
Matrix-derived combination effects influencing absorption, distribution, metabolism and excretion (ADME) of food-borne toxic compounds: implications for risk assessment
Ivonne M. C. M.
Rietjens
*a,
Bożena
Tyrakowska
b,
Suzanne J. P. L.
van den Berg
a,
Ans E. M. F.
Soffers
a and
Ans
Punt
a
aDivision of Toxicology, Wageningen University, Tuinlaan 5, NL-6703 HE Wageningen, The Netherlands. E-mail: ivonne.rietjens@wur.nl; Fax: +31-317-484931; Tel: +31-317-483971
bFaculty of Commodity Science, The Poznań University of Economics, al. Niepodległości 10, 61-875 Poznań, Poland
First published on 1st September 2014
Abstract
Absorption, distribution, metabolism and excretion (ADME) of food-borne toxic compounds may be influenced by other compounds or constituents present in the food. The present review presents an overview of evidence currently available on food matrix-derived combination effects influencing the ADME characteristics of food-borne toxic compounds and the possible implications for risk assessment. The results obtained indicate that interactions may occur at all levels of ADME and that the interactions may decrease but also increase the bioavailability and/or toxicity of the compounds of interest. The overview also illustrates that food matrix-derived combination effects should be considered on a case-by-case basis, taking into account especially the mode of action underlying the interactions and the dose dependency of the effects. Especially food matrix-derived combination effects that proceed by a reversible mode of action, such as for example binding to biotransformation enzymes or transport proteins, may be detected at concentrations used in in vitro assays and at dose levels used in animal bioassays but may be absent at dose levels representing realistic human intake. It is concluded that although food matrix-derived combination effects may exist, their detection in in vitro assays or in animal bioassays at high dose levels may not improve risk assessment practice because interactions observed may not be maintained at low realistic levels of intake. Insight in the mode of action underlying the interactions combined with physiologically based kinetic (PBK) modelling may prove a way to obtain better insight in whether interactions detected at high dose levels will still be relevant at more realistic lower intake levels, and thus to what extent these effects should be taken into account in the risk assessment for human exposure.
 Ivonne M. C. M. Rietjens | Professor Ivonne M. C. M. Rietjens is full professor in Toxicology at Wageningen University (WUR), Netherlands. She is a member of the Royal Netherlands Academy of Arts and Sciences (KNAW) and member of several national and international advisory committees. She is author of over 350 scientific publications in refereed journals. Her research focuses on food ingredients, including natural toxins and functional food ingredients, physiologically-based kinetic and dynamic models for low dose cancer risk extrapolation, the consequences of genetic polymorphisms and life style factors for individual sensitivity and risk assessment, and alternative methods for animal testing. |
 Bożena Tyrakowska | Bożena Tyrakowska completed her PhD on physicochemical properties of flavin derivatives at the Poznań University of Economics. In 1987–1989 she was working on enzymatic reactions in reversed micelles in the group of Professor Cees Veeger in the Department of Biochemistry of the Wageningen University, the Netherlands. Further cooperation with Professor Ivonne Rietjens focused her research on biotransformation of xenobiotics catalysed by cytochrome P-450, which resulted in D. Sci. degree, in the Farmacology Department of the Medical University, Poznań, in 1997. She works permanently at the Poznań University of Economics as a professor. Her research focuses on physicochemistry and toxicology of bioactive food ingredients. |
 Suzanne J. P. L. van den Berg | Suzanne J. P. L. van den Berg received her M.Sc. degree in Biomedical Sciences from the Radboud University Nijmegen with a major in Toxicology, a minor in Nutrition and Health and a second minor in Human Pathobiology. She received a Ph.D. in Toxicology from Wageningen University where she worked on testing and validating new concepts that could be of use for the risk and safety assessment of botanicals and botanical preparations containing genotoxic carcinogens. She currently works as a toxicological risk assessor food safety at TNO, the Netherlands. |
 Ans E. M. F. Soffers | Ans E.M.F. Soffers is technician on the department of Toxicology of Wageningen University since 2002. She is involved in literature searches, quantum mechanical calculations and PBK modelling. Before she worked in the field of clinical chemistry, hematology, human nutrition and biochemistry. |
 Ans Punt | Dr ir. Ans Punt is assistant professor at the division of Toxicology at Wageningen University. The goal of her research is the development of computational systems toxicology approaches that quantitatively integrate in vitro data to simulate functioning organisms. She successfully developed various physiologically based kinetic and dynamic models to simulate dose-dependent effects, species differences, human variation, and matrix modulation of bioactivation, detoxification, and biological effects of chemicals in intact organisms and supervises various PhD projects in this field. She is both a Dutch and European Registered Toxicologist (ERT). |
Introduction
An important aspect that should be taken into account when assessing the risk of food-borne toxic compounds is whether results from long-term animal studies with pure compounds dosed by gavage without the occurrence of the natural food matrix represent a good starting point for the risk assessment. For example, a slow or incomplete release of the ingredient from the matrix and/or inhibition of specific intestinal carriers involved in active uptake of an ingredient may result in reduced bioavailability of a compound as compared to the bioavailability of the same compound when dosed in a pure form by gavage. Schilter et al. (2003) already concluded that such a matrix interaction would raise serious questions about the use of toxicity data of the pure compound for risk assessment of the compound within the complex food matrix.1 In addition to interactions at the level of absorption, interactions may also occur at the level of distribution, metabolism or excretion, thereby also influencing bioavailability and toxicity. The present review presents an overview of data available on matrix-derived combination effects influencing absorption, distribution, metabolism and excretion (ADME) of food-borne toxic compounds, and evaluates to what extent and how these interactions should be taken into account in subsequent risk assessment.Absorption
Table 1 presents examples of matrix-derived combination effects on absorption of food-borne toxic compounds and their possible mode of action. From this overview it appears that food matrix-derived effects on absorption may result from effects of the food matrix on bioaccessibility of compounds and/or from interactions with processes underlying the actual transport across the intestinal barrier.| Matrix-derived combination effect | Possible mode of action |
|---|---|
| A significant cellular accumulation of ochratoxin A in Caco-2 cells was observed upon co-incubation with chrysin, quercetin, genistein, biochanin A, or resveratrol, all at concentrations that can be expected in the gastrointestinal tract.8 | Competitive inhibition of the MRP efflux pump in the apical membrane of the cells, proposed to be MRP2.20 Inhibition of the apical MRP2-mediated excretion of ochratoxin A from the intestinal cells back to the apical luminal side, would explain the increased cellular accumulation resulting in increased possibilities for transport of ochratoxin A to the basolateral side. |
| An increase of the transcellular transport of PhIP in Caco-2 cell monolayers upon addition of the flavonoid myricetin.9 This example is discussed in some more detail in the main text. | Myricetin inhibits the ABC transporter-mediated excretion of PhIP from the intestinal cells back to the apical luminal side, resulting in increased possibilities for transport of PhIP to the basolateral side.9 |
| The extracellular transport of benzo(a)pyrene (B(a)P) metabolites from Caco-2 cells exposed to 3-hydroxy-B(a)P was inhibited by co-exposure with luteolin.21 | Luteolin interacts with the transporter breast cancer resistant (BCRP) protein.21 |
| Intracellular accumulation of the drug vinblastine was increased in the presence of EGCG. In addition, EGCG potentiates the cytotoxicity of vinblastine in CHRC5 cells.22 | EGCG inhibits the binding and efflux of vinblastine by Pgp in the multidrug-resistant cell line CHRC5.22 The inhibitory effect of EGCG on Pgp was also observed in human Caco-2 cells.22 |
| The level of ferulic acid metabolites recovered in the urine of rats amounted to only 3% of the ingested dose when ferulic acid was provided in a complex cereal matrix of Triticum durum, whereas the metabolites represented 50% of the dose when ferulic acid was dosed as a pure compound.23–25 | Impaired release from the food matrix.23–25 |
| The bioavailability of β-carotene was reported to be one order of magnitude higher when provided as a pure compound added to food (e.g. salad dressing) than when present naturally in mixed vegetables.26 | Influence on release from the food matrix. |
| The bioavailability of carotenoids is higher from salads ingested with full fat than with fat free salad dressing.25 | Effect of fat on release from the food matrix. |
| The aqueous bioaccessible fraction of isoflavones determined in vitro using simulated oral, gastric and small intestinal digestion, was shown to be higher from foods containing fat and protein than from an isoflavone supplement consumed without food.27 | Effect of fat on release from the food matrix. |
| Absorption of [14C]-phenanthrene, [14C]-TCDD or [14C]-B(a)P administrated in milk to pigs with a direct relationship between the PAH absorption and fat absorption.28 | Role of dietary fats in the bioavailability of polycyclic aromatic hydrocarbons (PAHs).29 |
| Effects on the uptake of B(a)P by dietary fibre and beef.30,31 | Role of dietary fats in the bioavailability of polycyclic aromatic hydrocarbons (PAHs).29 |
| Influence of fatty acid lipid composition on PAH (7,12-dimethylbenz[a]anthracene) bioavailability.32 | Role of dietary fats in the bioavailability of polycyclic aromatic hydrocarbons (PAHs).29 |
| Rate of absorption of EGCG was higher in rats following the combined exposure to EGCG and decaffeinated green tea in comparison to the situation in which pure EGCG was intragastrically administered to the rats.33 | The absorption rate constant (Ka) of pure EGCG after intragastral administration was 1.4 ± 0.6 min−1 × 10−3 and Ka of EGCG from decaffeinated green tea was 5.0 ± 2.6 min−1 × 10−3. Mode of action not known. |
| Pomegranate juice was found to show higher antiproliferative, apoptotic and antioxidant activity in vitro than the individual polyphenols extracted from the fruit.34,35 | Mode of action has not been identified. |
| A plant matrix of cinnamon influenced the bioavailability of coumarin, albeit to a limited extent.36 The authors demonstrated that the relative extent of coumarin absorption (measured as urinary excretion of the coumarin main metabolite 7-hydroxycoumarin) from powder of cassia cinnamon or from rice pudding was only slightly lower (89% and 87% respectively) than that of isolated coumarin. Surprisingly, the extent of absorption of coumarin in cinnamon tea was slightly higher amounting to 105% of that of the isolated coumarin. | Theoretically this last observation may be explained by the fact that components of the cinnamon matrix that interfere with the absorption of coumarin are not transferred from the cinnamon powder to the tea during preparation. |
| Administration of green tea extracts to fasting dogs leads to a no-observed-adverse-effect-level (NOAEL) for EGCG being at least 10-fold lower than that derived from the study in pre-fed dogs.37 This may suggest that conditions in which green tea extracts are taken combined with food consumption may minimize possible risks. Sarma et al. (2008) even concluded that clinical pharmacokinetic and animal toxicological information indicated that consumption of green tea concentrated extracts on an empty stomach is more likely to lead to adverse liver effects than consumption in the fed state.38 | Mode of action has not been identified. |
| In a clinical study in healthy human volunteers it was demonstrated that greater bioavailability of free catechins can be achieved by taking high grade green tea polyphenol extract capsules on an empty stomach after an overnight fast.39 | Mode of action has not been identified. |
| The bioavailability of aflatoxins (i.e. aflatoxin B1 (AFB)) could be reduced by chlorophyllin present in green leafy vegetables (e.g. spinach).13–15 This example is discussed in some more detail in the main text. | Chlorophyllin was shown to form a strong non-covalent complex with AFB in vitro and it was suggested that this complex formation between chlorophyllin-like compounds and carcinogens having an at least partially planar aromatic structure may contribute to the chemopreventive activities associated with a high green vegetable intake.15 |
| Natural compounds and plant extracts reduce the toxic potential of many food-related toxins: AFB, fumonisins, and ochratoxin A produced by fungi; cholera toxin produced by Vibrio cholerae bacteria; Shiga toxins produced by E. coli bacteria; staphylococcal enterotoxins produced by Staphylococcus aureus bacteria; ricin produced by seeds of the castor plant Ricinus communis; and the glycoalkaloid α-chaconine synthesized in potato tubers and leaves.40 | Complex formation thereby limiting bioavailability. For example, apple juice inhibits the biological (toxicological) activity of the food-borne pathogen Staphylococcal enterotoxin A in vitro because phenolic compounds in the apple juice may reduce the toxicity by binding the toxic protein, decreasing its bioavailability.41 |
| The bioavailability of nickel in the matrix of scrambled eggs was considerably reduced compared to the situation where nickel in drinking water was ingested during fasting.19 This study is discussed in some more detail in the main text. | Complex formation between nickel and matrix components. |
| The bioavailability of ingested soluble lead (Pb) in adults had been found to vary from 2–10% when ingested with a meal to 60–80% when ingested after a fast.42 | Complex formation between lead and matrix components. |
| Fasting humans absorbed 40–50% of 203Pb taken in distilled water,43 irrespective of the addition of Pb carrier up to 100 mg per dose. When taken with tea or coffee, uptake averaged 14% and with beer 19%. Much lower uptakes, ranging from 3 to 7%, were found when 203Pb was taken with a meal. | Complex formation between lead and matrix components. |
| Consuming a balanced meal with added soluble 203Pb reduced lead uptake to 4% and the influence of the food lasted for up to 3 h after consuming a meal.44 | Constituents of food in the gastrointestinal tract decrease the ingested lead absorption, although the exact mechanisms by which this occurs are not entirely understood.44 |
| In an in vitro digestion model black coffee as well as green and black tea added to each of the fish meal samples (raw fish) significantly reduced mercury bioaccessibility by 50–60%, compared to raw fish mercury bioaccessibility. Corn starch addition did not show significant impact on mercury bioaccessibility. Moreover, it was shown that boiling and frying reduced mercury bioaccessibility by 40% and 60% respectively, when compared to raw fish.45 | Complex formation between mercury and matrix components. |
| Cadmium, chromium, copper, nickel, manganese, lead and zinc present in the finest size particles (clay fraction) of urban soils are not equally bioaccessible.46,47 This appeared to be of importance when evaluating the risks associated with geophagy, the practice of eating clay or soil, as practiced by for example children and pregnant and lactating women on parts of the African continent, in Asia, and in South and Central America.48 | Complex formation between metals and matrix components. |
| The bioavailability of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/F) in young Goettingen minipigs orally exposed to known amounts of PCDD/F either from soil (soil-bound) or as an extract of the same soil by solvent was different.49 Under the experimental conditions and compared to PCDD/F orally administrated in solvent, the soil matrix reduced the bioavailability of PCDD/F by about 70%. | Complex formation between (PCDD/F) and matrix components. |
| In an in vitro model of gastrointestinal digestion followed by uptake into Caco-2 cells significantly lower amounts of [14C]-B(a)P were present in Caco-2 cells from soil containing a higher percentage of organic matter compared to soil with a lower percentage of organic matter.50 | Complex formation between B(a)P and matrix components. |
| The bioavailability of methyleugenol when given orally in a pure form is likely to be higher than when it is given as a part of the food matrix. This was derived from the observation that when the bioavailability of methyleugenol from a gingersnaps and orange juice food matrix would be 13.8%, the experimental value observed in a human intervention study would accurately match the values predicted by a physiologically based kinetic (PBK) model.51 | Mode of action has not been identified. |
The transcellular transport of ingested food ingredients across the intestinal barrier is an important factor determining bioavailability upon oral intake. This transcellular transport of a chemical over the intestinal epithelium can be largely dependent on the activity of membrane bound active ATP binding cassette (ABC) transport proteins. The intestinal ABC transporters involved in the efflux of chemicals from the intestinal cells include P-glycoprotein (Pgp), Multidrug Resistance Proteins (MRPs) and Breast Cancer Resistance Protein (BCRP).2–5 These transporters are generally located specifically in the apical (intestinal luminal side) or basolateral (blood/plasma side) membrane of the enterocytes (Fig. 1).2,4,6 As a result, ABC transporters are involved in the efflux of bioactive compounds from the intestinal cells, either to the basolateral blood side, facilitating absorption, or back into the intestinal lumen, reducing bioavailability. Studies on the role of ABC transporters in oral bioavailability often focused on oral drugs, but some studies also focused on a possible role of ABC transporters in determining the bioavailability of food ingredients, including food-borne toxic compounds. For example, a review by Brand et al. (2006) reported the influence of flavonoids on ABC transporters and the resulting effects on the bioavailability of several food-borne toxins like the mycotoxin ochratoxin A, the pro-carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and of other food-borne bioactive ingredients such as for example epigallocatechin-3-gallate (EGCG).7–12
 | ||
| Fig. 1 Cellular location of intestinal ABC transporters. For references see Brand et al. (2006).7 | ||
Table 1 includes an overview of these and other examples including effects on the absorption of ochratoxin A, PhIP, benzo(a)pyrene (B(a)P) and the drug vinblastine.
The example on PhIP is especially of interest because it illustrates that matrix-derived combination effects can result not only in reduced absorption but may also result in an increase of the transport across the intestinal barrier. The transcellular transport of PhIP in Caco-2 cell monolayers was increased upon addition of the flavonoid myricetin, reflected by an increase in the transport of PhIP from the apical to the basolateral compartment, observed at physiologically relevant concentrations of PhIP and myricetin.9 Myricetin inhibits the ABC transporter-mediated excretion of PhIP from the intestinal cells back to the apical luminal side, resulting in increased possibilities for transport of PhIP to the basolateral side and a possible increased bioavailability of PhIP.9 In subsequent studies it was shown that other flavonoids including flavone, kaempferol, luteolin, quercetin, chrysoeriol and naringenin exert a similar effect on the transport of PhIP through Caco-2 monolayers.10
Based on the data available in the literature, it can be concluded that flavonoid-mediated inhibition of ABC transporters may affect the bioavailability of bioactive food ingredients and/or food-borne toxic compounds upon oral uptake. Flavonoids are present in a wide variety of foods of plant origin and botanical preparations and therefore it is likely that these compounds affect the bioavailability of substances of concern simultaneously occurring in food or a botanical preparation of interest.
The occurrence of a difference in the bioavailability of a compound when ingested within a food matrix or as a pure compound may also be due to impaired release from the food matrix. Table 1 also presents several examples for which such an effect on release of an active or toxic compound from the food matrix has been observed. Of special interest are studies reporting that the oral bioavailability of green tea catechins can be enhanced when consumed in the absence of food. Chow et al. (2005), for example, demonstrated in a clinical study in healthy human volunteers that greater bioavailability of free catechins can be achieved by taking high grade green tea polyphenol extract capsules on an empty stomach after an overnight fast.39 This dosing condition is expected to influence the biological effects of tea catechins.
Other examples reveal effects of the food matrix on the bioavailability of ferulic acid, β-carotene and other carotenoids, isoflavones, polycyclic aromatic hydrocarbons (PAHs) including B(a)P, dioxins and dibenzofurans, polyphenols including EGCG and other catechins, and coumarin (Table 1).
Another interesting example of interactive effects reducing the bioavailability can be found in the studies reporting that the bioavailability of aflatoxins (i.e. aflatoxin B1 (AFB) could be reduced by chlorophyllin present in green leafy vegetables (e.g. spinach).13–15 Chlorophyllin was shown to form a strong non-covalent complex with AFB in vitro and it was suggested that this complex formation between chlorophyllin-like compounds and carcinogens having an at least partially planar aromatic structure may contribute to the chemopreventive activities associated with a high green vegetable intake.15 The authors even demonstrated that concomitant exposure to AFB and chlorophyllin resulted in the inhibition of AFB-induced hepatocarcinogenesis in rainbow trout by chlorophyllin, as a result of the formation of a tight chemically stable molecular complex between chlorophyllin and AFB.13–15 Thus, chlorophyllin may reduce the DNA damage caused by AFB in vivo, by acting as an ‘interceptor molecule’ that blocks the absorption of AFB from the diet. Based on its protective effects and lack of any apparent toxicity in humans chlorophyllin was used in a clinical trial in China. Administration of chlorophyllin three times a day led to a 50% reduction in the median level of urinary excretion of aflatoxin-N7-guanine, which was used as a biomarker for systemic bioavailability.16 Chlorophyllin was also reported to bind to planar aromatic carcinogens such as B(a)P thereby significantly reducing B(a)P-DNA adduct formation in normal human mammary epithelial cells.17 Chlorophyllin was also demonstrated to be effective in the reduction of transplacental cancer risk if given with the PAH carcinogen dibenzo(a,1)pyrene.18
An example of a food matrix effect on the bioavailability of heavy metals was reported by Nielsen et al. (1999) for nickel.19 These authors studied the influence of food intake and gastric emptying/fasting on the absorption and retention of nickel from drinking water in eight male volunteers fasted overnight before being given nickel in drinking water combined, at different time intervals, with standardized 1400 kJ portions of scrambled eggs. The study demonstrated that the bioavailability of nickel in the matrix of scrambled eggs was considerably reduced compared to the situation where nickel in drinking water was ingested during fasting. Table 1 presents several additional examples where food matrix effects influence the bioavailability of metals including lead, mercury, cadmium, chromium, copper, nickel, manganese, and zinc.
Based on all examples presented in Table 1 and described above it can be concluded that the absorption of food-borne chemicals can be affected by food-matrix based combination effects via several modes of action including especially the impaired release from the food matrix and/or influence on transport proteins. These matrix-derived combination effects may result in reduced absorption, but, as in the case of the inhibition of intestinal apical ABC transporters may also result in increased absorption.
Distribution
Only a few studies were identified reporting on possible food matrix-derived combination effects at the level of distribution. Table 2 presents some examples of matrix-derived combination effects on distribution of food-borne toxic compounds and their possible mode of action.| Matrix derived combination effect | Possible mode of action |
|---|---|
| Distribution of EGCG was different when pure EGCG was administered intravenously to rats than when an equivalent dose of ECGC was given intravenously in combination with decaffeinated green tea.33 The plasma concentration of EGCG and the area under the plasma concentration versus time curve were lower following the intravenous exposure to pure EGCG. In addition, a larger distribution volume was observed following the treatment with pure EGCG. | Interactions at the level of activity of efflux transporters. |
| Effect of dietary fat on disposition and metabolism of fluoranthene, a food-borne PAH, revealed that fluoranthene DNA adduct formation in several target tissues of F-344 rats was higher when administrated through saturated fat compared to mono- and polyunsaturated fat groups.52 | Fat composition of the diet affects disposition and metabolism.52 |
| An increase in polyunsaturated fat content in the diet greatly elevated the conversion of B(a)P-7,8-dihydrodiol to its ultimate carcinogenic metabolite B(a)P-7,8-dihydrodiol-9,10-epoxide and also resulted in higher levels of DNA binding.53 | The type of lipids available in plasma and their levels may play a role in delivering absorbed PAHs to target organs and/or in oxidation and formation of metabolites via lipid peroxidation related-processes, which were suggested to be enhanced upon intake of a diet rich in polyunsaturated fatty acids. |
| The high density lipoprotein fraction facilitates B(a)P uptake into hepatocytes, whereas low-density lipoproteins inhibit the uptake.54 | After gastric instillation B(a)P is absorbed via the intestinal lymphatic drainage and transported to the vascular circulation sequestered within lipoproteins. Therefore, it is possible that variations in the lipoprotein composition induce differences in uptake of PAHs into the liver and other organs and may determine the differences not only in organ specific metabolism but also in organ specific DNA adduct formation. |
| The flavonoid baicalin has been observed to modulate the protein binding of the drug nifedipine, resulting in an increased Cmax of unbound nifedipine.55 | Competition for plasma transport by plasma proteins may influence distribution.55 Similar interactions between food ingredients influencing plasma protein binding have not been reported, but could theoretically occur. |
The limited number of examples available reflects that experimental evidence for the effect of matrix-derived combination effects on distribution of food-borne toxic compounds is scarce but it can be anticipated that interactions at the level of distribution may originate form interaction at the level of activity of different efflux transporters, resulting in effects on the distribution to specific target tissues, and/or affecting the elimination by secretion into bile and urine. In theory, the disposition of drugs and bioactive compounds may be affected through interactions with the activity of different efflux transporters, including Pgp and BCRP known to be involved in distribution to target tissues and/or elimination via secretion into bile and urine.39 Furthermore, dietary factors affecting lipid profiles may affect distribution and target tissue concentrations as well, thereby possibly inducing changes in subsequent metabolism and bioactivation and toxicity. Food–food interactions at the level of plasma protein binding have not been reported, but such interactions could theoretically occur similar to what has been reported for combined food–drug exposures.55
Metabolism
In addition to the possible alterations in the rate and extent of absorption and distribution, the food or botanical-derived matrix may have an effect on the metabolism of the active chemical substance(s) of interest. Table 3 presents examples of matrix-derived combination effects on metabolism of food-borne toxic compounds and their possible mode of action. The effects can occur directly at the level of both phase 1 and phase 2 metabolism, or be due to the influences on the level of expression of metabolic enzymes. Various examples of food–food or food–drug interactions at the level of metabolism exist.| Matrix derived combination effect | Possible mode of action |
|---|---|
| In a Salmonella typhimurium NM2009 system individual PAHs were shown to inhibit their own metabolism and metabolism of other carcinogens catalysed by cytochrome P450 (CYP)1A1, CYP1A2 and CYP1B1.81 | Interaction with phase 1 enzymes. |
| Daidzein, a principle isoflavone in soybean, has an inhibitory effect on the metabolism of the CYP1A2 substrates caffeine and theophylline.62 | Daidzein, in higher doses may inhibit CYP1A2 activity in vivo. |
| Luteolin inhibited B(a)P-induced expression and activity of CYP1A1 in Caco-2 cells exposed to B(a)P.21 | Inhibition of CYP1A1. |
| Dietary consumption of apiaceous and allium vegetables was shown to inhibit CYP1A2 activity in humans, and it has been demonstrated that some compounds in those vegetables (e.g., apigenin, psoralen, 5- and 8-methoxypsoralen) act as potent inhibitors of human CYP1A2 and significantly reduce human CYP1A2-mediated mutagenicity of AFB in a recombinant in vitro system.82 | Inhibition of CYP1A2. |
| Inhibitory effect of flavonoids (e.g. naringin, naringenin, kaempferol and quercetin) and furanocoumarins (e.g. bergamottin and 6′,7′-dihydroxybergamottin), occurring in grapefruit juice, on CYP3A4 activity.56–58 This example is discussed in some more detail in the main text. | Inhibition of CYP3A4. |
| Many diet-derived compounds have been shown to influence the biotransformation of AFB, and some efficiently protect against AFB-induced genotoxicity.61 | These observations have been related to inhibition of the activity of CYP1A2 and CYP3A4 the major enzymes involved in the bioactivation of AFB to its genotoxic epoxide metabolite exo-AFB-8,9-epoxide. |
| Dietary flavonoids (i.e. flavone, galangin tangeretin) showed a potent inhibition of CYP1A2 in vitro.61 | Inhibition of CYP1A2. |
| Nevadensin inhibits the sulfotransferase (SULT) mediated bioactivation of the allylalkoxybenzenes like estragole en methyleugenol, also known to be present in basil.65–68 This example is discussed in some more detail in the main text. | Inhibition of SULT enzymes. |
| St. John's wort can induce CYP3A4, and this increased CYP3A4 activity can result in a decrease of plasma levels of several prescription drugs including alprazolam, irinotecan and indinavir.74–77 In addition, St. John's wort may interfere with the efficacy of oral contraceptives.78,79 | Induction of expression of metabolic phase 1 enzymes. |
| Dietary compounds of different origin (e.g., constituents of brassica vegetables and hops) have been shown to modify expression of human hepatic enzymes involved in the oxidation of AFB.61 | Induction of expression of metabolic phase 1 enzymes. |
| In the mouse intestinal cell line Apc(−/+), and control Apc(+/+) cells the combined exposure to B(a)P and PhIP resulted in an increase of PhIP derived DNA adducts in the presence of B(a)P.83 | A B(a)P mediated increase in the expression of CYP1A enzymes which are involved in the bioactivation of PhIP may provide a mechanistic explanation for these observations. |
| Sulforafane protected animals from AFB-induced tumors, reduced AFB biomarkers in humans in vivo and reduced AFB adduct formation in human hepatocytes.61 | In human hepatocytes the protective effects of sulforafane were ascribed to repression of human hepatic CYP3A4 expression, rather than induction of protective GSTs.61 |
| Another major glucosinolate-derived compound present in broccoli, 3,3′-diindolylmethane (DIM), significantly increased AFB-related DNA damage.84 | DIM significantly increased DNA adduct formation, in a concentration-dependent manner, due to a significant up-regulation of CYP1A1 and CYP1A2 as well as down-regulation of GSTM1.84 |
| Inhibition of B(a)P or cyclophoshamide induced mutagenicity by an aqueous extract of the bark of Cinnamomum cassia, a food flavor.85 In the bone marrow chromosomal aberration assay and the micronucleus test in mice C. cassia extract significantly inhibited the mutagenicity of B(a)P and cyclophoshamide after pretreatment of the mice with the C. cassia extract orally for seven consecutive days. | The C. cassia pretreatment decreased CYP content but increased GSH content and the activity of GSH dependent antioxidant enzymes, including glutathione S-transferases (GSTs), glutathione reductase and glutathione peroxidase. C. cassia-mediated protection could be due to the induction of phase 2 enzymes involved in the detoxification pathways of B(a)P and cyclophoshamide and/or inhibition of phase 1 enzymes responsible for the bioactivation of these carcinogens.85 |
| Disposition and metabolism of fluoranthene in F-344 rats was influenced by dietary fat, and also for B(a)P an effect of the type of dietary fat on biotransformation was reported.52,86 | The expression and activities of CYP1A1, CYP1B1 and GSTs were more pronounced when B(a)P was administered through saturated fat, compared to its administration through unsaturated fat or tricaprylin, influencing the biotransformation profiles and the level of formation of B(a)P-DNA adducts. |
Many studies report on direct interactions at the level of phase 1 and phase 2 enzymes. These include effects on cytochromes P450 (CYPs) involved in phase 1 metabolism by PAHs, flavonoids like diadzein, lutein and apigenin, psoralen and 5- and 8-methoxypsoralen (Table 3).
Well known is the inhibitory effect of flavonoids (e.g. naringin, naringenin, kaempferol and quercetin) and furanocoumarins (e.g. bergamottin and 6′,7′-dihydroxybergamottin), occurring in grapefruit juice, on CYP3A4 activity.56–58 Resulting from the grapefruit juice-mediated inhibition of human CYP3A activity in the small intestine, the level of pre-systemic metabolism of prescription drugs mediated by CYP3A4 has been found to be reduced increasing their oral bioavailability.59 Although it seems to be widely recognized that the major flavonoid in grapefruit juice, naringin and its aglycone naringenin, significantly alter the clearance/elimination of CYP3A4-dependent drugs, due to inhibition of human CYP3A4 activity, it has also been reported that the importance of this interaction is dependent on individual patient susceptibility, the type and amount of grapefruit juice and other administration related factors.60 In spite of the fact that in vitro findings support the conclusion that naringenin and 6′,7′-dihydroxybergamottin may be the active ingredients, other studies indicated that these ingredients may contribute but are not the major active ingredients causing the grapefruit juice–drug interactions in human.59,60 These findings show the importance of in vivo validation of combination effects detected in in vitro model systems, since in vivo kinetics and bioavailability of active ingredients may be different than in the in vitro model.
Many diet-derived compounds have been shown to influence the biotransformation of one of the most potent known human dietary carcinogen AFB, and some efficiently protect against AFB-induced genotoxicity.61 These observations have been related to inhibition of the activity of CYP1A2 and CYP3A4 the major enzymes involved in the bioactivation of AFB to its genotoxic epoxide metabolite exo-AFB-8,9-epoxide.
Such interactions at the level of CYP1A2 and CYP3A4 mediated metabolism have been frequently encountered. Dietary flavonoids (i.e. flavone, galangin tangeretin) showed a potent inhibition of CYP1A2 in vitro.61 Daidzein, an isoflavone in soybean, inhibited CYP1A2 activity and modified the pharmacokinetics of CYP1A2-dependent drug elimination in healthy volunteers.62 As already indicated above it is widely recognized that the major flavonoid in grapefruit juice, naringin and its aglycone naringenin, significantly alter clearance of CYP3A4-dependent drugs, due to inhibition of human CYP3A4 activity. Naringenin was also found to be an effective inhibitor of AFB activation in a CYP3A4-dependent in vitro system.63 In line with this observation, grapefruit juice markedly reduced liver DNA damage in rats in vivo induced by AFB, and it was shown in the same study that hepatic CYP3A4 activity was significantly decreased after intake of grapefruit juice, whereas hepatic CYP1A and GST contents remained unchanged.64 These results suggest that grapefruit juice suppresses AFB-induced genotoxicity in rat liver through inhibition of the bioactivation of AFB and not through enhanced detoxification.
An example of a possible matrix effect that proceeds via direct interaction at the phase 2 enzymes can be found in the effects of the basil ingredient nevadensin on the sulfotransferase (SULT) mediated bioactivation of the allylalkoxybenzenes like estragole and methyleugenol, known to be also present in basil.65–68 The bioactivation of the allylalkoxybenzenes to their ultimate carcinogen requires the involvement of SULTs converting the 1′-hydroxymetabolite formed by CYPs to a 1′-sulfooxy metabolite that is the ultimate electrophilic and carcinogenic metabolite that can covalently bind to DNA (Fig. 2).69–71
 | ||
| Fig. 2 Bioactivation pathway of estragole indicating the inhibition by nevadensin at the level of SULT mediated conversion to the ultimate carcinogenic metabolite 1′-sulfooxyestragole. | ||
Jeurissen et al. (2008) demonstrated that the level of DNA binding of the proximate carcinogenic metabolite 1′-hydroxyestragole to DNA in vitro but also to DNA in intact HepG2 human hepatoma cells could be inhibited by a methanolic basil extract.72 The flavonoid nevadensin was identified as the major compound responsible for this observed in vitro inhibition of estragole bioactivation and subsequent DNA-adduct formation by the methanolic basil extract.65 A similar food-matrix derived inhibition of SULT mediated bioactivation was identified for the allylalkoxybenzene safrole present in herbs like mace by malabaricone C, also present in mace.73 In subsequent in vivo studies in which rats were simultaneously dosed with estragole and nevadensin or with safrole and malabaricone C containing mace extract it was shown that the SULT inhibitors also significantly inhibited DNA-adduct formation in vivo.66,73 These examples are especially of interest because these studies also reported the dose dependent behaviour of the interactions using physiologically based kinetic (PBK) modeling. Based on insight in the mode of action of the SULT inhibition, proceeding for nevadensin by reversible non-competitive type inhibition with a Ki value of 4 nM, the interaction could be incorporated in the Michaelis–Menten equations of the PBK models developed to predict the levels of formation of the reactive 1′-sulfooxy metabolite and subsequent DNA adduct formation in the liver.66 Upon incorporating this reversible mode of SULT inhibition by nevadensin or malabaricone C into the PBK model, the dose dependency of this matrix derived combination effect could be studied.66,73Fig. 3 presents an overview of the outcomes that can be obtained by such an analysis. This reveals that the matrix-derived combination effect will be significant at dose levels used in rodent bioassays, but that the effect is predicted to be only limited or even absent at realistic human exposure levels. This appeared due to the fact that nevadensin is a reversible non-competitive inhibitor and that upon realistic low dose exposure the concentrations reached in the liver will be lower than the Ki of 4 nM for SULT inhibition by nevadensin, thus not resulting in effective inhibition. Only upon high dose levels used in animal bioassays this Ki of 4 nM can easily be reached and significant inhibition can be detected. This result implies that when real food preparations would be tested in rodent bioassays the results obtained may not be representative for the human situation with low dose exposure and not necessarily provide a better starting point for risk assessment than testing the compound of concern in isolation. For the current example the results even indicate that the experiments with the pure compound may provide a better starting point for the risk assessment of low dose exposure than testing of basil itself, since at low dose levels the matrix-derived combination effect will be absent. This indicates that the incorporation of a matrix-derived combination effect in risk assessment should be done on a case-by-case basis taking into account mode of action-based analysis of the dose dependency of the interactions detected. Mode of action-based PBK models were shown to provide a useful tool to perform such analyses.
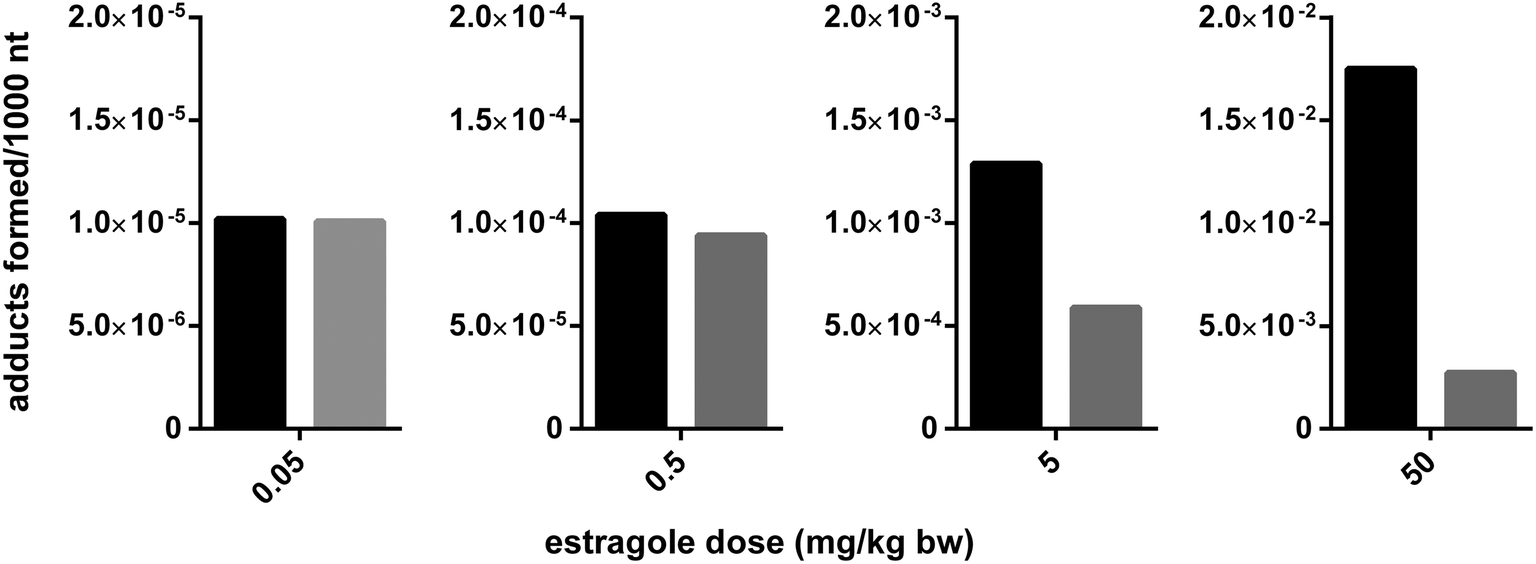 | ||
Fig. 3 PBK-model based prediction of the dose dependent food matrix-derived effect of the basil ingredient nevadensin on the DNA adduct formation in rats by the genotoxic carcinogen estragole also present in basil. Black bars represent the predicted DNA adduct formation in absence of nevadensin and the grey bars the level of DNA adduct formation in presence of nevadensin. The ratio of nevadensin to estragole was kept constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7, which reflects a possible ratio in basil, and the dose of both compounds was increased as indicated. The PBK model used was the model described by Alhusainy et al. (2013).66 :1.7, which reflects a possible ratio in basil, and the dose of both compounds was increased as indicated. The PBK model used was the model described by Alhusainy et al. (2013).66 | ||
An example of botanical ingredients that can interact with the pharmacokinetics of prescription drugs via induction of expression of metabolic enzymes are compounds present in St. John's wort. St. John's wort can induce CYP3A4, and this increased CYP3A4 activity can result in a decrease of plasma levels of several prescription drugs including alprazolam, irinotecan and indinavir.74–77 In addition, St. John's wort may interfere with the efficacy of oral contraceptives.78,79
Other dietary compounds of different origin (e.g., constituents of brassica vegetables and hops) have been shown to modify expression of human hepatic enzymes involved in the oxidation of AFB.61
Other examples include effects on CYP or glutathione S-transferase (GST) expression levels (Table 3). These examples illustrate that in addition to direct effects on biotransformation enzymes, metabolism of toxic compounds can also be affected upon combined exposure via enzyme induction.
Interaction at the level of DNA repair enzymes or genotoxicity reflects another type of interaction at the level of metabolism. Sanyal et al. (1997) investigated the effects of five food-borne possible antimutagens (cinnamaldehyde, tannic acid, vanillin, coumarin and caffeine) on spontaneous and heterocyclic amine (HCA)-induced micronucleus (MN) frequencies in human derived Hep-G2 cells. For all these compounds it has been claimed that they may act as antimutagens via interactions with DNA repair enzymes.80 In combination experiments with the HCA 2-amino-3-methylimidazo-[3,4-f]quinoline (IQ), post-treatment of the cells with the tested antimutagens resulted in a pronounced (75–90%) reduction in IQ induced MN formation. The largest effects were seen with vanillin, coumarin and caffeine which were active at concentrations <5 μg ml−1 concentrations which may be relevant for daily human exposure. Further experiments indicated that these compounds also attenuated the mutagenic effects of other HCAs including PhIP, MeIQ (2-amino-3,4-dimethylimidazo[4,5-f]quinoline), and MeIQx (2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline).80
Altogether the examples presented reveal that interactions at the level of metabolism may occur via interactions with both phase 1 and phase 2 enzyme activities, influencing detoxification and bioactivation of a toxic compound via either enzyme inhibition or influences on the level of enzyme expression. In addition, interactions have been reported at the level of DNA repair enzymes. Direct interactions with biotransformation enzymes often occur via reversible binding which implies that the effects will be dose-dependent and may be observed only at relative high dose levels, often applied in animal bioassays, but may no longer be relevant at low doses that better represent daily human intake. The examples also illustrate that in vivo validation of matrix-derived interactions detected in in vitro models is essential since due to limited bioavailability of active ingredients effects observed in vitro may not be relevant in vivo. This clearly illustrates that matrix-derived combination effects should be evaluated on a case-by-case basis taking mode of action, toxicokinetics as well as dose dependency of the relevant interactions into account. This also implies that although matrix-based combination effects may exist, their detection in animal bioassays at high dose levels may not improve risk assessment practice because interactions observed at the high dose levels in animal bioassays may not be maintained at lower, more realistic levels of intake. PBK modeling proved to be an adequate way to study these aspects.
Excretion
Combined exposure may not only affect the level of formation of the metabolite responsible for induction of adverse effects but may also alter formation of metabolites that can be excreted. Table 4 presents examples of matrix-derived combination effects on excretion of food-borne toxic compounds and their possible mode of action.| Matrix derived combination effect | Possible mode of action |
|---|---|
| Nevadensin may increase the excretion of the 1′-hydroxymetabolites of allylalkoxybenzenes.87–91 | The relative decrease in the formation of 1′-sulfoxymetabolites occurring in the presence of a botanical matrix containing SULT inhibitors (see section on metabolism), may result in a relative increase in the formation of other 1′-hydroxy metabolites such as 1′-hydroxyglucuronide and 1′-oxo metabolites. Such a shift in metabolism towards detoxification could subsequently lead to an increase in the biliary and urinary excretion of these metabolites.87–91 |
| In rats EGCG was found to be more rapidly eliminated following the intravenous or intragastric administration of pure EGCG compared to that following the concomitant exposure with decaffeinated green tea.33 In line with these findings, Johnson et al. (1999) reported that the mortality pattern in an oral rat study indicated that a green tea extract was more toxic than would be predicted based on its EGCG content alone.92 | Mode of action has not been identified. |
| When nickel was mixed into scrambled eggs or taken simultaneously with eggs a 10-fold lower amount of the administrated dose (2.5%) was excreted in comparison with the amount (25.8%) excreted in urine when eggs were taken 4 h prior to nickel containing drinking water.19 | Mode of action has not been identified. |
Often the modes of action underlying these modified excretion characteristics have not been identified. They could be a reflection of interactions at the level of absorption or metabolism but they may also reflect interaction at the level of ABC transport proteins that are involved in excretion toward bile and urine.
Discussion and conclusions
In the present review we collected examples available in literature of studies that demonstrate an influence on ADME characteristics of food-borne toxic compounds by other compounds or constituents present in the food. The results obtained indicate that interactions have been documented to occur at all ADME levels. However, what also became evident from this overview is that the actual mode of action underlying the reported effects on ADME characteristics has often not been elucidated. Mode of actions that were identified were mainly related to effects on transport proteins or on biotransformation enzymes either through direct inhibition or through effects on gene expression that affect enzyme activities. Other modes of action included effects on tissue distribution or biotransformation activities via dietary fat affecting tissue lipid composition, effects on bio-accessibility from the food matrix and/or effects on DNA repair enzymes. From the examples available it became also clear that the interactions may decrease but also increase the bioavailability and/or toxicity of the compounds of interest. The overview also illustrates that matrix-derived combination effects should be considered on a case-by-case basis, taking into account not only the mode of action of the interactions, but especially also their dose dependency. Matrix and combination effects detected at dose levels used in animal bioassays may turn out to be absent at dose levels representing realistic human intake. This can especially be the case for reversible interactions between inhibitors and biotransformation enzymes, transporter proteins or DNA repair enzymes. Given that these reversible interactions will be dependent on whether in vivo concentrations of the matrix-derived inhibitors will reach the relevant Ki values for the inhibition, it can be foreseen that inhibition will be detected at high dose levels used in animal experiments but may be less relevant at dose levels representing realistic human intake. This may also hold for the nuclear receptor mediated gene induction by compounds influencing the expression levels of biotransformation enzymes. This implies that although matrix-based combination effects may exist and can be demonstrated in in vivo experimental studies, their detection at high dose levels may not improve risk assessment practice because interactions observed at these high dose levels may not be maintained at lower, more realistic levels of intake. Insight in the mode of action underlying the interaction combined with PBK modelling may prove a way to obtain better insight in whether matrix-based interactions detected at high dose levels will still be relevant at more realistic lower intake levels, and thus to what extent these effects should be taken into account in the risk assessment for human exposure. The outcome of such a study may be that experimental data on the pure compound may even better predict what will happen at low dose levels than experimental data on the food of interest.From the examples presented it becomes clear that when a matrix effect is advocated to support the safety of a botanical or a botanical ingredient, data need to be provided that support the occurrence of the matrix effect in vivo at relevant levels of intake. This may best be achieved through mode of action based PBK modelling enabling extrapolation of matrix effects detected at high dose levels to low dose levels that better reflect estimated human daily intake.
Acknowledgements
BT acknowledges financial support from the SOIT foundation (the Foundation for Stimulation Of Innovation in Toxicology).References
- B. Schilter, C. Andersson, R. Anton, A. Constable, J. Kleiner, J. O'Brien, A. G. Renwick, O. Korver, F. Smit and R. Walker, Food Chem. Toxicol., 2003, 41, 1625–1649 CrossRef.
- L. M. S. Chan, S. Lowes and B. H. Hirst, Eur. J. Pharm. Sci., 2004, 21, 25–51 CrossRef CAS PubMed.
- J. Taipalensuu, H. Tornblom, G. Lindberg, C. Einarsson, F. Sjoqvist, H. Melhus, P. Garberg, B. Sjostrom, B. Lundgren and P. Artursson, J. Pharmacol. Exp. Ther., 2001, 299, 164–170 CAS.
- M. Takano, R. Yumoto and T. Murakami, Pharmacol. Ther., 2006, 109, 137–161 CrossRef CAS PubMed.
- C. Zimmermann, H. Gutmann, P. Hruz, J. P. Gutzwiller, C. Beglinger and J. Drewe, Drug Metab. Dispos., 2005, 33, 219–224 CrossRef CAS PubMed.
- C. G. Dietrich, A. Geier and R. P. J. Oude Elferink, Gut, 2003, 52, 1788–1795 CrossRef CAS PubMed.
- W. Brand, M. E. Schutte, G. Williamson, J. J. van Zanden, N. H. P. Cnubben, J. P. Groten, P. J. van Bladeren and I. M. C. M. Rietjens, Biomed. Pharmacother., 2006, 60, 508–519 CrossRef CAS PubMed.
- T. Sergent, S. Garsou, A. Schaut, S. De Saeger, L. Pussemier, C. Van Peteghem, Y. Larondelle and Y. J. Schneider, Toxicol. Lett., 2005, 159, 60–70 CrossRef PubMed.
- M. E. Schutte, J. J. M. van de Sandt, G. M. Alink, J. P. Groten and I. M. C. M. Rietjens, Cancer Lett., 2006, 231, 36–42 CrossRef CAS PubMed.
- M. E. Schutte, A. P. Freidig, J. J. M. van de Sandt, G. M. Alink, I. M. C. M. Rietjens and J. P. Groten, Toxicol. Appl. Pharmacol., 2006, 217, 204–215 CrossRef CAS PubMed.
- J. Hong, J. D. Lambert, S. H. Lee, P. J. Sinko and C. S. Yang, Biochem. Biophys. Res. Commun., 2003, 310, 222–227 CrossRef CAS PubMed.
- S. Zhang, X. Yang and M. E. Morris, Pharm. Res., 2004, 21, 1263–1273 CrossRef CAS.
- V. Breinholt, D. Arbogast, P. Loveland, C. Pereira, R. Dashwood, J. Hendricks and G. Bailey, Toxicol. Appl. Pharmacol., 1999, 158, 141–151 CrossRef CAS PubMed.
- V. Breinholt, J. Hendricks, C. Pereira, D. Arbogast and G. Bailey, Cancer Res., 1995, 55, 57–62 CAS.
- V. Breinholt, M. Schimerlik, R. Dashwood and G. Bailey, Chem. Res. Toxicol., 1995, 8, 506–514 CrossRef CAS.
- P. A. Egner, A. Munoz and T. W. Kensler, Mutat. Res., 2003, 523–524, 209–216 CrossRef CAS.
- C. Keshava, R. L. Divi, T. L. Einem, D. L. Richardson, S. L. Leonard, N. Keshava, M. C. Poirier and A. Weston, Environ. Mol. Mutagen., 2009, 50, 134–144 CrossRef CAS PubMed.
- D. J. Castro, C. V. Lohr, K. A. Fischer, K. M. Waters, B. J. M. Webb-Robertson, R. H. Dashwood, G. S. Bailey and D. E. Williams, Carcinogenesis, 2009, 30, 315–320 CrossRef CAS PubMed.
- G. D. Nielsen, U. Soderberg, P. J. Jorgensen, D. M. Templeton, S. N. Rasmussen, K. E. Andersen and P. Grandjean, Toxicol. Appl. Pharmacol., 1999, 154, 67–75 CrossRef CAS PubMed.
- V. Berger, A. F. Gabriel, T. Sergent, A. Trouet, Y. Larondelle and Y. J. Schneider, Toxicol. Lett., 2003, 140–141, 465–476 CrossRef CAS.
- H. Bothe, C. Gotz, N. Stobbe-Maicherski, E. Fritsche, J. Abel and T. Haarmann-Stemmann, Arch. Biochem. Biophys., 2010, 498, 111–118 CrossRef CAS PubMed.
- J. Jodoin, M. Demeule and R. Beliveau, Biochim. Biophys. Acta, 2002, 1542, 149–159 CrossRef CAS.
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- A. Adam, V. Crespy, M. A. Levrat-Verny, F. Leenhardt, M. Leuillet, C. Demigne and C. Remesy, J. Nutr., 2002, 132, 1962–1968 CAS.
- M. J. Rein, M. Renouf, C. Cruz-Hernandez, L. Actis-Goretta, S. K. Thakkar and M. da Silva Pinto, Br. J. Clin. Pharmacol., 2013, 75, 588–602 CAS.
- K. H. van Het Hof, C. E. West, J. A. Weststrate and J. G. A. J. Hautvast, J. Nutr., 2000, 130, 503–506 CAS.
- K. R. Walsh, Y. C. Zhang, Y. Vodovotz, S. J. Schwartz and M. L. Failla, J. Agric. Food Chem., 2003, 51, 4603–4609 CrossRef CAS PubMed.
- C. Laurent, C. Feidt, N. Grova, D. Mpassi, E. Lichtfouse, F. Laurent and G. Rychen, Chemosphere, 2002, 48, 843–848 CrossRef CAS.
- A. Ramesh, S. A. Walker, D. B. Hood, M. D. Guillen, K. Schneider and E. H. Weyand, Int. J. Toxicol., 2004, 23, 301–333 CrossRef CAS PubMed.
- I. K. O'Neill, A. C. Povey, S. Bingham and E. Cardis, Carcinogenesis, 1990, 11, 609–616 CrossRef PubMed.
- I. K. O'Neill, S. Bingham, A. C. Povey, I. Brouet and J. C. Bereziat, Carcinogenesis, 1990, 11, 599–607 CrossRef PubMed.
- J. M. Laher, G. A. Chernenko and J. A. Barrowman, Can. J. Physiol. Pharmacol., 1983, 61, 1368–1373 CrossRef CAS.
- L. Chen, M. J. Lee, H. Li and C. S. Yang, Drug Metab. Dispos., 1997, 25, 1045–1050 CAS.
- N. P. Seeram, L. S. Adams, S. M. Henning, Y. Niu, Y. Zhang, M. G. Nair and D. Heber, J. Nutr. Biochem., 2005, 16, 360–367 CrossRef CAS PubMed.
- T. Lavecchia, G. Rea, A. Antonacci and M. T. Giardi, Crit. Rev. Food Sci. Nutr., 2013, 53, 198–213 CrossRef CAS PubMed.
- K. Abraham, M. Pfister, F. Wohrlin and A. Lampen, Mol. Nutr. Food Res., 2011, 55, 644–653 CAS.
- R. A. Isbrucker, J. A. Edwards, E. Wolz, A. Davidovich and J. Bausch, Food Chem. Toxicol., 2006, 44, 636–650 CrossRef CAS PubMed.
- D. N. Sarma, M. L. Barrett, M. L. Chavez, P. Gardiner, R. Ko, G. B. Mahady, R. J. Marles, L. S. Pellicore, G. I. Giancaspro and T. Low Dog, Drug Saf., 2008, 31, 469–484 CrossRef PubMed.
- H. H. S. Chow, I. A. Hakim, D. R. Vining, J. A. Crowell, J. Ranger-Moore, W. M. Chew, C. A. Celaya, S. R. Rodney, Y. Hara and D. S. Alberts, Clin. Cancer Res., 2005, 11, 4627–4633 CrossRef CAS PubMed.
- M. Friedman and R. Rasooly, Toxins, 2013, 5, 743–775 CrossRef CAS PubMed.
- R. Rasooly, P. M. Do and M. Friedman, J. Agric. Food Chem., 2010, 58, 5421–5426 CrossRef CAS PubMed.
- M. H. Zia, E. E. Codling, K. G. Scheckel and R. L. Chaney, Environ. Pollut., 2011, 159, 2320–2327 CrossRef CAS PubMed.
- M. J. Heard, A. C. Chamberlain and J. C. Sherlock, Sci. Total Environ., 1983, 30, 245–253 CrossRef CAS.
- H. M. James, M. E. Hilburn and J. A. Blair, Hum. Toxicol., 1985, 4, 401–407 CrossRef CAS PubMed.
- O. Ouédraogo and M. Amyot, Environ. Res., 2011, 111, 1064–1069 CrossRef PubMed.
- F. Madrid, M. Biasioli and F. Ajmone-Marsan, Arch. Environ. Contam. Toxicol., 2008, 55, 21–32 CrossRef CAS PubMed.
- F. Madrid, E. Diaz-Barrientos and L. Madrid, Environ. Pollut., 2008, 156, 605–610 CrossRef CAS PubMed.
- N. M. Reeuwijk, W. N. M. Klerx, M. Kooijman, L. A. P. Hoogenboom, I. M. C. M. Rietjens and M. J. Martena, Food Addit. Contam., Part A, 2013, 30, 1535–1545 CrossRef CAS PubMed.
- J. Wittsiepe, B. Erlenkamper, P. Welge, A. Hack and M. Wilhelm, Chemosphere, 2007, 67, S355–S364 CrossRef CAS PubMed.
- L. Vasiluk, L. J. Pinto, Z. A. Walji, W. S. Tsang, F. A. P. C. Gobas, C. Eickhoff and M. M. Moore, Environ. Toxicol. Chem., 2007, 26, 387–393 CrossRef CAS.
- A. A. A. Al-Subeihi, B. Spenkelink, A. Punt, M. G. Boersma, P. J. van Bladeren and I. M. C. M. Rietjens, Toxicol. Appl. Pharmacol., 2012, 260, 271–284 CrossRef CAS PubMed.
- S. A. Walker, A. B. Addai, M. Mathis and A. Ramesh, J. Nutr. Biochem., 2007, 18, 236–249 CrossRef CAS PubMed.
- J. D. Gower and E. D. Wills, Chem. Biol. Interact., 1987, 63, 63–74 CrossRef CAS.
- D. L. Busbee, J. O. Norman and R. L. Ziprin, Arch. Toxicol., 1990, 64, 285–290 CrossRef CAS.
- Z. Y. Cheng, X. Tian, J. Gao, H. M. Li, L. J. Jia and H. L. Qiao, PLoS One, 2014, 9, e87234 Search PubMed.
- W. V. De Castro, S. Mertens-Talcott, A. Rubner, V. Butterweck and H. Derendorf, J. Agric. Food Chem., 2006, 54, 249–255 CrossRef CAS PubMed.
- P. C. Ho, D. J. Saville and S. Wanwimolruk, J. Pharm. Pharm. Sci., 2001, 4, 217–227 CAS.
- M. S. Arayne, N. Sultana and Z. Bibi, Pak. J. Pharm. Sci., 2005, 18, 45–57 CAS.
- D. G. Bailey, J. Malcolm, O. Arnold and J. D. Spence, Br. J. Clin. Pharmacol., 1998, 46, 101–110 CrossRef CAS.
- D. G. Bailey, J. H. Kreeft, C. Munoz, D. J. Freeman and J. R. Bend, Clin. Pharmacol. Ther., 1998, 64, 248–256 CrossRef CAS.
- K. Gross-Steinmeyer and D. L. Eaton, Toxicology, 2012, 299, 69–79 CrossRef CAS PubMed.
- W. X. Peng, H. D. Li and H. H. Zhou, Eur. J. Clin. Pharmacol., 2003, 59, 237–241 CrossRef CAS PubMed.
- F. P. Guengerich and D. H. Kim, Carcinogenesis, 1990, 11, 2275–2279 CrossRef CAS PubMed.
- M. Miyata, H. Takano, L. Q. Guo, K. Nagata and Y. Yamazoe, Carcinogenesis, 2004, 25, 203–209 CrossRef CAS PubMed.
- W. Alhusainy, A. Paini, A. Punt, J. Louisse, A. Spenkelink, J. Vervoort, T. Delatour, G. Scholz, B. Schilter, T. Adams, P. J. van Bladeren and I. M. C. M. Rietjens, Toxicol. Appl. Pharmacol., 2010, 245, 179–190 CrossRef CAS PubMed.
- W. Alhusainy, A. Paini, J. H. J. van den Berg, A. Punt, G. Scholz, B. Schilter, P. J. van Bladeren, S. Taylor, T. B. Adams and I. M. C. M. Rietjens, Mol. Nutr. Food Res., 2013, 57, 1969–1978 CAS.
- A. A. A. Al-Subeihi, W. Alhusainy, A. Paini, A. Punt, J. Vervoort, P. J. van Bladeren and I. M. C. M. Rietjens, Food Chem. Toxicol., 2013, 59, 564–571 CrossRef CAS PubMed.
- S. J. P. L. van den Berg, V. Klaus, W. Alhusainy and I. M. C. M. Rietjens, Food Chem. Toxicol., 2013, 62, 32–40 CrossRef CAS PubMed.
- E. W. Boberg, E. C. Miller, J. A. Miller, A. Poland and A. Liem, Cancer Res., 1983, 43, 5163–5173 CAS.
- K. Randerath, R. E. Haglund, D. H. Phillips and M. V. Reddy, Carcinogenesis, 1984, 5, 1613–1622 CrossRef CAS PubMed.
- R. W. Wiseman, E. C. Miller, J. A. Miller and A. Liem, Cancer Res., 1987, 47, 2275–2283 CAS.
- S. M. F. Jeurissen, A. Punt, T. Delatour and I. M. C. M. Rietjens, Food Chem. Toxicol., 2008, 46, 2296–2302 CrossRef CAS PubMed.
- E. Martati, R. Boonpawa, J. H. J. van den Berg, A. Paini, A. Spenkelink, A. Punt, J. Vervoort, P. J. van Bladeren and I. M. C. M. Rietjens, Food Chem. Toxicol., 2014, 66, 373–384 CrossRef CAS PubMed.
- B. J. Gurley, S. F. Gardner, M. A. Hubbard, D. K. Williams, W. B. Gentry, Y. Cui and C. Y. W. Ang, Drugs Aging, 2005, 22, 525–539 CAS.
- J. S. Markowitz, J. L. Donovan, C. L. DeVane, R. M. Taylor, Y. Ruan, J. S. Wang and K. D. Chavin, JAMA, 2003, 290, 1500–1504 CrossRef CAS PubMed.
- R. H. J. Mathijssen, J. Verweij, P. de Bruijn, W. J. Loos and A. Sparreboom, J. Natl. Cancer Inst., 2002, 94, 1247–1249 CrossRef CAS PubMed.
- S. C. Piscitelli, A. H. Burstein, D. Chaitt, R. M. Alfaro and J. Falloon, Lancet, 2000, 355, 547–548 CrossRef CAS.
- S. D. Hall, Z. Wang, S. M. Huang, M. A. Hamman, N. Vasavada, A. Q. Adigun, J. K. Hilligoss, M. Miller and J. C. Gorski, Clin. Pharmacol. Ther., 2003, 74, 525–535 CrossRef CAS PubMed.
- P. A. Murphy, S. E. Kern, F. Z. Stanczyk and C. L. Westhoff, Contraception, 2005, 71, 402–408 CrossRef CAS PubMed.
- R. Sanyal, F. Darroudi, W. Parzefall, M. Nagao and S. Knasmuller, Mutagenesis, 1997, 12, 297–303 CrossRef CAS.
- T. Shimada and F. P. Guengerich, Chem. Res. Toxicol., 2006, 19, 288–294 CrossRef CAS PubMed.
- S. Peterson, J. W. Lampe, T. K. Bammler, K. Gross-Steinmeyer and D. L. Eaton, Food Chem. Toxicol., 2006, 44, 1474–1484 CrossRef CAS PubMed.
- E. L. Jamin, A. Riu, T. Douki, L. Debrauwer, J. P. Cravedi, D. Zalko and M. Audebert, PLoS One, 2013, 8, e58591 CAS.
- K. Gross-Steinmeyer, P. L. Stapleton, J. H. Tracy, T. K. Bammler, S. C. Strom, D. R. Buhler and D. L. Eaton, Toxicol. Sci., 2009, 112, 303–310 CrossRef CAS PubMed.
- N. Sharma, P. Trikha, M. Athar and S. Raisuddin, Mutat. Res., 2001, 480–481, 179–188 CrossRef CAS.
- D. L. Diggs, J. N. Myers, L. D. Banks, M. S. Niaz, D. B. Hood, L. J. Roberts 2nd and A. Ramesh, J. Nutr. Biochem., 2013, 24, 2051–2063 CrossRef CAS PubMed.
- A. Anthony, J. Caldwell, A. J. Hutt and R. L. Smith, Food Chem. Toxicol., 1987, 25, 799–806 CrossRef CAS.
- T. R. Fennell, J. A. Miller and E. C. Miller, Cancer Res., 1984, 44, 3231–3240 CAS.
- A. Punt, A. P. Freidig, T. Delatour, G. Scholz, M. G. Boersma, B. Schilter, P. J. van Bladeren and I. M. C. M. Rietjens, Toxicol. Appl. Pharmacol., 2008, 231, 248–259 CrossRef CAS PubMed.
- A. Punt, A. Paini, M. G. Boersma, A. P. Freidig, T. Delatour, G. Scholz, B. Schilter, P. J. van Bladeren and I. M. C. M. Rietjens, Toxicol. Sci., 2009, 110, 255–269 CrossRef CAS PubMed.
- E. Solheim and R. R. Scheline, Xenobiotica, 1973, 3, 493–510 CrossRef CAS PubMed.
- W. D. Johnson, R. L. Morrissey, J. A. Crowell and D. L. McCormick, Toxicologist, 1999, 48, 57–58 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |