
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced electron mobility in crystalline thionated naphthalene diimides†
Lisa M.
Kozycz
a,
Chang
Guo
b,
Joseph G.
Manion
a,
Andrew J.
Tilley
a,
Alan J.
Lough
a,
Yuning
Li
*b and
Dwight S.
Seferos
*a
aLash Miller Chemical Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dseferos@chem.utoronto.ca
bDepartment of Chemical Engineering and Waterloo Institute for Nanotechnology (WIN), University of Waterloo, 200 University Avenue West, Waterloo, Ontario N2L 3G1, Canada. E-mail: yuning.li@uwaterloo.ca
First published on 1st October 2015
Abstract
A series of five thionated naphthalene diimides (NDIs) with linear alkyl chains was synthesized and the optoelectronic, self-assembly, and device properties were studied. When tested in organic thin-film transistors, the electron mobilities of the thionated derivatives are three orders of magnitude higher than the non-thionated parent analogue, with the highest mobility measured for cis-S2 (μmax = 7.5 × 10−2 cm2 V−1 s−1). In contrast to branched chain PDIs and NDIs, the electron mobility does not increase appreciably with degree of thionation, and the average mobilities are quite consistent ranging from 3.9 × 10−2 to 7.4 × 10−2 cm2 V−1 s−1 for one to three sulfurs.
Introduction
While there have been significant advances in the design of π-conjugated p-type materials for organic electronics over the last 20 years, research on complementary n-type materials has gained interest only recently.1–5 High-mobility π-conjugated n-type materials are needed for use as acceptor materials in organic photovoltaics (OPVs) and in n-channel organic thin-film transistors (OTFTs).6–12 As with OPVs, there are many more examples of p-type (i.e. hole transporting) OTFTs than n-type (i.e. electron transporting).13–15 Design criteria for new π-conjugated n-type materials for transistor applications include (1) lowering the LUMO level for efficient electron injection and (2) invoking strong intermolecular interactions for efficient electron transport in the solid-state.16,17Many of the best-performing n-type materials for both OPVs and OTFTs contain an electron withdrawing carbonyl group in the form of an amide or imide functionality,4,18–20 such as diketopyrrolopyrrole (DPP),21,22 thieno[3,4-c]pyrrole-4,6-dione (TPD),23 perylene diimide (PDI),24 and naphthalene diimide (NDI).25,26 A far less studied electron deficient functional group is the thioimide, that is an imide group where the two carbonyl oxygens are replaced with sulfur. Previously, this functionality had been mainly studied in biological chemistry, however, recently there has been interest in thionating n-type materials for organic electronics.27–32 This so-called thionation typically results in a lowering of the LUMO energy and increase in electron affinity, satisfying the first design criterion for an ideal n-type OTFT material. Furthermore, thionation gives rise to the possibility of S–S contacts, which can increase intermolecular interactions, satisfying the second design criterion outlined above.
We recently showed that thionation leads to an increase in thin film transistor electron mobility by two orders of magnitude when the four diimide oxygens are replaced with four sulfurs in PDI small molecules.33 We were interested in expanding the scope of the thionation reaction to smaller NDIs, as well as replacing the branched 3-hexylundecyl side chain used in the PDI study with a linear alkyl chain so that the solid-state packing and self-assembly properties could be studied.
Herein, we describe the synthesis, characterization, self-assembly, and thin film transistor performance of a series of linear chain thionated NDIs (Fig. 1). We find that the electron mobilities of the thionated derivatives are three orders of magnitude higher than the non-thionated parent analogue, with the highest mobility measured for cis-S2 (μmax = 7.5 × 10−2 cm2 V−1 s−1). Unlike the series of branched chain PDIs and NDIs, we find that the electron mobility does not increase appreciably with degree of thionation, and the average mobilities are quite consistent ranging from 3.9 × 10−2 to 7.4 × 10−2 cm2 V−1 s−1 for one to three sulfurs. This work demonstrates that high performance can be achieved with derivatives having low degrees of thionation, which have higher yields, improved solubility, and require less intensive syntheses than the higher thionated analogues, in contrast to what was previously thought.
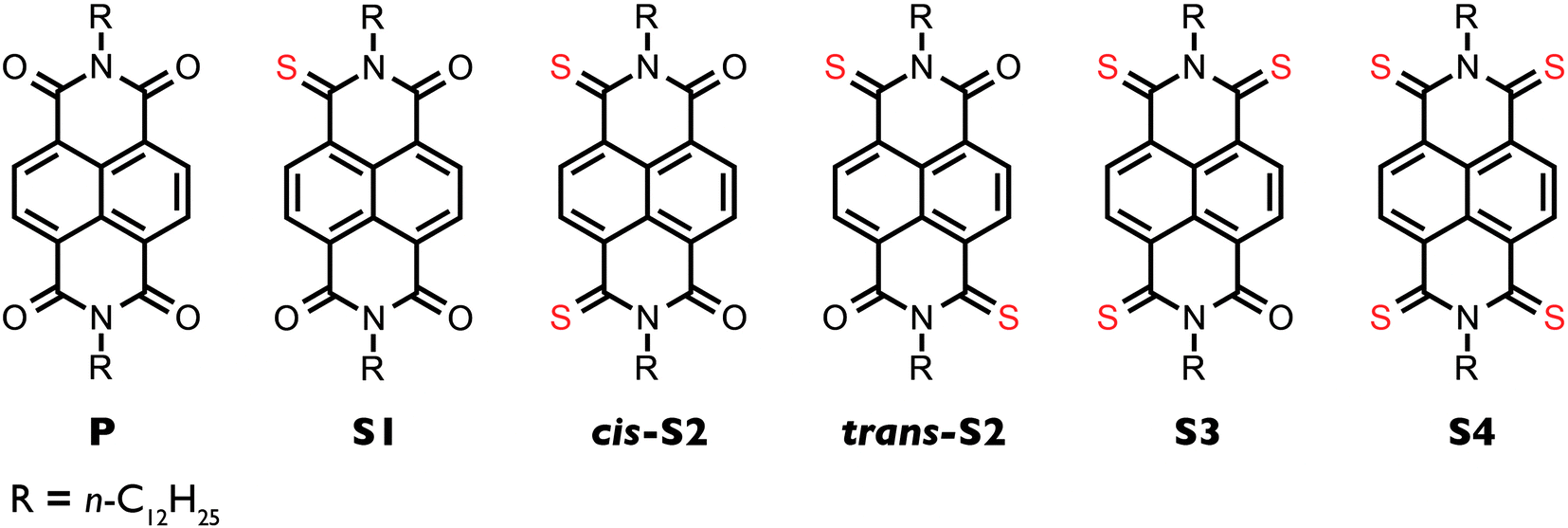 | ||
| Fig. 1 Chemical structures of the six N,N′-di(dodecyl)-4,5,8,9-naphthalene diimide derivatives. | ||
Results and discussion
Synthesis
The non-thionated parent NDI (P) was synthesized from naphthalene-4,5,8,9-tetracarboxylic dianhydride and n-dodecylamine according to literature procedure.34 The crude product was obtained in high yield and was used in the next step without further purification. Although characterization and device fabrication for P were done using the product that was not subject to chromatography, spectroscopic evidence (1H NMR; see ESI†) indicated that P was as pure as the other samples. Thionation was achieved by treating P with recrystallized Lawesson's reagent (LR) in refluxing toluene. The degree of thionation was controlled by varying the reaction time and stoichiometry. When less than one equivalent of LR was used and the reaction time was 17 h, the major product is S1 (each equivalent of LR is capable of delivering two sulfur atoms per reaction). When three equivalents of LR were used and the reaction time was 17 h, the major products are cis-S2 and trans-S2. When five equivalents of LR were used and the reaction time was 48 h, the major products are S3 and S4. Due to a decrease in solubility with increasing degree of thionation, the compounds must be separated by a series of chloroform columns on both silica gel and aluminum oxide. Similarly to the analogous thionated PDIs, both cis and trans isomers of the doubly thionated compound are obtained, as confirmed by gCOSY NMR (Fig. S6 and S9, ESI†). The geminate isomer is also present in very minor amounts but could not be isolated. It is important to note that in contrast to the study on 2-ethylhexyl NDIs, we were able to obtain the S4 derivative. The previous report suggests that the inability to synthesize the S4 compound may be due to the low LUMO energy of the S3 decreasing its reactivity towards Lawesson's reagent;32 however, our results suggest that further thionation is, in fact, possible. This difference in reactivity may be a steric effect, such that the branching at the 2-position in the ethylhexyl derivative may decrease the rate of the fourth sulfur substitution, which does not occur in the dodecyl derivative due to the absence of branching.Optoelectronic properties
The absorption spectra in both solution and solid-state red-shift with increasing degree of thionation, similarly to the thionated PDIs (Fig. 2).29 This red-shift is attributed to an increase in the HOMO energy and decrease in LUMO energy, as confirmed by cyclic voltammetry and density functional theory calculations (Table 1). The solution spectra are consistent with the 2-ethylhexyl analogues disclosed in a 2011 patent by Polyera,35 and in a recent publication by Zhang and co-workers.32 The low energy shoulder near the onset of absorption in S3 and S4 is likely due to the formally forbidden (n, π*) transition which is the S1 excited state for all six derivatives, as determined by TD DFT calculations (see ESI†).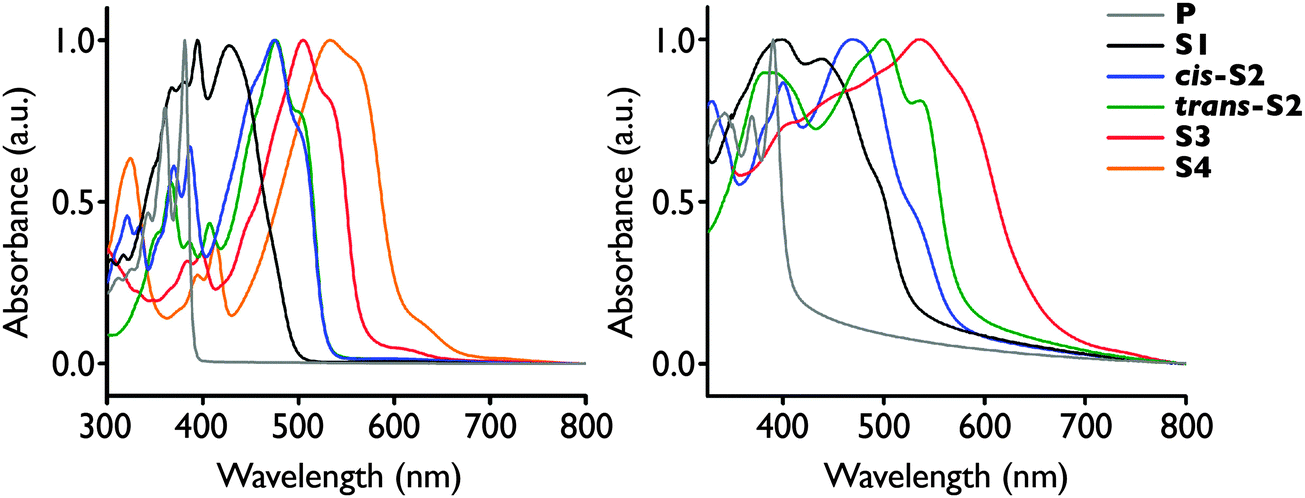 | ||
| Fig. 2 Optical absorption spectra of P-S4 in 10−5 M chloroform solution (left) and solid-state (right). Thin films were spin-coated from 5 mg mL−1 chloroform solution at 2000 rpm onto glass substrates. The compound is denoted by the line colour. | ||
| E 1/2 −1 (V) | E 1/2 −2 (V) | E HOMO , (eV) | E LUMO , (eV) | |
|---|---|---|---|---|
| a Calculated energies are shown in brackets next to the experimentally obtained values. Calculations were performed using DFT applying the B3LYP functional and 6-311G basis set. b E HOMO = ELUMO + Eopt,solng. c E LUMO = −e(Ered,onset + 4.8) where Ered,onset is the onset of reduction versus ferrocene obtained from cyclic voltammetry.36 d A second reduction peak was not observed. | ||||
| P | −1.11 | −1.53 | −6.97 (−7.42) | −3.79 (−3.87) |
| S1 | −0.92 | −1.30 | −6.38 (−6.71) | −3.95 (−4.09) |
| cis-S2 | −0.79 | −1.11 | −6.46 (−6.71) | −4.11 (−4.27) |
| trans-S2 | −0.80 | −6.50 (−6.69) | −4.15 (−4.25) | |
| S3 | −0.65 | −6.47 (−6.62) | −4.30 (−4.44) | |
In the solid-state, the spectra become much broader and the onset of absorption is red-shifted for P-S3 signifying a narrowing of the optical HOMO–LUMO gap consistent with more extensive exciton delocalization in the solid-state. Thin films of S4 could not be formed due to its low solubility in organic solvents and poor adhesion to the substrate. In contrast to solution, the solid-state spectra of the two S2 isomers have quite distinct shapes and energies, with the spectrum of the trans isomer red-shifted approximately 0.15 eV relative to the cis isomer. This is consistent with the stark difference in colour of the two isomers in powder form, where cis-S2 is a dark yellow-brown, while trans-S2 is bright red. Furthermore, the trans-S2 spectrum possesses more vibronic structure than that of cis-S2, suggesting it may be more ordered in the solid-state.
Cyclic voltammetry was performed in 0.1 M TBAPF6 in dry methylene chloride solution in an argon atmosphere to determine the redox properties of the compounds (Fig. S12, ESI†). P, S1, and cis-S2 exhibit typical reversible two-electron reductions, while only one reduction peak is observed for trans-S2 and S3, likely due to the low solubility of these compounds and film formation at the electrodes. The LUMO levels were estimated from the onset of the first reduction peak (Table 1). Oxidation peaks were not observed due to the oxidation of the electrolyte and/or solvent in the same potential region. HOMO energies were therefore calculated by subtracting the LUMO energy from the optical bandgap also measured in solution. It was found that the LUMO energies decrease with increasing degree of thionation, consistent with density functional theory calculations and analogous to the thionated PDIs. The HOMO energies of the thionated derivatives increase relative to the parent compound, however remain relatively constant over the range of sulfur incorporation. Electrochemistry was not performed on S4 due to poor solubility.
Thermal properties
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were performed on the compounds to determine their thermal transitions and thermal stability (Fig. S13 and S14, ESI,† respectively). For DSC, the compounds were sealed in aluminum hermetic pans and subject to two heating and cooling cycles between 20 and 200 °C at a heating/cooling rate of 10 °C min−1. The DSC curves of P-S3 are characteristic of a typical liquid crystalline material, with the two main heating transitions corresponding to the crystalline to liquid-crystalline and liquid-crystalline to isotropic liquid transitions for each compound. The transitions observed for the parent compound are consistent with previous reports in the literature,37 while the deviation from these transitions in the thionated compounds is likely due to their different strengths of intermolecular interactions and solid-state packing. A second heating transition is not observed for S4 below 200 °C, but likely does occur at higher temperatures (DSC was not performed above 200 °C to avoid thermal decomposition). It is also interesting that the only transitions observed for S3 and S4 are during the first heating cycles, after which no further transitions are observed. The experiment was repeated for S3 and S4 with a total of four heating and cooling cycles, and no significant transitions were observed during the additional two cycles. From TGA less than 1% of each compound degrades below 200 °C and therefore the absence of transitions after the first heating cycle is not due to decomposition in this case. A possible explanation is that these compounds become very thermodynamically stable after the initial heating cycle, however further analysis of this is beyond the scope of this investigation.The thermal stability of the compounds is relatively good, with 5% weight loss occurring between 260 and 360 °C. The lower thermal stability of the thionated compounds relative to the non-thionated parent is consistent with the analogous thionated PDIs, and is likely due to the weaker C–S thionyl bond relative to the carbonyl C–O.33 It should be noted that the thermal stability of the thionated NDIs (S4: Td = 297 °C) is much better than the thionated PDIs (S4: Td = 175 °C), while no branched chain S4 has been reported. Although thermal stability is somewhat decreased with thionation, the photostability of the compounds is quite good, in contrast to what was reported for thionated DPP and TPD small molecules. In that work, Leclerc and co-workers found that several of the thionated compounds decomposed over a few hours even when kept in the dark.28 In our work, we do not observe any evidence of decomposition after storing the compounds in ambient conditions over several months.
Solid-state properties
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). When this method was applied to the less soluble derivatives, the rate of crystallization increased, resulting in crystals too small to be suitable for X-ray analysis. Single crystals of trans-S2 were thus obtained by slow cooling a saturated o-dichlorobenzene solution from 120 °C to room temperature. S3 was crystallized similarly by slow cooling a saturated 1,2,4-trichlorobenzene solution. Attempts at growing single crystals of S1 and S4 suitable for X-ray analysis using numerous techniques were unsuccessful.
:1 v/v). When this method was applied to the less soluble derivatives, the rate of crystallization increased, resulting in crystals too small to be suitable for X-ray analysis. Single crystals of trans-S2 were thus obtained by slow cooling a saturated o-dichlorobenzene solution from 120 °C to room temperature. S3 was crystallized similarly by slow cooling a saturated 1,2,4-trichlorobenzene solution. Attempts at growing single crystals of S1 and S4 suitable for X-ray analysis using numerous techniques were unsuccessful.
All four compounds lie across crystallographic inversion centers and therefore there is only half of the molecule in the asymmetric unit and the other half is related by symmetry. Since cis-S2 and S3 do not have molecular inversion symmetry, the absolute positions of the sulfur and oxygen sites in the crystal lattice cannot be determined and the sites for O1,O2/S1,S2 are refined without any distance restraints. For the sake of crystallographic analysis, it is assumed that the sulfur and oxygen sites are disordered 50:50. Although this assumption makes the C–O and C–S bond distances somewhat unreliable, important information can still be determined regarding the packing of the molecules.
P and cis-S2 belong to the triclinic crystal system and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, while trans-S2 and S3 belong to the monoclinic crystal system and P21/n space group (Table S1, ESI†). Although the n-dodecyl side-chains adopt an all-trans conformation in all four crystal structures, the side-chains are almost completely interdigitated in the trans-S2 and S3 structures but show no overlap at all in either P or cis-S2 (Fig. 3). The side-chains in the trans-S2 derivative show the greatest degree of overlap and most compact structure, which is consistent with it having the shortest d-spacing of the series observed by PXRD (vide infra), as well as the highest density (Fig. S17 and Table S1, ESI†).
space group, while trans-S2 and S3 belong to the monoclinic crystal system and P21/n space group (Table S1, ESI†). Although the n-dodecyl side-chains adopt an all-trans conformation in all four crystal structures, the side-chains are almost completely interdigitated in the trans-S2 and S3 structures but show no overlap at all in either P or cis-S2 (Fig. 3). The side-chains in the trans-S2 derivative show the greatest degree of overlap and most compact structure, which is consistent with it having the shortest d-spacing of the series observed by PXRD (vide infra), as well as the highest density (Fig. S17 and Table S1, ESI†).
 | ||
| Fig. 3 Long-range crystal packing motif of (a) P, (b) cis-S2, (c) trans-S2, and (d) S3. Hydrogens have been omitted for clarity. | ||
All four derivatives crystallize in parallel planes, however, the NDI cores are not cofacial but are displaced along both the short (Dy) and long (Dx) axes of the molecule (Fig. 4). This type of slip-stacked structure is common for NDIs with both linear and cyclic alkyl chains.38,39 Furthermore, the slip-stacked motif has been used as an alternative strategy for developing high efficiency PDI-based acceptors for organic photovoltaics. It was shown that substitution at the PDI 2,5,8,11-positions (“headland positions”) leads to a slip-stacked packing structure which is known to prevent excimer formation that is prevalent in cofacial dimers.40
 | ||
| Fig. 4 Crystal structures of (a) P, (b) cis-S2, (c) trans-S2, and (d) S3 viewed from the top of the molecular plane (top) and along the long molecular axis (bottom). N-dodecyl chains and hydrogens have been omitted for clarity. Sulfur and oxygen sites are disordered 50:50 in cis-S2 and S3 so both atoms are shown. | ||
Both the slip-stacking and π–π stacking distances were extracted from the crystallographic data and show an increase in long axis slip and decrease in short axis slip of 1.18 Å and 0.61 Å, respectively, going from P to cis-S2 to S3 (Table S2, ESI†). The trans-S2 isomer seems to deviate from this trend, as it has the largest long axis slip and a very small short axis slip of just 1.18 Å. The π–π stacking distance for P, cis-S2 and trans-S2 is very similar, but decreases by about 0.3 Å for the S3 derivative. This result is somewhat surprising, as a more pronounced decrease in π–π stacking distance was expected with increasing degree of thionation. The major effect, however, seems to be on the slip-stacking distances, which suggests that thionation does have a significant effect on intermolecular packing. It is also significant that substitution of oxygen with a larger sulfur atom does not seem to perturb the NDI core planarity, which is important for retaining strong π–π interactions.
Perhaps the most interesting result of the single crystal analysis is the significant difference between the long range crystal packing of the two S2 derivatives. While the cis-S2 derivative crystallizes similarly to the non-thionated P compound, the trans-S2 isomer is more similar to triply thionated S3. This helps explain the observed differences in the solubility of the compounds, where P and cis-S2 have similar and significantly higher solubilities than trans-S2 and S3. When considering the intermolecular short contacts of the four derivatives, only trans-S2 and S3 have short contacts between atoms other than carbon and hydrogen that are less than the sum of the van der Waals radii. In the trans-S2 crystal packing structure, there are four sulfur–oxygen contacts per molecule each with a distance of 3.32 Å, while in the S3 packing motif there are four contacts per molecule with distances of 3.42 Å, which are unspecified sulfur–sulfur, sulfur–oxygen or oxygen–oxygen interactions. There are no sulfur–sulfur, sulfur–oxygen or oxygen–oxygen short contacts (i.e. intermolecular distances less than the sum of the van der Waals radii) in the P or cis-S2 long range packing structures. These contacts may be responsible for causing the large differences in crystal packing structure between P and cis-S2, and trans-S2 and S3, and well as the differences in solubility.
It is known that the crystallization method can have an effect on the resultant crystal structure and therefore it is possible that the observed differences between the derivatives is due to differences in crystallization method rather than degree of thionation. Unfortunately, we were unable to find any common method that resulted in X-ray quality single crystals for all of the derivatives, despite numerous attempts. It is likely, therefore, that the successful methods described above resulted in the most stable crystal structures in each case, and thus comparison between the crystal structures should be valid.
 | ||
| Fig. 5 AFM images of P-S3 thin films as-cast (top) and after annealing at 200 °C for 30 min under nitrogen (bottom). P, S1, and cis-S2 were spin-cast onto glass substrates from 8 mg mL−1 and trans-S2 and S3 from 5 mg mL−1 CHCl3 solutions, all at 50 °C. | ||
Self-assembly
Again due to the use of the linear side chains, we were able to study the self-assembly properties of the thionated compounds, as perylene diimides with linear side-chains have been shown to assemble into nanobelts or nanoribbons using solution-based techniques.41–43 Examples of micro/nanowires using bulkier groups such as perfluorophenyl44 or spirobifluorene45 have also been reported, however PDIs having branched alkyl chains can only assemble into zero-dimensional nanospheres.42 These types of one-dimensional structure form as a result of strong π–π interactions between the planar aromatic cores, which facilitate face-to-face packing and anisotropic growth. Nanowires of oligothiophene-functionalized naphthalene diimides have been used as acceptor materials in all-nanowire BHJ solar cells using P3HT nanowires as the donor.46 However, to the best of our knowledge, this is the first report of small molecule core-unfunctionalized NDI micro/nanowires.The 1D solution based self-assembly of P-S4 was performed using the so-called phase transfer method.42 Briefly, a larger amount (10:1 v/v) of methanol (poor solvent) was slowly transferred atop a chloroform (good solvent) solution (0.3 mM) of NDI in a test tube and left undisturbed overnight. Self-assembly occurs at the interface due to the slow mixing of the solvents and decreased solubility of the materials in the mixed solvent phase. Crystals of trans-S2, S3 and S4 begin to form instantaneously, while those of cis-S2 form within minutes, and those of P and S1 over several hours. This is likely due to the solubility of the various compounds, such that decreased solubility results in an increased rate of crystallization. The crystals were then removed via pipette and drop-cast onto glass substrates and the solvent was evaporated in air.
Optical micrographs reveal the formation of long fiber-like structures, which upon magnification appear to be one-dimensional microbelts (Fig. 6). Although all six compounds self-assemble into these 1D structures, their dimensions and uniformity seem to change with degree of thionation. There appear to be three main types of structures which can be classified as (1) microbelts, (2) crystalline microbelts and (3) dendrites. The crystalline microbelts are the widest structures and are the exclusive structure formed by the non-thionated P compound (widths 20–80 μm). Crystalline microbelts with slightly narrower, more monodisperse widths are formed by cis-S2 (15–55 μm), and even narrower ones are formed by S3 and S4 (∼20 μm). A small number of the crystalline microbelts are formed by S1 with widths of 50–80 nm. Interestingly, these wider structures are completely absent in trans-S2, which forms thinner microbelts exclusively (∼1.5 μm). The width of these microbelts is similar to those formed by cis-S2 and S4, and slightly thinner than those formed by S3 (∼7.5 μm). The width of both types of S3 and S4 structure can be increased by decreasing the concentration of the chloroform solution, however no significant change with concentration is observed for the other compounds. This suggests that the dimensions can be controlled by the rate of self-assembly, since decreasing the concentration of the initial solution effectively decreases the rate of crystallization. The dendritic structures are formed exclusively by S1 and appear as several microbelt “arms” growing from a single microbelt structure, all with similar widths. Although it is very difficult to visualize the entire length of an isolated structure, the average length appears to be several hundred micrometers for all three classes of structure, leading to aspect ratios (length/width) between 10:1 and 100:1. The self-assembled structures were investigated under higher magnification using AFM and SEM to confirm their belt-like structure (Fig. S15 and S16, ESI†). Both methods confirm that the average widths of the cis-S2 and trans-S2 microbelt structures are around 1.4 μm while AFM reveals their height is around 0.4 μm, leading to an aspect ratio of the belt cross-section (width/height) of 3.5. The S3 belts are wider (average width 7.3 μm) and taller (average height 1.3 μm) with an aspect ratio of 5.6. The S4 structures have the smallest widths (1.2 μm) and heights (0.4 μm). In some areas, the structures appear twisted with the edge lifted up, allowing further confirmation of the belt-like morphology. The larger crystalline structures were too large to be imaged by AFM, however SEM of the P compound reveals large microbelt structures.
 | ||
| Fig. 6 Optical micrographs of self-assembled P-S4 structures at 5× magnification. | ||
To investigate the crystallinity of these structures, powder X-ray diffraction (PXRD) was conducted on the self-assembled materials. Samples were prepared by gently scraping material off the glass substrates used for optical microscopy and onto a SiO2 wafer for PXRD analysis. A strong peak was observed around 3–4° corresponding to a side-chain interdigitation distance of 23 to 35 Å for the various compounds (Fig. S17, ESI†). It should be noted that the values obtained for the P and cis-S2 compounds using powder and single crystal X-ray diffraction differ slightly, despite the crystals being formed by similar methods. This discrepancy is likely due to differences in sample preparation for the two methods, or to differences in instrument sensitivity. The samples for PXRD were drop-cast onto a glass substrate and the solvent evaporated in air. The sample was then scrapped off the substrate and the powder redeposited onto a SiO2 wafer. In contrast, the crystals were individually removed from suspension and mounted for single crystal X-ray analysis. Additional unidentified peaks are present in the cis-S2, trans-S2, and S3 patterns, suggesting that they may be more highly crystalline than the singly thionated and non-thionated compounds. Unfortunately no π–π stacking peak was observed for any of the compounds, however the distances are expected to be similar to those measured for the single crystals.
Transistor performance
To evaluate the charge transport properties of the NDIs, bottom gate, bottom contact OTFTs were fabricated on n++ Si/SiO2 substrates with patterned gold source and drain electrodes under nitrogen. NDI thin films were spin coated from 10 mg mL−1 chloroform solutions at a spin speed of 5000 rpm for 60 s. Devices were annealed at 30, 100 or 150 °C for 20 min. No performance was observed for the parent compound upon annealing at 100 or 150 °C, in contrast to what has been previously reported for the compound.37 This discrepancy may be due to slight differences in device configuration and fabrication conditions, or in compound purity. The S3 device annealed at 150 °C also showed no performance, possibly due to the lower thermal stability of the higher thionated compounds (vide supra). Devices could not be prepared from trans-S2 or S4 due to their poor solubility and film-forming ability.Output and transfer curves show that all derivatives exhibit n-type behavior (Fig. 7 and Fig. S18, ESI†). Electron mobilities (μ) were calculated in the saturation regime at a drain-source voltage (VDS) of 60 V, and we found that the thionated derivatives have electron mobilities and on–off ratios that are three orders of magnitude higher than the non-thionated parent compound (μavg = 3.0 × 10−5 cm2 V−1 s−1; Ion/Ioff = 103) (Table 2).
 | ||
| Fig. 7 Output (a and c) and transfer (b and d) characteristics for the champion OTFTs fabricated from P (a and b) annealed at 30 °C and cis-S2 (c and d) annealed at 100 °C on DDTS-treated SiO2 substrates. Output and transfer characteristics of S1 and S3 can be found in Fig. S18 in the ESI.† | ||
| T (°C) | μ e,avg (cm2 V−1 s−1) | μ e,max (cm2 V−1 s−1) | I on/Ioff | V T | |
|---|---|---|---|---|---|
| a No performance observed after annealing at 100 or 150 °C. b No performance observed after annealing at 150 °C. | |||||
| P | 30 | 3.0 × 10−5 | 3.5 × 10−5 | 103 | 44 |
| S1 | 30 | 6.5 × 10−2 | 7.2 × 10−2 | 106 | 31 |
| 100 | 4.4 × 10−2 | 5.6 × 10−2 | 106 | 29 | |
| 150 | 2.2 × 10−2 | 2.6 × 10−2 | 106 | 35 | |
| cis-S2 | 30 | 2.1 × 10−2 | 2.2 × 10−2 | 106 | 28 |
| 100 | 7.4 × 10−2 | 7.5 × 10−2 | 106 | 19 | |
| 150 | 1.9 × 10−2 | 2.0 × 10−2 | 106 | 27 | |
| S3 | 30 | 3.9 × 10−2 | 4.7 × 10−2 | 106 | 39 |
| 100 | 2.2 × 10−3 | 2.2 × 10−3 | 106 | 31 | |
The previous studies on the branched chain thionated PDIs and NDIs revealed increasing mobilities and on–off ratios with increasing degree of thionation. In contrast, we observe electron mobilities on the same order of magnitude (10−2 cm2 V−1 s−1) for all of the linear chain thionated derivatives, and a constant on–off ratio of 106, which is higher than the best values obtained for both the branched chain PDIs (Ion/off ∼ 104 for S3) and NDIs (Ion/off ∼ 105 for trans-S2 and S3). In the previous works, the increase in performance was largely attributed to the decrease in LUMO level with increasing degree of thionation, which should improve electron injection. In this work, however, we do not observe a similar correlation, as the performance remains relatively constant despite the decrease in LUMO energy. One possible explanation is that the linear side chain enables the most favorable packing for all of the thionated derivatives (which may differ amongst the series), while in the branched series the packing is more dependent on the degree of thionation such that the most favorable packing can only be achieved with the maximum number of sulfurs present. Alternatively, it may be that the decrease in LUMO energy from −3.8 eV for P to −4.0 eV or below for the thionated derivatives is sufficient to increase the mobility to the same value for all three thionated compounds. Regardless, this result is quite promising as it suggests that in the case of the linear chain thionated NDIs, only one sulfur substitution is required to reach the highest mobility of the series.
Finally, it should be noted that the best mobility of the devices using the lowest temperature annealing step was obtained for the S1 derivative. This result is desirable because although arylene diimide derivatives with varying degrees of thionation can be synthesized, as the degree of thionation is increased, the yield and solubility of these higher thionated derivatives is decreased. Furthermore, in order to synthesize these higher thionated derivatives, a large excess of thionating agent is required which lowers the atom economy and complicates the purification. The realization of maximum electron mobility in linear side chain S1 thus highlights a potential design strategy for the development of high mobility thionated arylene diimide derivatives that require only minimal sulfur incorporation to achieve maximum electron mobility.
Conclusions
A series of five thionated naphthalene diimide derivatives were synthesized in a single-step reaction from the parent NDI derivative and their optoelectronic, self-assembly, and device properties were studied. Thionation results in a decrease in LUMO level and increase in intermolecular interactions, likely due to strong S–S contacts. Thermogravimetric analysis reveals that all compounds are thermally stable up to 250 °C, and although thermal stability does decrease slightly with thionation, the compounds are significantly more stable than other thionated species that have recently been reported in the literature. Due to their linear side-chain, all compounds are crystalline, as confirmed by differential scanning calorimetry, single crystal and powder X-ray diffraction, and undergo solution based self-assembly into microbelt-like structures.Thin film transistor electron mobilities of the thionated derivatives (S1, cis-S2, and S3) were three orders of magnitude higher than the non-thionated derivative (P). The high electron mobilities (∼10−2 cm2 V−1 s−1) and on–off ratios (106) were relatively constant for thionated derivatives having one, two or three sulfurs, in contrast to previous reports of branched chain PDIs and NDIs, where the device performance increases with increasing degree of thionation. This result is promising as it suggests that the highest performance can be achieved with derivatives having low degrees of thionation when a linear alkyl chain is used, which have higher yields, improved solubility, and require less thionating reagent than the higher thionated derivatives. The difference in trends between the linear and branched series is likely due to the linear alkyl chain facilitating more favorable solid-state packing even at low sulfur incorporation, which is only possible for the higher thionated derivatives having branched alkyl side chains. These results motivate the continued development of thionated n-type small molecules for organic electronics applications. Furthermore, the results imply that the degree of thionation may not be the only factor to consider when designing these types of materials, and that several other factors including alkyl chain length and branching position may also contribute to improved device performance. Finally, other chemical modifications such as introduction of functional groups at the core positions may be used in combination with thionation to realize high performance materials.
Experimental
Materials
All reagents were used as received unless otherwise noted. 1,4,5,8-Naphthalenetetracarboxylic dianhydride, n-dodecylamine, and Lawesson's reagent were purchased from Sigma-Aldrich. Lawesson's reagent was recrystallized from boiling toluene (10 g per 100 mL) prior to use.Instrumentation and methods
NMR spectra of the compounds were recorded on a Varian Mercury 400 spectrometer unless otherwise specified. Absorption spectra were recorded on a Varian Cary 5000 UV-vis-NIR spectrophotometer. The thin-films used for absorbance spectroscopy were spin-coated from solutions in chloroform (5 mg mL−1) at 2000 rpm for 60 seconds onto glass substrates that had been washed with methanol. Electrochemistry was performed with a BASi Epsilon potentiostat. A standard three-electrode cell with a 2.0 mm diameter Pt button working electrode, a Ag/Ag+ reference electrode, and a Pt wire counter electrode was used, with a ferrocene internal standard and a 100 mV s−1 scan rate. Measurements were conducted in a nitrogen-filled glovebox in dry dichloromethane solution against ferrocene/ferrocenium (Fc/Fc+) in the presence of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) electrolyte. LUMO levels were estimated from cyclic voltammetry reduction onset potentials and converted to the vacuum scale using the equation ELUMO = −e(Ered + 4.8), referenced to the ferrocene/ferrocenium redox couple.36 HOMO levels were estimated by adding the ELUMO to the solution state optical bandgap. TGA was performed using an SDT Q600 instrument at a heating rate of 10 °C min−1 under nitrogen in an aluminum pan. DSC was performed on a DSC Q100 TA instrument at a heating rate of 10 °C min−1 under nitrogen in an aluminum hermetic pan.Microstructures were assembled through slow crystallization at the interface between chloroform (good solvent) and methanol (poor solvent). Briefly, methanol was slowly transferred atop a 0.3 mM chloroform solution of the NDI derivative (10:1 v/v methanol:chloroform) in a test tube. The mixture was left undisturbed until crystal formation was complete (solution became colorless). The crystals were transferred via pipette onto a glass slide for further analysis.
Density functional theory calculations
Geometry optimizations were performed using the Gaussian 09 program,47 employing the Becke Three Parameter Hybrid Functionals Lee–Yang–Parr (B3LYP) level of theory.48,49 The standard 6-311G basis set was used. From the optimized geometry the first twenty singlet transitions were calculated using time-dependent DFT.50Synthesis
Acknowledgements
This work was supported by NSERC, DuPont, and the A. P. Sloan Foundation. L.M.K. is grateful for an NSERC CGS D. A.J.T. would like to thank the Connaught Global Challenge Award for a postdoctoral fellowship. The authors thank Tyler Schon for assistance with electrochemistry, Ilya Gourevich for assistance with SEM, and Abdolkarim Danaei for assistance with PXRD.Notes and references
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- A. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2013, 2, 1201–1213 Search PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- X. Gao, C.-A. Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697–3699 CrossRef CAS PubMed.
- Y. Hu, X. Gao, C.-A. Di, X. Yang, F. Zhang, Y. Liu, H. Li and D. Zhu, Chem. Mater., 2011, 23, 1204–1215 CrossRef CAS.
- F. Zhang, Y. Hu, T. Schuettfort, C.-A. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed.
- M. Nakano, I. Osaka, D. Hashizume and K. Takimiya, Chem. Mater., 2015, 27, 6418–6425 CrossRef CAS.
- M. Mamada, H. Shima, Y. Yoneda, T. Shimano, N. Yamada, K. Kakita, T. Machida, Y. Tanaka, S. Aotsuka, D. Kumaki and S. Tokito, Chem. Mater., 2015, 27, 141–147 CrossRef CAS.
- M.-L. Yeh, S.-Y. Wang, J. F. Martínez Hardigree, V. Podzorov and H. E. Katz, J. Mater. Chem. C, 2015, 3, 3029–3037 RSC.
- J.-Y. Hu, M. Nakano, I. Osaka and K. Takimiya, J. Mater. Chem. C, 2015, 3, 4244–4249 RSC.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J.-L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Y. He, W. Hong and Y. Li, J. Mater. Chem. C, 2014, 2, 8651–8661 RSC.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- Y.-J. Hwang, B. A. E. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenekhe, Adv. Mater., 2015, 27, 4578–4584 CrossRef CAS PubMed.
- Y. Ie, S. Jinnai, M. Nitani and Y. Aso, J. Mater. Chem. C, 2013, 1, 5373–5380 RSC.
- S. Lévesque, D. Gendron, N. Bérubé, F. Grenier, M. Leclerc and M. Côté, J. Phys. Chem. C, 2014, 118, 3953–3959 Search PubMed.
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CAS.
- F. S. Etheridge, R. Fernando, J. A. Golen, A. L. Rheingold and G. Sauvé, RSC Adv., 2015, 5, 46534–46539 RSC.
- T.-F. Yang, S.-H. Huang, Y.-P. Chiu, B.-H. Chen, Y.-W. Shih, Y.-C. Chang, J.-Y. Yao, Y.-J. Lee and M.-Y. Kuo, Chem. Commun., 2015, 51, 13772–13775 RSC.
- W. Chen, J. Zhang, G. Long, Y. Liu and Q. Zhang, J. Mater. Chem. C, 2015, 3, 8219–8224 RSC.
- A. J. Tilley, C. Guo, M. B. Miltenburg, T. B. Schon, H. Yan, Y. Li and D. S. Seferos, Adv. Funct. Mater., 2015, 22, 3321–3329 CrossRef PubMed.
- Y. Miyake, T. Nagata, H. Tanaka, M. Yamazaki, M. Ohta, R. Kokawa and T. Ogawa, ACS Nano, 2012, 6, 3876–3887 CrossRef CAS PubMed.
- J. Quinn, W. Zheng, Z. Chen, H. Usta, C. Newman, H. Yan and A. Facchetti, US Pat., 062 365–2010, 2011 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- M. Ichikawa, Y. Yokota, H.-G. Jeon, G. de R. Banoukepa, N. Hirata and N. Oguma, Org. Electron., 2013, 14, 516–522 CrossRef CAS PubMed.
- D. Shukla, S. F. Nelson, D. C. Freeman, M. Rajeswaran, W. G. Ahearn, D. M. Meyer and J. T. Carey, Chem. Mater., 2008, 20, 7486–7491 CrossRef CAS.
- T. Kakinuma, H. Kojima, M. Ashizawa, H. Matsumoto and T. Mori, J. Mater. Chem. C, 2013, 1, 5395–5401 RSC.
- P. E. Hartnett, A. Timalsina, H. S. S. R. Matte, N. Zhou, X. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, R. Oitker, H. Chen, J. Zuo and L. Zang, J. Am. Chem. Soc., 2005, 127, 10496–10497 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847–2853 CrossRef CAS PubMed.
- J. Lambrecht, T. P. I. Saragi and J. Salbeck, J. Mater. Chem., 2011, 21, 18266–18270 RSC.
- J. Lambrecht, T. P. I. Saragi, K. Onken and J. Salbeck, ACS Appl. Mater. Interfaces, 2011, 3, 1809–1812 CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, J. Mater. Chem., 2012, 22, 24373–24379 RSC.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1421895–1421898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc02753e |
| This journal is © The Royal Society of Chemistry 2015 |