Carboxylated-QD | PEG-QD | HDA-QD |
|---|---|---|---|
| MPV17 | 2.1592 | 2.693 | 2.141 |
| TXRND1 | 4.259 | 3.2307 | 3.2677 |
| NOS2 | 2.3465 | A | 3.4064 |
| BNIP3 | 3.1613 | 2.3164 | 1.8128 |
| APOE | −3.4093 | −14.4014 | −3.3213 |
| GPX4 | −2.265 | −5.8284 | −2.1611 |
| PRDX2 | −2.6564 | −3.5019 | −6.2409 |
Finally, of particular interest was the positive correlation between the uptake pattern of the 3 different QD under study and alterations in their transcriptional gene activity. Thus, the order of greater cellular uptake and a higher number of alterations in the transcriptional levels of genes involved in oxidative stress signalling pathways was as follows: carboxylated- > PEG- > HDA-QD.
Discussion
QD have unique optical and electrical features that can be exploited to generate valuable novel tools for biological and medicinal applications in areas such as improving diagnoses, prognosis and treatment of disease.19 Importantly, all these diagnostic and therapeutic interventions using QD-based technology, require internalisation of the material to render them beneficial for their specific applications. Cellular internalisation can be dependent upon functional groups attached to QD, which influence nanoparticle-membrane interactions leading to differential cellular uptake. However, surface modification and differential uptake can also have an impact on the toxicity of these nanoparticles. This study therefore exposed THP-1 cells differentiated into macrophages to CdSe/ZnS QD with varying surface functional groups to assess subsequent uptake and cellular response.With regard to the cellular uptake of QD, Image Stream analysis demonstrated that the differentiated THP-1 cells internalized carboxylated-QD to a much greater extent than PEG and HDA capped QD. This observation corresponds to reports in the literature that indicate carboxylated QD were more readily internalized by macrophages and HEK cells as compared to PEG QD.20,21 The differential pattern of cellular uptake of the QD can also be influenced by the protein corona that forms at the NP surface in the extracellular environment. The content of this protein corona is determined by the physico-chemical properties of the NP (in this case QD) and an array of biomolecules including proteins, nutrients and growth factors, in the extracellular environment. The NP-protein corona not only affects NP size and its surface properties, but could potentially influence cellular interactions, including QD-cell adhesion and intracellular uptake.22 Thus, the differing surface functional groups could lead to variations in protein corona forming at the QD surface, which in turn result in the specific uptake profiles observed in the present investigation.
Cellular uptake can drastically modify the inherent toxicity profile of a given nanomaterial and therefore, the ensuing safety considerations if any, need to be addressed to minimise any potential toxicological hazards associated with any nanomaterials under study.23 Therefore, following the cellular uptake investigation, it was important to determine if the THP-1 cells experienced toxicity or disturbances to their cell cycle dynamics as a consequence of the internalised NPs. There was no noticeable toxicity following a 24 h treatment with each of the three QD assessed but when the exposure time was increased to 72 h some cytotoxicity was observed. With HDA-QD, a significant decrease in cell viability was observed that was more predominant in 10% serum containing media, which was intriguing, given their low cellular uptake (that was below the detection limits of the technologies utilised). An important point to make is that the observed cytotoxicity seemed to be governed by serum concentration i.e. significant cytotoxicity in only 10% serum containing medium and not in 2% serum medium. A plausible explanation for these observations is based on different agglomeration behaviour. The QD peak size was 6 nm in media with 10% serum, where significant cytotoxicity was observed; in contrast, the peak size was 150 nm in 2% serum containing medium, where there was lesser cytotoxicity. It is therefore possible that the agglomeration in the latter experimental condition could have resulted in fewer QD-cell interactions that might have been the underpinning reason for the observed toxicity seen in 10% vs. 2% serum containing media. Moreover, these agglomerations could have well protected the HDA to QD linkage from being broken over time and releasing the QD core in 2% serum containing medium (where far less cytotoxicity was observed), as compared to the HDA to QD linkage in 10% serum containing medium (which showed significant cytotoxicity).
In contrast, the carboxylated-QD demonstrated no reduction in cell viability when treated for the extended 72 h treatment, despite the high cellular uptake. This contradicts some observations in the literature where carboxylated-QD were toxic to a range of cell lines including Vero cells (African green monkey kidney cells), Hela cells, human primary hepatocytes, human embryonic kidney cells and rat hepatocytes.21,24,25 It is possible that THP-1 may be more resilient than these other cell types, but may also be due to slight material differences coupled to variation in the test systems applied. In contrast, PEG QD demonstrated a significant dose-dependent reduction in THP-1 cell viability after a 72 h treatment. To date, there have been very few studies that have assessed PEG-QD, but similar observations have been reported in human epidermal keratinocytes, where a 40% reduction in cell viability was found following a 48 h exposure at 20 nM.26 Thus, it appears prolonged exposure may be required to observe an impact on cell viability.
In addition to cytotoxicity, cell cycle disruption was also examined following exposure of differentiated THP-1 to all three test QD. Interestingly, none of the test QD affected the phase distribution throughout the cell cycle (G0/G1 vs. S vs. G2/M phases) despite the varying degrees of uptake observed. Consequently, the presence of the carboxylated- and PEG-QD inside the cells did not interfere with the normal cell cycle dynamics. There are few studies in the literature that have examined the consequence of QD uptake on cell cycle phase distribution, but exposure of CdSe-core QD to preneoplastic epidermal (JB6) cells were found to increase cell percentages in G1 phase while decreasing the proportion of cells in S and G2 phases following a 24 h treatment.27 In this report, the CdSe-core QD also demonstrated severe cytotoxicity. These finding are however, not surprising because the QD applied to the cells was uncoated and thus lacked a protective shell; the cells were therefore, directly exposed to the highly toxic CdSe core that would be expected to cause cytotoxicity. It has been demonstrated that uncoated CdSe QD inhibit Rho-associated kinase (ROCK) activity necessary for the attenuation of ROCK-c-Myc signaling in cervical carcinoma Hela cells.28 This inhibition results in cell cycle arrest in the G1 phase of HeLa cell, which explains why Kong and colleagues found a greater cell population in the G1 phase in their report.27 However, in contrast, the QD used in the present investigation were coated with ZnS and had a functional group attached to their surfaces, which provides additional protection from the QD core that was clearly sufficient to prevent the QD from interfering with cell cycle progression.
Nanomaterials may not always cause toxicity, but their internalisation by pro-inflammatory cells, such as THP-1 cells, can trigger an immune reaction with the subsequent inflammatory response causing oxidative stress in the biological environment. If the immune cells are unable to adequately remove the invading particles because they are biopersistent, a chronic inflammatory response could result, which in turn may cause secondary genotoxicity in the surrounding epithelial tissue.29 Therefore, in order to shed light on this aspect of toxicity, the cytokine and chemokine release by differentiated THP-1 cells following exposure to the three QD was assessed.
In contrast to IL-1β, differentiated THP-1 cells expressed high levels of IL-8 when exposed to all QD for 2 and 4 h treatment times regardless of the quantity of serum in the media. However, IL-8 expression returned to control level with all QD samples by 8 h suggesting a very fast inflammatory burst in response to the QD. A similar observation has also been made in model macrophages (J774A.1) and colonic epithelial cells (HT29) when treated with CdTe-QD at exposures between 10−7 to 10−3 μg ml−1.30 In this study, the authors also reported that IL-8 was not elevated at doses below 10−7 μg ml−1 suggesting a threshold inflammatory response.30 Additionally, CdSe/CdS QD (QD621) capped with PEG applied to human epidermal keratinocyte (HEK) cells induced significant increased levels of IL-8 from 2.5 μM to 10 μM and IL-6 at doses from 1.25 μM to 10 μM.31 Thus, it is evident that a range of QD have the capacity to induce an inflammatory response. IL-8 induction correlated with the uptake levels of the carboxylated- and PEG-QD, where cells exposed to the carboxylated-QD demonstrated a slightly higher IL-8 induction than the PEG-QD. However, HDA-QD exposed samples also demonstrated quite a high IL-8 induction despite the fact that there was no observed cell uptake. It is possible that the HDA-QD may be exerting an effect via an extracellular inflammatory signaling pathway or may be degrading in the cell culture conditions to influence the IL-8 response of the THP-1 cells in the absence of their internalization. Interestingly, for all three QD the level of IL-8 induction was generally lower in cells exposed in the presence of 2% FBS than 10% serum, indicating that serum concentration also affected the observed inflammatory response.
IL-8 is known to be a neutrophil activating protein (NAP- 1) and neutrophil chemotactic factor (NCF).32,33 Not only does IL-8 activate neutrophils, but in fact it also activates lymphocytes, fibroblast and other cells. Additionally, IL-8 induces phagocytosis, which could indicate that when THP-1 cells were exposed to QD, the response was an increase in IL-8 and thus a subsequent increase in QD phagocytosis. IL-8, which is produced by EGF stimulation, is not only responsible for recruiting cells, but an induction of IL-8 could also cause cell proliferation (initiating carcinogenesis) or angiogenesis via EGFR, PI3 K, Akt, and Erk signal pathway activation.34 Thus, the induction of IL-8 by QD exposure to THP-1 cells could have important pathological implications in vivo, particularly if the inflammatory response is maintained long-term because of the biopersistance of these materials.
As the ELISA study indicated that an inflammatory response did arise following exposure of the THP-1 cells to the QD, it was of importance to further examine the resultant inflammatory response. For this purpose, a gene expression study was conducted to examine the transcriptional profile of 84 genes involved in inflammation and oxidative stress (as chronic immune response can lead to the generation of reactive oxygen species (ROS) and oxidative stress). Interestingly, some gene expression changes were common to all QD assessed. The four commonly up-regulated genes were MPV17, TXRND1, NOS2 and BNIP3.
MPV17 was only just over 2-fold up regulated following exposure to all three QD assessed and it encodes for a mitochondrial inner membrane protein. Over-production of MPV17 causes high levels of ROS intracellularly, which indicates that MPV17 is responsible for the production of ROS.35 Thus, up-regulation of this gene could play a general role in driving oxidative stress in the THP-1 cells as a consequence of exposure to the QD. Thioredoxin reductase 1 (TXNRD1) was also up regulated to a slightly higher extent in all treated samples. Interestingly, this molecule has been associated with apoptosis and oxidative stress responses in BEAS-2B when treated with ZnO nanoparticles.36 TXNRD1 also plays a role in protecting cells from oxidative stress. As an increase in TXNRD1 gene expression was observed in all treated samples in the present study, it suggests oxidative stress had been induced and the THP-1 cells were responding by up-regulating TXNRD1 to counteract the adverse affects. Another reactive oxygen species metabolism related gene, BNIP3, showed 2–3-fold increases in expression; the encoded protein interacts with the E1B 10 kDa protein and plays a role in protecting the cell against viral induced cell death. Interestingly, it has previously been reported that ZnO increased the expression of BNIP3 when exposed to BEAS-2B cells, which caused apoptosis and an oxidative stress response.36 The cytotoxicity data presented in this manuscript demonstrated a decrease in cell viability when treated with HDA and PEG-coated QDs and no decrease with carboxylated QDs, which suggests that BNIP3's role may be more related to an oxidative stress response in the latter case.
NOS2 also demonstrated a significant up-regulation (∼2–4 fold increase) following exposure to HDA-QD and carboxylated QDs. NOS2 and its gene product, inducible NOS (iNOS) can generate nitric oxide (NO), which is known to be directly involved in redox reactions, oxidative stress and tissue damage.37 It is believed that NO is an important pro-inflammatory factor as it leads to cell injury or even death.38 Thus, its up-regulation following exposure to the HDA CdSe/ZnS QD could be associated with the observed cytotoxicity in THP-1 cells.
Among the down-regulated genes, glutathione peroxidase 4 (GPX4) gene was down-regulated by all three QD and encodes a protein that catalyzes the reduction of hydroperoxides, lipid peroxide and organic hydroperoxides reduced by glutathione; therefore acting as the cellular defense against toxic oxidant species.39 Interestingly GPX4 is a first line antioxidant defence against ROS and nitrogen species (RNS) in the airway epithelial surfaces.40 Its expression is increased in asthma patients to protect cells from ROS and RNS.40 Peroxiredoxin 2 (PRDX2) acts similarly to GPX4 as it is also a defense against oxidative damage. Thus, the decrease in both PRDX2 and GPX4 gene expression levels after the exposure to QD indicates that these QD affect the glutathione system resulting in reduced defenses in the THP-1 cells against oxidative stress. Another gene, Apolipoprotein E (APOE), which is linked to modifications of the systemic and brain inflammatory responses,41 was 3-fold down regulated in cells exposed to both HDA and carboxylated QDs, but was suppressed by nearly 14-fold following exposure to the PEG-QD. The APOE gene is associated with cholesterol uptake, as its major lipoprotein component (very low-density lipoprotein (VLDL)) stimulates the transfer of excess cholesterol to liver for processing. The APOE expression level is also known to control macrophage response during inflammation, which could explain why APOE was down-regulated in the present study following exposure to all QD assessed.42
Of particular interest in the gene expression study, was that the transcriptional profiles corresponded with the level of cellular uptake, as both the level of cellular uptake and the degree of expression changes followed the order of: carboxylated > PEG > HDA-QD. Consequently, the gene expression profile is suggestive of an oxidative stress based environment arising in the cells as a result of exposure to the QD. Thus, this investigation clearly demonstrates that although all 3 QD are capable of inducing an inflammatory and oxidative stress response, there is clearly variation in the degree of molecular change according to surface chemistry, which correlates with the degree of cellular uptake.
Conclusions
The outcomes of this study have illustrated how immune responsive cells react following exposure to a range of CdSe/ZnS QD. The findings have demonstrated that CdSe/ZnS QD with varying surface functional groups exposed to differentiated THP-1 do induce slightly different responses; the QD pattern of uptake was: carboxylated > PEG > HDA-QD. Although minimal cytotoxicity was induced by the QD and no cell cycle perturbations were observed, the induction of IL-8 by all three QD and the altered oxidative stress related gene expression profiles demonstrated that CdSe/ZnS QD do cause an inflammatory response in THP-1 cells. This is an important observation, because if the immune cells are unable to adequately remove the biopersistent invading particles in vivo, a chronic inflammatory response could result, which in turn may have the potential to promote secondary genotoxicity in the surrounding epithelial tissue.Experimental
Cell culture
The human monocytic cell line derived from peripheral blood of an acute leukaemia infant male patient (THP-1) was used (gifted from Cardiff Metropolitan University) and cultured in RPMI 1640 medium with 10% FBS, 1% filtered non-essential amino acids, 1% L-glutamine and 1% Sodium Pyruvate. THP-1 cells were sub-cultured when the cell growth reached 80% confluence, which was 0.8–1.2 × 106 cells ml−1. Prior to each experiment, flasks were seeded with 1.5 × 105 and/or 3 × 105 THP-1 cells ml−1 depending on the experiment performed.THP-1 cells were differentiated into macrophages using 100 ng ml−1 of phorbol 12-myristate 13-acetate (PMA) in 10 ml of RPMI-1640 culture medium containing THP-1 cells and incubated for 24 h. The differentiated adherent cells were then washed twice with PBS and 10 ml of fresh culture RPMI-1640 media was added.
QD nanoparticles and sample preparation
CdSe/ZnS hexadecylamine (HDA) coated QD were purchased from Sigma Aldrich, UK, while carboxylated CdSe/ZnS and amino polyethylene glycol (PEG) coated quantum dots were from Invitrogen, UK. The emission maxima of each QD were 590 nm for the HDA-QD and 585 nm for the carboxyl- and PEG-QD. Prior to cell exposure carboxyl- and PEG-QD were suspended in water, while the HDA-QD were suspended in 1% DMSO in phosphate buffer solution (PBS). All suspensions were sonicated for 10 min immediately prior to introduction into the cell cultures at a final concentration of 0, 5, 10, 15 and 20 nM. Dose range selection for such studies are also made more difficult by the fact that QD are not being used in specific applications that are associated with human exposure, thus it is currently not possible to determine how doses selected relate to true human exposure scenarios. However, the dose range used in our project was quite typical as a similar dose range (5–20 nM) was also used by other studies on CdSe/ZnS QD capped with PEG, Carboxyl and Polyethylene.21,43 However molar concentrations conversions to mass/volume metrics assuming a primary particle radii of 2.25 nm, diameter of 4.5 nm and cadmium selenide density of 5.82 g cm−2 are also shown in ESI† Table S1.DLS analysis
The hydrodynamic diameter and the zeta potential of the QDs (at the concentration of 15 nM) were measured with a Malvern 4700 system (Malvern instruments Limited, UK) in water and RPMI-1640 medium with or without 2% and 10% fetal bovine serum. Data are presented as the average of 30 readings (10 readings per replicate).Transmission electron microscopy
The QDs were prepared for TEM and also assessed by Elemental Dispersive X-ray (EDX) as described previously.23Cellular uptake: image stream analysis
Differentiated THP-1 cells were exposed to QD for 24 h. THP-1 cells were then trypsinized and washed in PBS, before fixing in 1–1.5 ml of FACS FIX (BD-Bioscience/USA) for 30 min. Following centrifugation at 200 g for 10 min, the FACS FIX was discarded and 5 ml of PBS was added. Five thousand cells per replicate were analyzed on the Image Stream (Amnis Corporation/UK). Each sample was prepared in duplicate, resulting in a total of 10000 cells analysed for QD uptake per sample. Data were analysed using the Ideas v5 software (Amnis Corporation).
Relative population doubling (RPD)
Differentiated THP-1 cells exposed to QD for the appropriate time period and to determine cell viability, the relative population doubling (RPD) calculation was applied as previously described23:where: Population Doubling = [log (Post-treatment cell number ÷ Initial cell number)] ÷ log
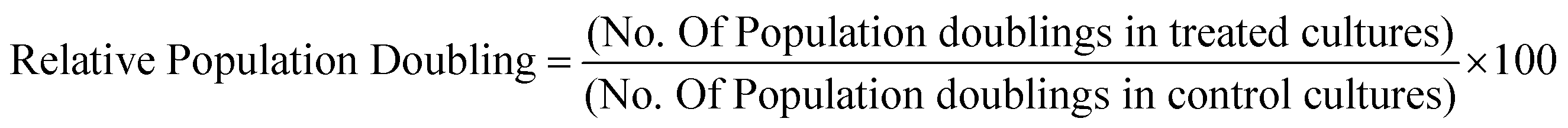 2.
2.
Enzyme-linked immunosorbent assay (ELISA)
Following THP-1 differentiation, QD samples were applied in the presence of 10% or 2% FBS containing media, for 2, 4 or 8 h treatment. The supernatant was then collected after each time point in 1 ml eppendorfs and immediately stored at −20 °C prior to conducting the ELISA assay. IL-1β and IL-8 ELISAs (R&D Systems/UK) were conducted as described in the manufacturers’ protocol, with each sample performed in triplicate per plate and two replicate plates per dose range assessed.Cell cycle analysis
Differentiated THP-1 cells were exposed to QD for 24 h. Following washing with PBS, cells were trypsinised, fixed in 70% ice cold ethanol, then transferred to PBS containing RNaseA. Hoescht DNA stain was applied to the treated THP-1 cells for 45–60 miutes prior to imaging on the BD FACSAria Flow Cytometer system (BD Biosciences/UK) and analysed for cell cycle phase distribution with the FACSDiva v6 1.3 software.Gene expression analysis (PCR arrays)
QD were exposed to differentiated THP-1 cells for 24 h. RNA was extracted using the RNeasy Kit (Qiagen, Crawley, UK), then cDNA was synthesised with the RT2 First Strand Kit (Qiagen/UK). The expression pattern of a panel of 84 genes was subsequently analysed by real-Time PCR using the 96-well RT2 PCR Human Oxidative Stress and Antioxidant Defense PCR Array (PAHS-065A; SABioscience Qiagen Company/UK). Gene arrays expression data analysis was carried out using the Bio-Rad IQ5 software and the SABiosciences PCR Arrays Data Analysis web based software according to manufacturer's instructions.Statistical analysis
All experiments were conducted in triplicate and analyse for statistical significance with a one-way ANOVA. The tests were considered statistically significant if P < 0.05.Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council (EPSRC) for the funding that supported this research (grant application number EP/H008683/1). We gratefully acknowledge the excellent electron microscopy support provided by Dr Andy Brown and Dr Nicole Hondow at the Institute of Materials Research, University of Leeds, who provided all TEM images presented. Additionally we wish to acknowledge the generous financial support provided to Dr Abdullah Al-Ali by the State of Kuwait Ministry of Defence, which supported his contribution to this research.References
- H. Lin and R. H. Datar, Natl. Med. J. India, 2006, 19, 27–32 Search PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef.
- R. A. Tripp, R. Alvarez, B. Anderson, L. Jones, C. Weeks and W. Chen, Int. J. Nanomedicine, 2007, 2, 117–124 CrossRef CAS.
- P. Tallury, A. Malhotra, L. M. Byrne and S. Santra, Adv. Drug Delivery Rev., 2010, 62, 424–437 CrossRef CAS PubMed.
- E. L. Bentzen, F. House, T. J. Utley, J. E. Crowe Jr. and D. W. Wright, Nano Lett., 2005, 5, 591–595 CrossRef CAS PubMed.
- B. Mukhopadhyay, M. Martins, R. Karamanska and D. Russell, Tetrahedron Lett., 2009, 50, 886–880 CrossRef CAS PubMed.
- Y. Zheng, D. Tan, Z. Chen, C. Hu, Z. J. Mao, T. P. Singleton, Y. Zeng, X. Shao and B. Yin, J. Nanosci. Nanotechnol., 2014, 14, 4014–4021 CrossRef CAS PubMed.
- R. S. Yang, L. W. Chang, J. P. Wu, M. H. Tsai, H. J. Wang, Y. C. Kuo, T. K. Yeh, C. S. Yang and P. Lin, Environ. Health Perspect., 2007, 115, 1339–1343 CrossRef CAS PubMed.
- J. M. Barnett, J. S. Penn and A. Jayagopal, Methods Mol. Biol., 2013, 1026, 45–56, DOI:10.1007/978-1-62703-468-5_4.
- X. Zhang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438–4443 CrossRef CAS PubMed.
- N. Singh, B. Manshian, G. J. Jenkins, S. M. Griffiths, P. M. Williams, T. G. Maffeis, C. J. Wright and S. H. Doak, Biomaterials, 2009, 30, 3891–3914 CrossRef CAS PubMed.
- F. A. Kauffer, C. Merlin, L. Balan and R. Schneider, J. Hazard. Mater., 2014, 268, 246–255 CrossRef CAS PubMed.
- S. Tang, Y. Wu, C. N. Ryan, S. Yu, G. Qin, D. S. Edwards and G. D. Mayer, Chemosphere, 2014, 120C, 92–99 Search PubMed.
- V. Lee, R. S. McMahan, X. Hu, X. Gao, E. M. Faustman, W. C. Griffith, T. J. Kavanagh, D. L. Eaton, J. K. McGuire and W. C. Parks, Nanotoxicology, 2014, 1–8 Search PubMed.
- N. Fujiwara and K. Kobayashi, Curr. Drug Targets: Inflammation Allergy, 2005, 4, 281–286 CrossRef CAS.
- S. J. Byrne, S. A. Corr, T. Y. Rakovich, Y. K. Gun'ko, Y. P. Rakovich, J. F. Donegan, S. Mitchell and Y. Volkov, J. Mater. Chem., 2006, 16, 2896–2902 RSC.
- J. Lee, Y. Choi, Y. Cho and R. Song, J. Nanosci. Nanotechnol., 2013, 13, 417–422 CrossRef CAS PubMed.
- J. D. Clogston and A. K. Patri, Methods Mol. Biol., 2011, 697, 63–70 CAS.
- P. Zrazhevskiy and X. Gao, Nano Today, 2009, 4, 414–428 CrossRef CAS PubMed.
- M. J. Clift, B. Rothen-Rutishauser, D. M. Brown, R. Duffin, K. Donaldson, L. Proudfoot, K. Guy and V. Stone, Toxicol. Appl. Pharmacol., 2008, 232, 418–427 CrossRef CAS PubMed.
- L. W. Zhang and N. A. Monteiro-Riviere, Toxicol. Sci., 2009, 110, 138–155 CrossRef CAS PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- N. Singh, G. J. Jenkins, B. C. Nelson, B. J. Marquis, T. G. Maffeis, A. P. Brown, P. M. Williams, C. J. Wright and S. H. Doak, Biomaterials, 2012, 33, 163–170 CrossRef CAS PubMed.
- L. W. Zhang, L. Zeng, A. R. Barron and N. A. Monteiro-Riviere, Int. J. Toxicol., 2007, 26, 103–113 CrossRef CAS PubMed.
- A. Shiohara, A. Hoshino, K. Hanaki, K. Suzuki and K. Yamamoto, Microbiol. Immunol., 2004, 48, 669–675 CrossRef CAS PubMed.
- J. P. Ryman-Rasmussen, J. E. Riviere and N. A. Monteiro-Riviere, J. Invest. Dermatol., 2007, 127, 143–153 CrossRef CAS PubMed.
- L. Kong, T. Zhang, M. Tang and Y. Pu, J. Nanosci. Nanotechnol., 2012, 12, 8258–8265 CrossRef CAS PubMed.
- L. Chen, G. Qu, C. Zhang, S. Zhang, J. He, N. Sang and S. Liu, Integr. Biol., 2013, 5, 590–596 RSC.
- R. P. Schins, Inhalation Toxicol., 2002, 14, 57–78 CrossRef CAS PubMed.
- K. C. Nguyen, V. L. Seligy and A. F. Tayabali, Nanotoxicology, 2013, 7, 202–211 CrossRef CAS PubMed.
- L. W. Zhang, W. W. Yu, V. L. Colvin and N. A. Monteiro-Riviere, Toxicol. Appl. Pharmacol., 2008, 228, 200–211 CrossRef CAS PubMed.
- M. D. Miller and M. S. Krangel, Crit. Rev. Immunol., 1992, 12, 17–46 CAS.
- M. Baggiolini and I. Clark-Lewis, FEBS Lett., 1992, 307, 97–101 CrossRef CAS.
- Y. Zhang, L. Wang, M. Zhang, M. Jin, C. Bai and X. Wang, J. Cell Physiol., 2012, 227, 35–43 CrossRef CAS PubMed.
- R. M. Zwacka, A. Reuter, E. Pfaff, J. Moll, K. Gorgas, M. Karasawa and H. Weiher, EMBO J., 1994, 13, 5129–5134 CAS.
- C. C. Huang, R. S. Aronstam, D. R. Chen and Y. W. Huang, Toxicol. in Vitro, 2010, 24, 45–55 CrossRef CAS PubMed.
- C. A. Colton, D. M. Wilcock, D. A. Wink, J. Davis, W. E. Nostrand and M. P. Vitek, J. Alzheimer's Dis., 2008, 15, 571–587 CAS.
- X. Gao, R. Ray, Y. Xiao and P. Ray, Basic Clin. Pharmacol. Toxicol., 2008, 103, 255–256 CrossRef CAS PubMed.
- Q. Shen, S. Chada, C. Whitney and P. E. Newburger, Blood, 1994, 84, 3902–3908 CAS.
- S. A. Comhair, P. R. Bhathena, C. Farver, F. B. Thunnissen and S. C. Erzurum, FASEB J., 2001, 15, 70–78 CrossRef CAS PubMed.
- J. R. Lynch, W. Tang, H. Wang, M. P. Vitek, E. R. Bennett, P. M. Sullivan, D. S. Warner and D. T. Laskowitz, J. Biol. Chem., 2003, 278, 48529–48533 CrossRef CAS PubMed.
- A. Gafencu, M. Robciuc, E. Fuior, V. Zannis, D. Kardassis and M. Simionescu, J. Biol. Chem., 2007, 282, 21776–21785 CrossRef CAS PubMed.
- T. Rong Kuo, C. Lee, S. Lin, C. Dong, C. Chen and H. Tan, Chem. Res. Toxicol., 2011, 24, 253–261 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tx00175c |
| ‡ Joint first authors (A. Al-Ali & N. Singh). |
| This journal is © The Royal Society of Chemistry 2015 |