
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally activated LTA(Li)–Ag zeolites with water-responsive photoluminescence properties†
Eduardo
Coutino-Gonzalez
a,
Wouter
Baekelant
a,
Didier
Grandjean
b,
Maarten B. J.
Roeffaers
c,
Eduard
Fron
a,
Mohammad S.
Aghakhani
b,
Nicolas
Bovet
d,
Mark
Van der Auweraer
a,
Peter
Lievens
b,
Tom
Vosch
d,
Bert
Sels
*c and
Johan
Hofkens
*a
aDivision of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: johan.hofkens@chem.kuleuven.be
bLaboratory of Solid State Physics and Magnetism, Department of Physics and Astronomy, KU Leuven, Celestijnenlaan 200D, 3001 Leuven, Belgium
cCentre for Surface Chemistry and Catalysis, Department of Microbial and Molecular Systems, KU Leuven, Kasteelpark Arenberg 23, 3001 Leuven, Belgium. E-mail: bert.sels@biw.kuleuven.be
dNano-Science Center, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
First published on 21st October 2015
Abstract
Silver–zeolite composites are interesting materials with unique optical properties such as high external quantum efficiencies and large Stokes shifts. The selective formation of luminescent silver clusters within zeolite scaffolds can be achieved by varying silver guest and zeolite host conditions. Nevertheless, at present, the controlled synthesis of Ag–zeolite composites with responsive optical properties remains a challenge. In this report, silver–zeolite composites displaying a dynamical emission color change with respect to their water content were synthesized using LTA zeolites containing lithium cations as counter-balancing agents. An intense blue emission was encountered in partially hydrated LTA(Li)–Ag composites, at low silver loadings, whereas a green/yellow emission was observed in their fully hydrated state. The materials synthesized in this report possess high external quantum efficiencies, up to 62%, compared to their close analogues having Na, K, and Ca as counter-balancing ions. Due to the remarkable dynamical change in emission color depending on the hydration level of LTA(Li)–Ag composites, the use of these materials as luminescence-based humidity sensors is suggested.
Introduction
Oligoatomic metal clusters with sub-nanometer sizes have become an important topic in the nanoscience field over the past years due to their unique catalytic and optical properties.1,2 However, at this level of structural organization the controlled synthesis and stabilization of clusters with well-defined sizes, shapes, and electronic properties becomes a challenge. To overcome this problem different synthesis approaches based on the self-assembly of the metal cluster precursors in confined environments have been proposed.3,4 The selective production of a particular metal cluster species can be achieved by varying the metal-guest and confinement structure-host properties. For instance, among biological scaffolds, the luminescence of silver clusters can be tuned over the whole visible range when encapsulated in DNA strands.5–10 The use of block copolymers as stabilizing agents for the fabrication of silver nanoclusters with tunable emission colors has been also demonstrated.11,12 Moreover, template-mediated strategies, in which glassy materials and metal organic frameworks are utilized to confine silver nanoclusters have also been developed.13–16Due to their meso- and microporosity, high cation exchange capacity, and high degree of crystallinity, zeolites have been proposed as suitable scaffolds for the synthesis of well-defined oligoatomic metal clusters.17–19 The formation of highly luminescent silver exchanged zeolites with emission colors spanning the whole visible range has been recently reported.20–23 These materials displayed a high chemical and photo-stability, however, a dependence of their luminescence properties with respect to the water content was observed. The exposure to high humidity environments led to a quenching of the luminescence of certain Ag–zeolite composites.23 Such process is fully reversible and a subsequent drying by mild heating showed the reestablishment of their original luminescent properties. A similar behavior in which the absorption properties of silver exchanged zeolites was strongly related to their hydration levels has been previously reported.24–26 Due to this peculiar phenomenon, the use of these materials as water vapor sensors was then suggested.26,27
Among the different counter-balancing cations that are mostly encountered in LTA zeolites, lithium possesses particular characteristics. The high affinity with which Li cations coordinate to oxygen28 and the distortions that these cations can induce in the LTA zeolite framework29 are of particular interest for the stabilization of metal guests within LTA zeolites. For instance, there are indications that Li cations in Li-exchanged LTA are much more resistant toward reduction.30 Therefore it has been suggested that Li cations may facilitate the stabilization of metal clusters within zeolite cavities, without strong direct involvement in chemical reactions with the metal guest.31 Some of these metal guests are quite unusual, such as the rubidide (Rb−) ion, in Rb-loaded LTA(Li) zeolites.32 Moreover, because of the small size of the lithium cation, it can occupy locations buried deep in the face of the sodalite cage of LTA zeolites, allowing the electric field generated to be nearly completely shielded by the surrounding oxygen atoms of the zeolite framework.33
Herein, we report the fabrication of highly luminescent silver clusters using LTA zeolites partially exchanged with Li cations as confinement scaffold, henceforth referred as LTA(Li). Following a ship-in-a-bottle approach, we partially exchanged the counter-balancing cations (Na, Li) with silver cations and after a subsequent heat-treatment the formation of highly luminescent composites was achieved by (partial) reduction of the Ag+ ions.20 A strong relationship between the color of the photoluminescence and the hydration level was observed in these phosphors, as e.g. in luminescent LTA(Na, K, Ca)–Ag and FAU(Na)–Ag zeolite composites. Nevertheless, the luminescence quenching, reported in previous studies,23 was replaced by a dynamical emission color change (from blue to yellow) with respect to the water content in LTA(Li)–Ag samples possessing low silver loadings. This dynamical change of the emission color in LTA(Li)–Ag zeolite composites depending on their hydration level could be potentially used for monitoring humidity levels at micro and macro scales. Moreover, these materials displayed higher external quantum efficiencies, up to 62%, compared to their analogues having Na, K, and Ca as counter-balancing cations.
Experimental
Sample preparation
The LTA zeolite (Union Carbide) partially exchanged with lithium cations was obtained by exchanging the parent zeolite (having sodium as counter-balancing cations) with a highly concentrated LiNO3 (10 M, Sigma Aldrich 99%) solution, following the procedure described by Yahiro and collaborators.34 LTA zeolite (1 gram) in its sodium form was suspended in the aqueous LiNO3 (500 mL) solution, the pH of the suspension was adjusted to 9 by adding lithium hydroxide solution (0.1 M, Sigma Aldrich 98%). The sample was agitated overnight using an end-over-end shaker, then filtered and washed several times with MilliQ water. This procedure was repeated 4 times to obtain a higher degree of cation exchange.The LTA(Li) was subsequently exchanged with Ag cations following the procedure fully described elsewhere.20 LTA(Li) zeolites (500 mg) were re-suspended in an aqueous silver nitrate solution (500 mL, 0.04 to 0.5 mM, Sigma-Aldrich, 99% purity), the suspension was stirred for 2 hours in the dark. The powder was recovered by filtration using a Büchner filter and washed several times with milliQ water, then the sample was calcined overnight at 450 °C (5 °C min−1) following 2 steps of 15 minutes each at 100 and 150 °C to prevent any damage in the zeolite structure during the calcination process. After heat treatment the sample was cooled under ambient conditions, allowing the sample to reach a fully hydrated state, and stored in the dark for further analysis. The structural formula for fully dehydrated LTA zeolites, after normalization of the total amount of T atoms (Si, Al) in the unit cell to 24, is (M12+)[Si12Al12O48] where M represents the counter-balancing ion. The amount of silver cations that can be exchanged in LTA zeolites ranges from 1 to 12 per normalized unit cell, we therefore prepared samples containing low, intermediate and high silver loadings to study the effect of the silver content on their luminescence properties. We denoted the samples as follows; LTA(Li)–Agx, where x represents the amount of silver atoms per normalized unit cell (1–12).
Steady state luminescent characterization
For the excitation–emission characterization, the heat-treated Ag containing LTA(Li) samples were placed in a quartz cuvette (1 mm path length) and sealed by a Teflon stopper. Emission and excitation spectra were recorded using an Edinburgh Instruments FLS 980 fluorimeter (corrected for wavelength dependence throughput and sensitivity of the detection channel). For every excitation wavelength, the emission was collected starting 30 nm above the excitation wavelength up to 800 nm using a 5 nm step size for the excitation wavelength. The signal above 410 nm was measured using a 400 nm long pass glass filter to avoid interference from second-order excitation peaks, the measured intensities were corrected for the transmittance of the long pass filter. The emission was collected in “front face mode” through the quartz cuvette and sent to a monochromator and PMT for detection. From the separate emission spectra at varying excitation wavelengths, the two-dimensional excitation–emission matrices were constructed, the raw data were corrected for background and noise and were interpolated to a resolution of 1 nm × 1 nm (two-dimensional excitation–emission plots of the non-interpolated data can be found in Fig. S8, ESI†). For the in situ measurements at different temperatures we designed and built a heating-cell (Fig. S4, ESI†), this device was coupled to the integrating sphere (Labsphere, 100 mm diameter) which was connected to the fluorimeter (Edinburgh Instruments FLS 980) through fiber optics. The samples were left at the measurement temperature for 1 hour for stabilization, and then cooled down to room temperature under vacuum (1 × 10−4 bar pressure) to measure their excitation–emission profiles.External quantum efficiency (EQE) measurements
The detailed procedure is described elsewhere.35 Briefly, external quantum efficiencies were measured using an integrating sphere (Labsphere, 100 nm diameter) coupled to a commercial spectrofluorimeter (Edinburgh Instruments FLS 980 fluorimeter). The device provides high reflectance in the UV-visible and near infrared region. The sphere accessories were made from Teflon (sample holders). Excitation was provided by a xenon lamp with a monochromator and directed to the sample inside the sphere via an optical fiber. The emission was collected through an optical fiber and sent to a photon multiplier tube (PMT) after passing a monochromator.Luminescent time-resolved characterization
Micro-second time-resolved luminescence measurements were performed with a home-built setup consisting of a Nd:YAG laser (Quanta-Ray INDI-40, Spectra Physics) equipped with an optical parametric oscillator OPO (Basiscan/120, GWU-Lasertechnik). The laser system is able to deliver 8 ns pulses at a repetition rate of 10 Hz and can be tuned over a spectral range (signal and idler) of 410–2500 nm. The OPO was pumped with 355 nm pulses, its signal tuned to 520 nm and focused on a 1 mm thick BBO crystal where the second harmonic (260 nm) was generated. The resulting laser pulse was filtered for the residual 520 nm light, collimated and focused on the sample by a 150 mm focal length lens and a small part of this light was sent to a fast photodiode to generate a trigger signal. A right angle configuration between excitation and light collection paths was used. A SpectroPro-300i monochromator/spectrograph was used to disperse the emitted light and select the desired wavelength. The luminescence signal was detected by a PMT (Hamamatsu, R928). The transient electrical signal was amplified and sent to a computer controlled oscilloscope. Homemade Labview based software was used to control, trigger the instruments, read, average, and store the transient data.The fluorescence decay times at the nanosecond time scale were determined by time-correlated single photon counting (TCSPC), using the setup that has been described in detail previously.20 Briefly, the frequency-tripled and doubled output (266 & 420 nm, 8.18 MHz, 2 ps FWHM) of a mode-locked Ti:sapphire laser (Tsunami, Spectra Physics) was used as excitation source. The linearly polarized excitation light was rotated to a vertical direction by the use of a Berek compensator (New Focus) in combination with a polarization filter and directed onto the sample. The sample consisting of LTA(Li)–Agx composites in powder form was placed in a quartz cuvette (1 mm path length) and sealed by a Teflon stopper, and then mounted on the device. The emission was collected under 90° with respect to the incident light and guided through a polarization filter that was set at the magic angle (54.7°) with respect to the polarization of the excitation beam. The fluorescence was spectrally resolved by a monochromator (Sciencetech 9030, 100 nm focal length, wavelength accuracy 0.3 nm), and detected by a microchannel plate photomultiplier tube (MCP-PMT, R3809U-51, Hamamatsu). A time-correlated single photon timing PC module (SPC 830, Becker & Hickl) was used to obtain the fluorescence decay histogram in 4096 channels. The decays were recorded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts in the peak channel, in time windows of 10 ns corresponding to 2.4 ps per channel and analyzed individually with a time-resolved fluorescence analysis (TRFA) software. The full width at half-maximum (FWHM) of the IRF was typically in the order of 42 ps. The quality of the fits was judged by the fit parameters χ2 (<1.2), Zχ2 (<3) and the Durbin–Watson parameter (1.8 < DW < 2.2) as well as by visual inspection of the residuals and autocorrelation function.36 The fluorescent decays were individually analyzed as multi-exponential decays, in terms of decay times (τi) and their associated pre-exponential factors (Ai).
000 counts in the peak channel, in time windows of 10 ns corresponding to 2.4 ps per channel and analyzed individually with a time-resolved fluorescence analysis (TRFA) software. The full width at half-maximum (FWHM) of the IRF was typically in the order of 42 ps. The quality of the fits was judged by the fit parameters χ2 (<1.2), Zχ2 (<3) and the Durbin–Watson parameter (1.8 < DW < 2.2) as well as by visual inspection of the residuals and autocorrelation function.36 The fluorescent decays were individually analyzed as multi-exponential decays, in terms of decay times (τi) and their associated pre-exponential factors (Ai).
X-ray absorption measurements
X-ray absorption data were collected on beamline DUBBLE (BM26A) at the ESRF (European Synchrotron Radiation Facility, Grenoble, France), operating under beam conditions of 6 GeV, 200 mA, (7/8 + 1) filling mode. The beamline is equipped with a Si(111) double-crystal monochromator and a vertically focusing Si mirror that suppresses higher harmonics. A 13 mm diameter pellet (prepared with a mechanical press, 1.5 tons) under ambient conditions (fully hydrated sample), a sealed quartz cuvette and a sealed glass capillary containing the partially hydrated samples, as well as a sealed glass capillary with the fully dehydrated sample were measured in transmission mode at the Ag K-edge (25514 eV). The ionization chambers were filled with Ar/He gas mixtures. The dataset was collected up to 13 Å−1 with acquisition times of about 40 minutes (5 to 25 s per point). Three spectra were averaged to improve the signal to noise ratio to an optimal level.Thermogravimetric analysis
Thermogravimetric analysis was performed on a Q500 TGA device (TA instruments). Approximately 20 mg of heat-treated LTA(Li)–Agx zeolite composites were loaded in a platinum sample holder. The sample was heated using a temperature rate of 5 °C min−1 to 650 °C under a constant nitrogen flow of 90 mL min−1.Elemental analysis
Elemental analysis was performed by X-ray photoelectron spectroscopy (XPS) using a Kratos Axis UltraDLD instrument, fitted with a monochromatic AlKα X-ray source (energy = 1486.6 eV, power = 150 W) and a charge neutralizer to avoid charging of the samples. XPS is a surface sensitive technique which gives the chemical composition of roughly the top 10 nm of the sample. The data were analyzed using the commercial software CasaXPS and its associated library of sensitivity factors, and the binding energy calibration was performed using the adventitious carbon C 1s set at 285 eV. Pass energies of 160 eV and 20 eV were used for wide scans and high resolution scans respectively. Peaks monitored were Na 1s at 1072.2 ± 0.1 eV, Si 2p at 102 ± 0.1 eV, Al 2p at 74.2 ± 0.1 eV, Li 1s at 55.8 ± 0.1 eV, Na 2s at 64.0 ± 0.1 eV and Ag 3d5/2 at 368.6 ± 0.1 eV. To calculate the initial Na/Li ratio, Na 2s/Li 1s peaks were used due to their close binding energies, whereas for the monitoring of Na atomic percentage the Na 1s peak was utilized, since this peak is more intense and presents less overlap with the Ag peaks.Diffuse reflectance spectroscopy and in situ Fourier transformed infrared (FT-IR) spectroscopy
An UV-Vis-NIR Lambda 950 Perkin Elmer spectrophotometer equipped with a 150 mm diameter integrating sphere (coated with Spectralon) was used to record the DRS spectra. The samples were placed in a Teflon sample holder and covered with a quartz plate for the measurements.Self-supporting zeolite wafers were prepared (∼10 mg, sample thickness about 50 μm) and loaded into the measurement cell of a NICOLET 6700 FT-IR device (400–4000 cm−1). The cell was kept under vacuum conditions (<1 × 10−3 bar) and the temperature was adjusted with a EUROTHERM temperature controller (1 °C accuracy) and monitored with a thermocouple. The spectra were recorded at each specific temperature using a thermal ramp of 5 °C min−1 up to 450 °C. The FT-IR spectra consist of an average of 128 measurements with a wave number range between 1500 and 4000 cm−1, and with a spectral resolution of 2 cm−1. The spectra were corrected for the background at each measurement temperature. The OMNIC software was used for controlling the device and data processing.
Results and discussion
Optical characterization of LTA(Li)–Ag zeolite composites
The photoluminescence characterization of heat-treated Ag–LTA zeolite composites and the effect of the counter-balancing cations (K, Na & Ca) on their optical properties have been described previously.20,35 In these Ag–LTA zeolites, containing K, Na, and Ca as counter-balancing cations, the formation of green and yellow (emission maxima between 530 and 570 nm, excitation range between 320 and 380 nm) emitting species was mainly observed at low and intermediate silver loadings (Ag1–Ag6) in most of the samples. Whereas the presence of red emitting species (emission maxima between 670 and 710 nm, excitation range between 420 and 450 nm) was mainly found in Ag–LTA zeolite composites with higher silver contents (Ag6–Ag12),20 independently of the counter-balancing cations. The nature of the counter-cations used (K, Na, and Ca) in the LTA zeolites did not display a remarkable effect on the external quantum efficiency (EQE) of the silver loaded zeolites (Ag1 to Ag12 per unit cell).35 When monovalent cations were employed (LTA-K and LTA-Na), EQE values between 3 and 16% were recorded, whereas in Ag–zeolite composites containing divalent cations (LTA-Ca), EQE's ranging from 1 to 10% were obtained. It has been proposed, based on X-ray analysis, electron spin resonance (ESR) experiments, theoretical studies, and high resolution transmission electron microscopy (HR-TEM) evidence,37–43 that partially reduced hexa-atomic (Ag6n+) clusters are preferentially formed in the sodalite cavities of LTA zeolites containing high silver loadings. Whereas for LTA zeolites with a low silver content, smaller clusters species (Ag3n+, Ag4m+) have been postulated as the species responsible for their characteristic optical properties.Here, we used LTA zeolites partially exchanged with lithium cations LTA(Li) as starting materials to investigate the effect of Li cations on the luminescence properties of oligoatomic silver clusters in LTA zeolites. The elemental analysis of the starting LTA(Li) zeolite material, as determined by X-ray photoelectron spectroscopy (XPS), showed a silicon to aluminum ratio (Si/Al) close to one and a lithium to sodium ratio (Li/Na) of about 0.5, indicating a 33% of Na replacement by Li cations.
In Fig. 1 the picture of the series of luminescent LTA(Li)–Ag composites, in their fully hydrated state, with different silver contents [LTA(Li)–Ag1–12] is displayed. The formation of green, yellow, and red emitting Ag–zeolite composites was observed depending on the silver content, which is in agreement with previous results.20 At low silver loadings, green emission is dominant for LTA(Li)–Ag1 and LTA(Li)–Ag2. Yellow emission was found in the samples LTA(Li)–Ag3 and LTA(Li)–Ag4, and finally red emission was observed in samples with higher silver contents, LTA(Li)–Ag5–12. A more detailed investigation of the excitation–emission profiles of the LTA(Li)–Ag samples was carried out by analyzing their two-dimensional excitation–emission plots (Fig. 2). The emission profiles of the luminescent LTA(Li)–Ag composites are similar to those found previously for Ag-exchanged (Na, K and Ca) LTA zeolites,20 however a consistent blue shift of about 60 nm in the excitation profiles of LTA(Li)–Ag composites with low and intermediate silver loadings, LTA(Li)–Ag1–6, was observed. A small but significant red shift in the maximum of the emission profiles from 570 to 600 nm was noticed with respect to the silver content in LTA(Li)–Ag1–4 samples when excited between 250–280 nm. The emission signals presented an elongated profile with a FWHM ∼ 100 nm. As a function of the excitation wavelength, a splitting of the elongated emission signal recorded at 600 nm, into two well defined spots at 250 and 280 nm was recorded in samples with intermediate silver loadings, LTA(Li)–Ag4–6. At lower silver loadings, this splitting, although still present is less pronounced. From LTA(Li)–Ag5 on an emission band with a maximum between 680 and 700 nm appeared upon excitation at 420 nm. Upon further increasing the silver loading, the intensity of this emission signal (680–700 nm) was increased, becoming the major band in LTA(Li)–Ag9. Simultaneously a decrease of the emission band between 570 and 600 nm was detected. Furthermore from LTA(Li)–Ag6 on a second short wavelength emission band with maximum about 500 nm was observed. This emission occurred by excitation at ∼380 nm and ∼470 nm.
 | ||
| Fig. 1 Picture of LTA(Li)–Agx samples, in their fully hydrated state, with different silver contents under 254 nm (from x = 1–6) and 366 nm illumination (from x = 7–12). | ||
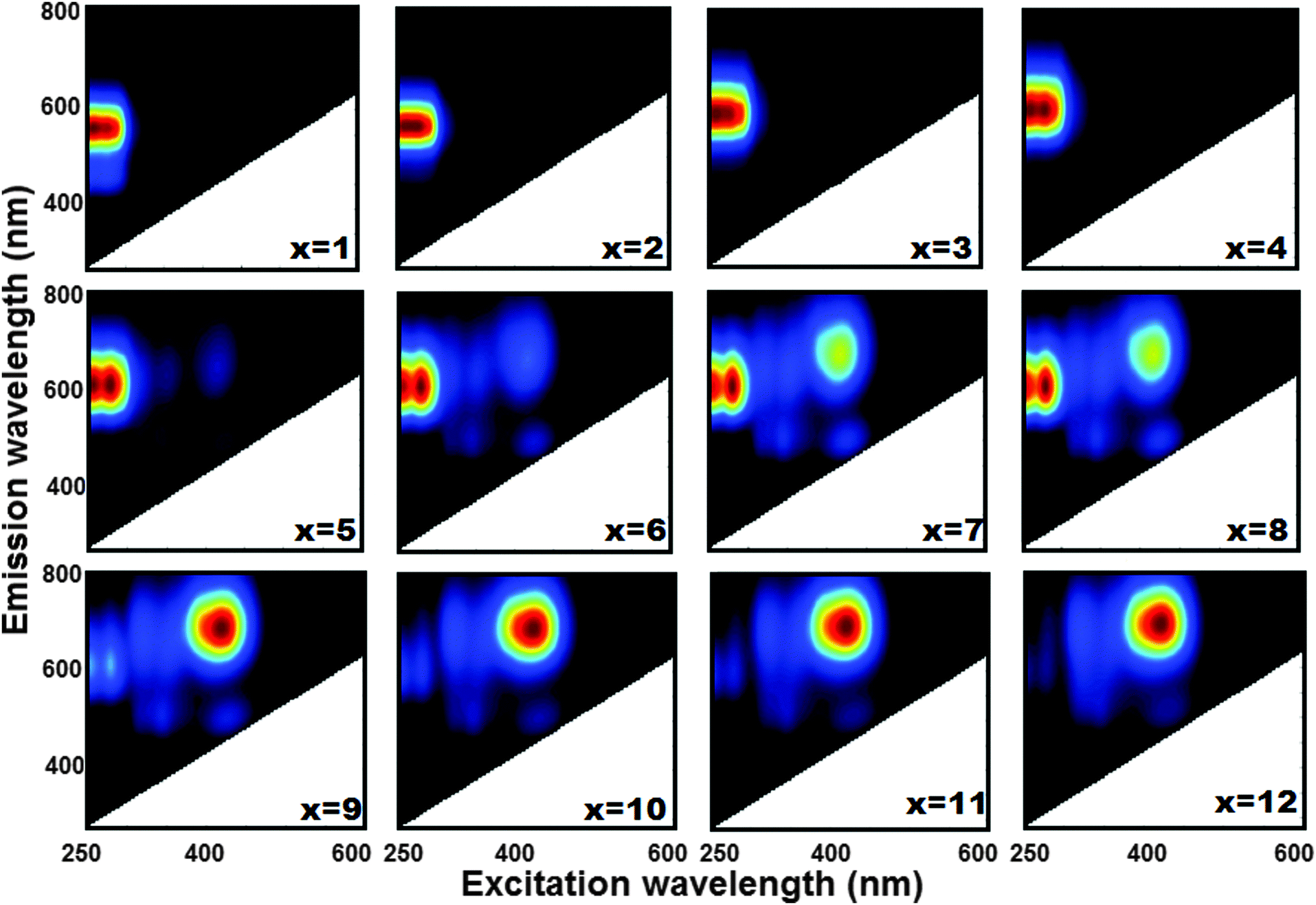 | ||
| Fig. 2 Two dimensional excitation–emission profiles of luminescent LTA(Li)–Agx samples in their fully hydrated state. | ||
We recorded diffuse reflectance spectra (DRS) of LTA(Li)–Ag composites with different silver contents (Fig. S12, ESI†) to examine the different absorbing species present in the samples. We observed a sharp absorption band in the UV region (230–280 nm) in LTA(Li)–Ag1; this band became broader (200–350 nm) and two new signals appeared at 380 and 480 nm in samples containing higher silver loadings [LTA(Li)–Ag4,7,12]. This is fully in agreement with a study reported elsewhere,25 in which the appearance of absorption bands in the UV and visible region, depending on the silver loading, in Ag-exchanged LTA zeolites (having Na cations as counter-balancing agents) was related to the presence of Ag atoms at specific sites in the zeolite framework. For LTA(Li)–Ag1–6 the range of the short wavelength absorption band corresponds with the major band in the excitation scans in Fig. 2, although the splitting of the band in two maxima at respectively 250 and 280 nm is not observed in the DRS spectra. This could be due to additional absorption bands of light-absorbing non-luminescent Ag species located in this region.25 At higher silver loadings (Ag7–Ag12), the bands which are observed under those conditions in the excitation scans (Fig. 2) in the region between 380 and 500 nm are also present in the DRS spectra. However, contrary to the photoluminescence spectra the 200–300 nm band remains the most important at all silver loadings.
Fig. 3 displays the EQE's of the LTA(Li)–Ag composites, which were determined using three different excitation wavelengths (260, 280, and 420 nm) based on the excitation–emission profiles of the examined samples (Fig. 2), as a function of the silver loading. For LTA(Li)–Ag1–8 samples, where excitation occurred at 260 and 280 nm, the highest EQE values (62 and 46% when excited at 280 and 260 nm, respectively) were recorded for LTA(Li)–Ag1. Under those conditions a systematic decrease of the EQE's was observed for the samples LTA(Li)–Ag2–8, upon increasing the silver content. This might be due to the presence of other non-emissive silver species in samples containing higher silver loadings, which although absorbing excitation light do not contribute to the emission and hence have a negative effect on the overall EQE, as suggested in a previous study.35
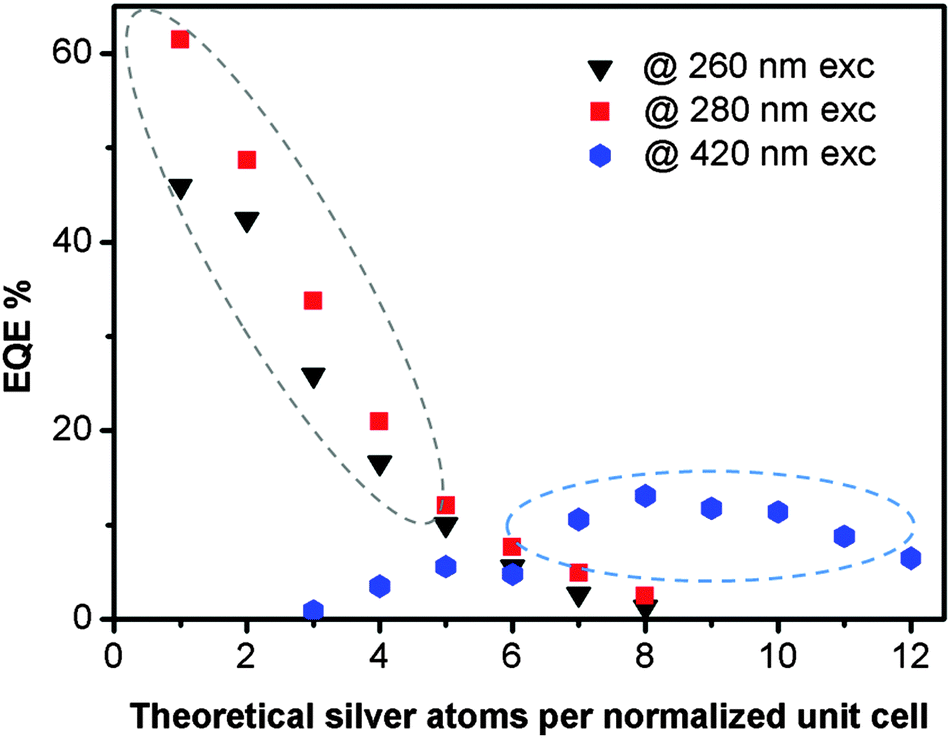 | ||
| Fig. 3 External quantum efficiencies measured for LTA(Li)–Agx samples, in their fully hydrated state, for different excitation wavelengths. In the region indicated by the dotted gray ellipse the presence of Li and Na cations in the zeolite framework was observed by XPS, whereas in the region marked with a blue dotted ellipse only Na cations were found. | ||
The EQE values of LTA(Li)–Ag zeolite composites with low and intermediate silver loadings are much higher than those measured under the same conditions on related LTA(Na)–Ag samples (Table S2, ESI†), showing the large effect of the Li cations on the emission properties of luminescent silver clusters in LTA(Li) zeolites. Among several studies it has been suggested that Li cations may facilitate the stabilization of metal clusters within zeolite cavities, without strong direct involvement in chemical reactions with the metal guest.30–33 This has been mainly attributed to the great affinity with which Li cations coordinate to oxygen from the zeolite framework and from the interstitial water.28
In samples having high silver contents (Ag7–Ag12) upon excitation at 420 nm, EQE values up to 13% were measured for the emission detected between 600 and 800 nm; such values are similar to those found for the analogous LTA(Na)–Ag7–12 composites reported elsewhere.35 The different dependence of the EQE's of the emission observed upon short and long wavelength excitation on the silver loadings corresponds with the different behavior of the DRS and excitation spectra when the silver loading is increased. From the elemental analysis performed by XPS (Fig. S3, ESI†), we noticed that at low and intermediate silver loadings (Ag1–Ag4) the presence of Li cations is still significant. Whereas for samples having more than 6 Ag cations per normalized unit cell only Na cations remained present. This is fully in agreement with the selectivity series for the exchange of univalent ions (Ag ⋙ Cs > Rb > K > Na > Li) in LTA zeolites.44
The steady-state absorption and emission spectra clearly indicate the presence of different emitting species. To obtain a better insight into their spectroscopic properties, picosecond time-correlated single photon counting (TC-SPC) measurements were performed on the luminescent LTA(Li)–Ag samples. We used two different excitation (266 and 420 nm) and four detection wavelengths (390, 470, 580 and 685 nm) based on excitation–emission profiles (Fig. 2) of the different composites. Hydrated samples with low-to-intermediate silver loadings (Ag1 to Ag6) with emission signals ranging from 500 to 600 nm (green and yellow emitting species) show three decay components of 60 ps, 800 ps and 3.5 ns, respectively (Table S3 and Fig. S13, ESI†) upon 266 nm excitation. The amplitude of the component with the longest decay time was always larger than 84%. For the samples with higher silver loadings (Ag7 to Ag12) where mainly emission in the 600–800 nm region is observed, two main decay components of 90 ps (2 to 11% amplitude) and 5.2 ns (89 to 95% amplitude) were retrieved for the emission at 685 nm upon 420 nm excitation. These values are similar to those found upon analyzing the luminescence decays of silver nanoclusters confined in zeolites,20,22 DNA,6 and polymer scaffolds.45 For LTA(Li)–Agx samples with low silver loadings blue emission (emission maxima at 390 and 470 nm, see Fig. S9, ESI†) was observed upon 260 nm excitation in case of partial hydration. The fluorescence decay was investigated for the series of LTA(Li)–Ag1–4 samples and analyzed as a sum of three exponentials (Table S4, ESI†). At 390 nm detection the samples decay with 2.2 ns (91 to 75% amplitude), 433–580 ps (7 to 20% amplitude) and a component faster than the time resolution of the setup (<42 ps, 1–5% amplitude). Comparable decay components were also retrieved at 470 nm detection. Here, the longest component found in the range 2.4–3.2 ns is most likely a combination of the longest decays found for blue (2.2 ns) and yellow species (4.1 ns). In addition to the decay components obtained by the TCSPC technique, two long living components with decay times of 24 and 270 μs were recorded at low silver content, LTA(Li)–Ag1 sample, for the emission at 580 nm upon excitation at 256 nm (Fig. S15, ESI†). These long decay times suggest that, either a part of the emitting species has a spin-multiplicity different from the ground state or that an equilibrium between an emitting species having the same spin-multiplicity as the ground state and a longer-lived non-emitting excited species occurs.
For the fully hydrated LTA(Li)–Ag composites the water content, as estimated by thermogravimetric analysis (TGA), amounted to 19% (Fig. S7, ESI†). Remarkably, a strong dependence of the optical properties of LTA(Li)–Ag samples, with respect to their water content was observed mainly at low silver loadings (Ag1 to Ag4). While an intense blue emission (Fig. 4) was encountered in partially hydrated samples (up to 2% water content), a green/yellow emission was found in fully hydrated LTA(Li)–Ag composites. We selected LTA(Li)–Ag1 as a model sample for further investigation of this phenomenon, due to the most pronounced color change and higher EQE's observed for this sample (Fig. 4). LTA(Li)–Ag1 displayed EQE's of 21 and 62% for the blue and green/yellow emitters, at respectively partial and full hydration.
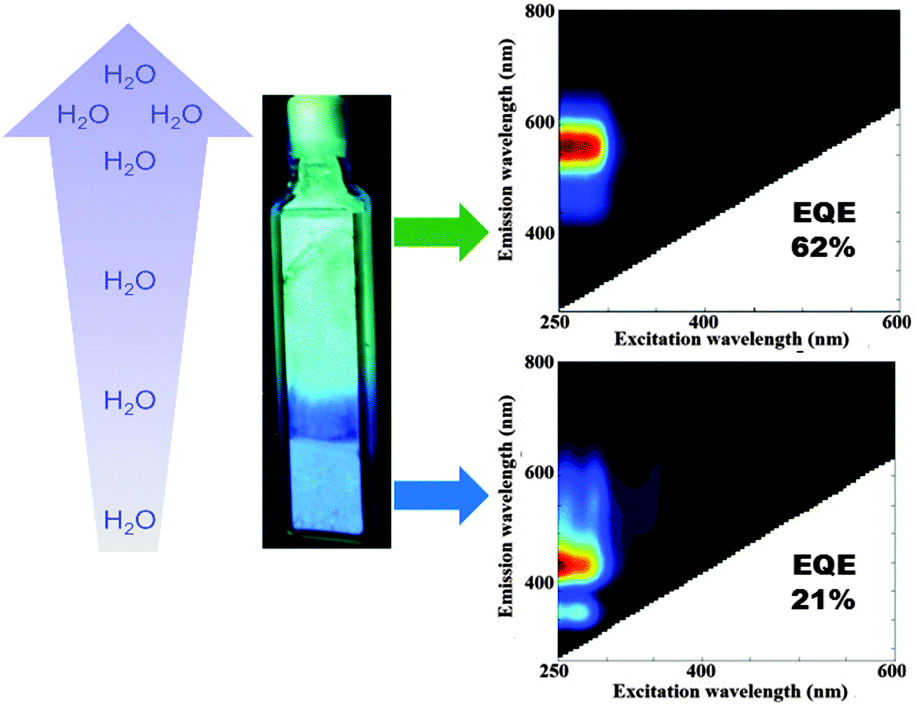 | ||
| Fig. 4 Water dependence of the emission color observed in LTA(Li)–Ag1 composites. In fully hydrated samples (19% water content) a green emission was found, whereas in partially hydrated samples (2% water content) blue emission was recorded. The water gradient observed in the cuvette was generated by leaving the material exposed to air under ambient conditions. | ||
Emission color to water content dependence on LTA(Li)–Ag samples containing low silver loadings
We used a home-made heating cell (Fig. S4, ESI†) coupled to a commercial fluorimeter to investigate, in a systematic way, the changes in the optical properties of LTA(Li)–Ag1 composites with respect to the water content. The luminescent fully hydrated LTA(Li)–Ag1 sample (in pellet form) was mounted on the heating cell that was tightly sealed using a quartz window. Two-dimensional excitation and emission profiles were recorded on the sample after heat-treatment at different temperatures: room temperature, 50, 75, 100, 200, 300, and 400 °C. The sample was left at the desired temperature for 1 hour for stabilization, then cooled down to room temperature under vacuum conditions (1 × 10−4 bar) before proceeding with the excitation–emission measurements.In Fig. 5 a compilation of excitation–emission profiles of LTA(Li)–Ag1 at different temperatures is shown. The correlation between the different temperatures used and the water content in LTA(Li)–Ag1 as determined by TGA analysis was investigated and is presented in Fig. S7 (ESI†). At room temperature (about 200 water molecules per normalized unit cell) we observed the presence of a broad emission band centered at about 590 nm (yellow emission) when the sample was excited at 260 and 280 nm. This emission band was slightly blue shifted by about 30 nm when the sample was heat-treated at 50 °C (about 180 water molecules per normalized unit cell) giving rise to a green emission at about 560 nm. This green emission was also observed in the samples heat-treated at 75 and 100 °C (about 120 and 50 water molecules per normalized unit cell, respectively), however a decrease of the intensity of the emitting species excited at 260 nm was observed.
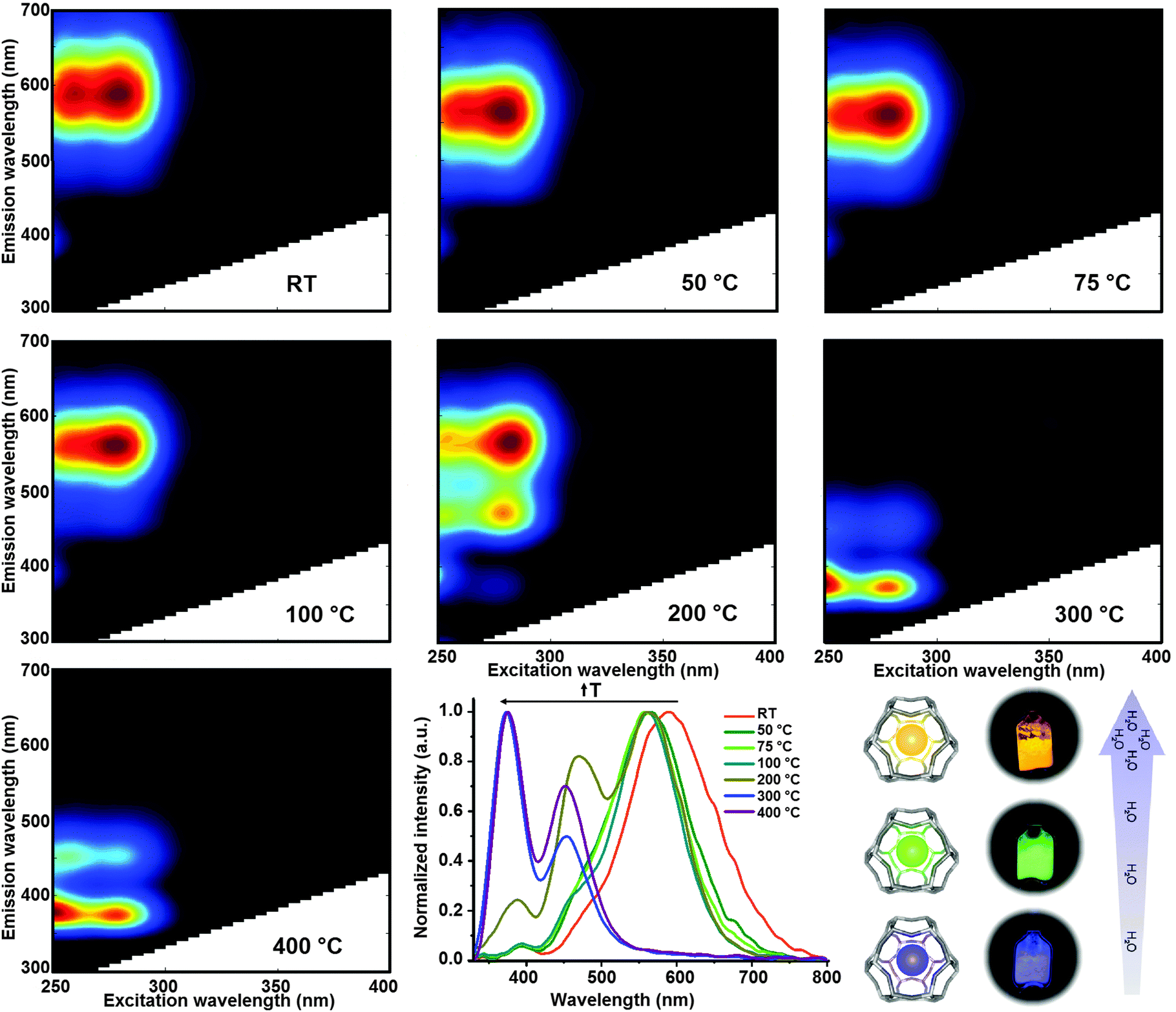 | ||
| Fig. 5 Excitation–emission profiles of LTA(Li)–Ag1 after heat-treatment at different temperatures; the samples were kept at the desired temperature for 1 hour, then cooled down to room temperature to perform the measurements. A compilation of the normalized emission profiles obtained upon excitation at 260 nm is also shown (low-middle panel). Additionally, a scheme displaying the color change of real samples with respect to the water content is depicted in the low-right panel. | ||
The most remarkable changes were recorded in the sample heat-treated at 200 °C (about 20 water molecules per normalized unit cell), in which a splitting of the emission signal into three well-defined emission spots situated at about 560, 470 and 390 nm, was found while maintaining similar excitation profiles with maxima at 260 and 280 nm excitation, at this temperature a mixture of green and blue emitting species was encountered. Further heat-treatment of the LTA(Li)–Ag1 sample at 300 °C (about 10 water molecules per normalized unit cell) triggered the disappearance of the emission signal at 560 nm and a blue shift of the 470 and 390 nm bands to 450 and 375 nm was observed; at this stage only blue emitting species were found. At 400 °C (about 3 water molecules per normalized unit cell) similar emission and excitation profiles were recorded as for the sample treated at 300 °C. A significant drop of the blue emission signal, with maxima at 450 and 370 nm, was observed when the sample was heat-treated at 450 °C under constant vacuum (highly dehydrated sample) for 1 hour; at this temperature, only one water molecule per unit cell is expected (Fig. S7 and S9, ESI†).
A summary of the evolution of the different emission bands with respect to pretreatment temperature and hence to the water content is presented in Fig. 5 (low panel middle), in which the normalized emission profiles of LTA(Li)–Ag1, when excited at 260 nm, are depicted. Additionally, a video displaying the color changes of the sample can be found in the ESI† (Video S1). A difference of approximately 12% in water content in LTA(Li)–Ag1 was reflected in a 120 and 200 nm blue shift of the luminescence and a visible change of emission color from yellow (fully hydrated state) to blue (partially hydrated state).
Structural investigation of low silver loaded LTA(Li)–Ag composites
To obtain structural information related to the different silver cluster species observed in LTA(Li)–Ag1 composites and their interactions with the zeolite scaffold, we performed an analysis of the sample, with different hydration levels, by means of extended X-ray absorption fine structure (EXAFS) measurements. In Fig. 6 and 7 the χ(k) k3-weighted EXAFS data and the corresponding phase-corrected Fourier transform (FT) best fits of LTA(Li)–Ag1 samples with different hydration levels are shown. Details of the curve-fitting and a table with the fitting parameters are given in the ESI† (Table S5). Among several possible EXAFS models that have been applied in the study of Ag-exchanged LTA zeolites in literature, the model based on a three-shell structure46 (Ag–Ag, Ag–O, and Ag–Al/Si) appeared to be the most appropriate to be applied in our study. In this model we used a virtual mixed Al/Si site corresponding to a 0.5Si:0.5Al occupancy that better reflects the Si/Al ratio of the LTA zeolites. As our results demonstrate that the change in the luminescent properties of LTA(Li)–Ag1 composites is directly correlated with the hydration level of the Ag–zeolite sample, the first O shell in the EXAFS model has been split in two separate shells corresponding to the Ag–O distance found for Ag cations occupying the center of the S6R (Fig. S2, ESI†) of the sodalite cage in LTA zeolites and to the typical Ag–OH2 distance found in hydrated Ag–zeolite based materials.47
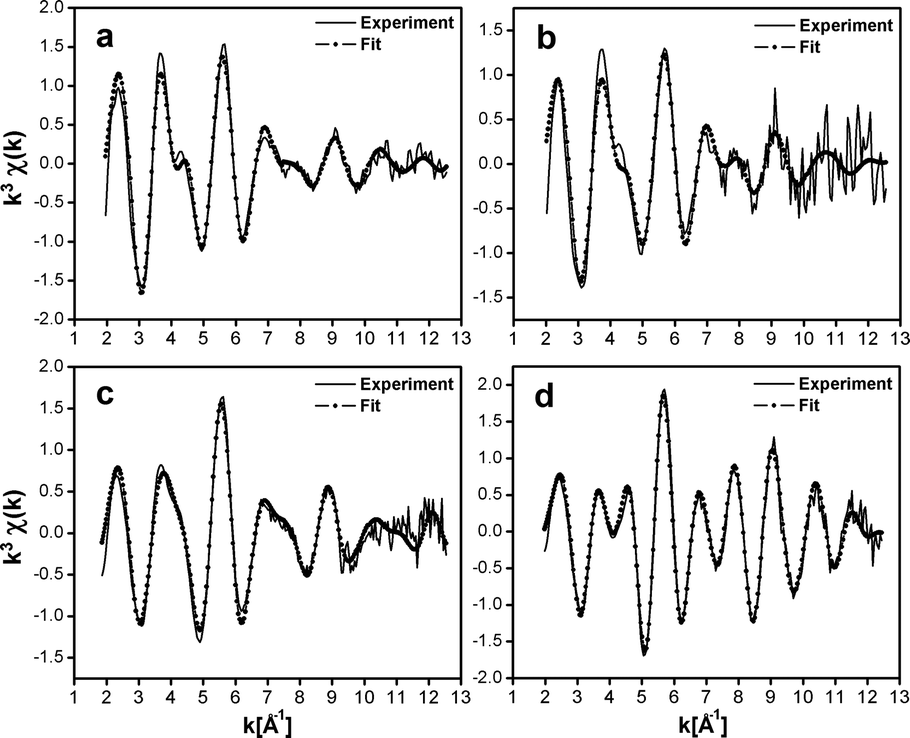 | ||
| Fig. 6 Ag K-edge k3-weighted EXAFS spectra of LTA(Li)–Ag1 composites, (a) fully hydrated (pellet, ∼19% water content), (b) partially dehydrated (quartz cuvette, ∼2% water content), (c) partially dehydrated (glass capillary, ∼1% water content), (d) fully dehydrated (glass capillary). | ||
 | ||
| Fig. 7 Phase corrected FTs of the Ag K-edge k3-weighted EXAFS of LTA(Li)–Ag1 composites (a) fully hydrated (pellet, ∼19% water content), (b) partially dehydrated (quartz cuvette, ∼2% water content), (c) partially dehydrated (glass capillary, ∼1% water content), (d) fully dehydrated (glass capillary). | ||
The first peak in the FT could be fitted with a dual O contribution consisting of 1.3 to 1.4 O at 2.28 to 2.31 Å (N1, Table S3, ESI†) corresponding to the Ag–O coordination, and 0.7 to 1.3 O at 2.44 to 2.47 Å (N5, Table S5, ESI†) corresponding to the Ag–OH2 coordination in the fully and partially hydrated samples.
This dual oxygen shell model was further confirmed by the absence of the O shell at the Ag–OH2 distance in the fully dehydrated sample, where only the presence of an Ag–O contribution at 3.22 Å (N5, Table S5, ESI†) corresponding to a direct long bond distance to the oxygen atoms of the sodalite framework was observed. This clear variation of the coordination number of the Ag–OH2 shell going from 1.3 in the hydrated sample, to 1.1 and 0.7 in the partially dehydrated materials, and finally 0 in the fully dehydrated composite is in very good agreement with the level of hydration that was measured by TGA and FT-IR (Fig. S7 and S8, ESI†).
The second peak in the FT is a multipeak composed of an Ag–Si/Al contribution (N2, Table S5, ESI†) with a relatively stable distance varying from 3.22 to 3.28 Å among the four samples. In addition to the large Ag–Si/Al contribution an Ag–Ag contribution (N3, Table S5, ESI†) could also be detected in the second FT peak. In contrast with the other shells, the coordination and the bond distances of this Ag–Ag shell show large variations among the different samples investigated (hydrated, partially hydrated and fully dehydrated), and ranged from 2.81 Å to 2.58 Å and 1.7 to 0.9, for the bond distances and coordination numbers, respectively. This contribution might correspond to the silver atoms forming oligomeric clusters located inside the sodalite cage. A calculation based on the silver coordination of 3.1, 1.6, and 1.8, suggests that the clusters nuclearity is about 4 in the hydrated LTA(Li)–Ag1 sample, and close to 3 in the partially dehydrated and fully dehydrated LTA(Li)–Ag1 composites (see Section S8 of the ESI† for the details of the calculation). These nuclearities are in line with those assigned for silver clusters in partially Ag exchanged LTA(Na) zeolites examined by electron spin resonance (ESR).41 The unusually large Debye–Waller factors (Table S5, ESI†) associated with this Ag–Ag shell (from 0.02 to 0.07 Å−2) demonstrates the large level of static disorder affecting most of these silver clusters.
Additionally, two contributions of Ag–Li (N4, Table S5, ESI†) and Ag–Ag shells (N6 and N7, Table S5, ESI†) were added to our model as they improved significantly the quality of the fit. The relatively large Ag–Li coordination varying from 1.1 to 1.4 and the short distances from 2.88 to 3.05 Å among the samples, having different hydration levels, show the particular configuration between silver and lithium in LTA(Li)–Ag1 composites. Whereas the long Ag–Ag shell distances from 3.37 to 3.96 Å (N6 & N7, Table S5, ESI†) might correspond to Ag–Ag distances between silver atoms forming the clusters and silver atoms located in the S6R of the sodalite cage (Fig. S2, ESI†).
The vibrational dynamics of water molecules within zeolitic environments strongly depend on the host structure and the balance between guest–guest and guest–host interactions. The interactions between two water molecules and between water molecules and the zeolite framework through hydrogen bonding, and between water molecules and extra-framework cations through ion–dipole interactions have been previously reported using Fourier transformed-infrared (FT-IR) spectroscopy.48,49 Therefore, the dynamics of the behavior of water as a function of temperature within the crystalline structure of LTA(Li)–Ag1 composites was investigated by vibrational spectroscopy. The temperature-dependent FT-IR spectra of LTA(Li)–Ag1 under vacuum conditions measured from 25 up to 450 °C are displayed in Fig. S8 (ESI†). We noticed that at the beginning of the thermal treatment, water molecules weakly bonded to the zeolite framework were released preferentially, whereas at higher temperatures the strongly bonded water molecules coordinated to the extra-framework cations/clusters were liberated (Fig. S8, ESI†).
The materials presented in this report are potential candidates to be used as luminescence-based humidity sensors. For instance, luminescent LTA(Li)–Ag composites could be used as moisture sensors (in the low relative humidity range) in gloveboxes, in which an absolutely water free environment is needed. Small contamination with water will cause a change in the emission color from blue to yellow in LTA(Li)–Ag composites indicating a leakage in the system. The differences in humidity could be easily monitored, without the need of highly sophisticated instrumentation. Furthermore, the versatility of LTA(Li)–Ag composites to be incorporated into different substrates could facilitate the detection systems and decrease the fabrication costs.
Conclusions
The synthesis and characterization of novel LTA(Li)–Ag zeolites displaying better luminescence performance, compared to the already described LTA(Na)–Ag samples, and water-reponsive photoluminescence properties is presented for the first time in this study. Yellow, green and blue emitters were observed in LTA(Li)–Ag zeolite composites with the same composition (low silver loadings) but different hydration levels. The structural characterization carried out by EXAFS suggested a difference in the nuclearity of the emitting clusters. In green/yellow emitting LTA(Li)–Ag composites a nuclearity of about 4 was observed whereas in the blue emitting materials a nuclearity of approximately 3 was found, indicating a decrease in the cluster size with respect to the water content. Moreover, a strong interaction between the lithium cations and the Ag species was also observed by EXAFS and XPS measurements. Further experimental and theoretical research is ongoing to unravel the mechanisms of silver cluster formation and to correlate the structure to the emissive properties of silver clusters confined in LTA(Li) zeolites. Finally, the systematic evaluation of the luminescence properties of these materials with respect to their water content was performed. These composites displayed different emission colors with respect to their water contents, fully hydrated LTA(Li)–Ag composites (19% water content) presented a yellow emission, whereas partially hydrated samples (17–2% water content) showed green and blue emission and finally samples with less than 1% water content displayed only blue emission. This shows the great potential that these new type of luminescent Ag–zeolite composites might have as blue emitting materials and as luminescence-based humidity sensors at macro and micro scale.Acknowledgements
The authors gratefully acknowledge financial support from the Belgian Federal government (Belspo through the IAP VI/27 and IAP-7/05 programs), the European Union's Seventh Framework Programme (FP7/2007-2013 under grant agreement no. 310651 SACS), the Flemish government in the form of long-term structural funding “Methusalem” grant METH/08/04 CASAS, the ‘Strategisch Initiatief Materialen’ SoPPoM program, and the Fund for Scientific Research Flanders (FWO) grant G.0349.12; the KU Leuven Research Funds (IDO/07/011, GOA 2011/3), and the bioSYNergy (University of Copenhagen's Excellence Programme for Interdisciplinary Research). Access to the ESRF (DUBBLE-experiment 26-01-998) was arranged through the general support of the FWO for the use of central facilities. We thank Dr Dipanjan Banerjee and Dr Alessandro Longo (DUBBLE beamline) for their assistance and advice during the EXAFS measurements. We are indebted to Bjorn Dieu and the mechanical workshop of the KU Leuven for helping with the design and construction of the heating cell used in this study.Notes and references
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594 RSC.
- A. Corma, P. Concepcion, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. Lopez-Quintela, D. Bucetta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775 CrossRef CAS PubMed.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401 CrossRef CAS.
- M. Ramanathan, S. M. Kilbey, Q. Ji, J. P. Hill and K. Ariga, J. Mater. Chem., 2012, 22, 10398 RSC.
- S. Choi, R. M. Dickson and J. H. Yu, Chem. Soc. Rev., 2012, 41, 1867 RSC.
- T. Vosch, Y. Antoku, J. C. Hsiang, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616 CrossRef CAS PubMed.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207 CrossRef CAS PubMed.
- Y. Junhua, S. Choi and R. M. Dickson, Angew. Chem., Int. Ed., 2009, 48, 318 CrossRef PubMed.
- S. M. Copp, D. Schultz, S. Swasey, J. Pavlovich, M. Debord, A. Chiu, K. Olsson and E. Gwinn, J. Phys. Chem. Lett., 2014, 5, 959 CrossRef CAS PubMed.
- J. Sharma, C. H. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280 RSC.
- I. Diez, M. I. Kanyuk, A. P. Demchenko, A. Walther, H. Jiang, O. Ikkala and R. H. A. Ras, Nanoscale, 2012, 4, 4434 RSC.
- I. Diez, H. Jiang and R. H. A. Ras, ChemPhysChem, 2010, 11, 3100 CrossRef CAS PubMed.
- V. K. Tikhomirov, T. Vosch, E. Fron, V. D. Rodriguez, J. J. Velazquez, D. Kirilenko, G. Van Tendeloo, J. Hofkens, M. Van der Auweraer and V. V. Moshchalkov, RSC Adv., 2012, 2, 1496 RSC.
- A. Royon, K. Bourhis, M. Bellec, G. Papon, B. Bousquet, Y. Deshayes, T. Cardinal and L. Canioni, Adv. Mater., 2010, 22, 5282 CrossRef CAS PubMed.
- M. Eichelbaum and K. Rademann, Adv. Funct. Mater., 2009, 19, 2045 CrossRef CAS.
- R. Ameloot, M. B. J. Roeffaers, G. De Cremer, F. Vermoortele, J. Hofkens, B. Sels and D. E. De Vos, Adv. Mater., 2011, 23, 1788 CrossRef CAS PubMed.
- L. Jing, A. Ceren, D. Nigel and B. C. Gates, Angew. Chem., Int. Ed., 2012, 51, 5842 CrossRef PubMed.
- C. Leiggener and G. Calzaferri, Chem. – Eur. J., 2005, 11, 7191 CrossRef CAS PubMed.
- G. Calzaferri, C. Leiggener, S. Glaus, D. Schurch and K. Kuge, Chem. Soc. Rev., 2003, 32, 39 RSC.
- G. De Cremer, E. Coutino-Gonzalez, M. B. J. Roeffaers, B. Moens, J. Ollevier, M. Van der Auweraer, R. Schoonheydt, P. A. Jacobs, F. C. De Schryver, J. Hofkens, D. E. De Vos, B. F. Sels and T. Vosch, J. Am. Chem. Soc., 2009, 131, 3049 CrossRef CAS PubMed.
- G. De Cremer, B. F. Sels, J. Hotta, M. B. J. Roeffaers, E. Bartholomeeusen, E. Coutino-Gonzalez, V. Valtchev, D. E. De Vos, T. Vosch and J. Hofkens, Adv. Mater., 2010, 22, 957 CrossRef CAS PubMed.
- E. Coutino-Gonzalez, D. Grandjean, M. B. J. Roeffaers, K. Kvashnina, E. Fron, B. Dieu, G. De Cremer, P. Lievens, B. F. Sels and J. Hofkens, Chem. Commun., 2014, 50, 1350 RSC.
- G. De Cremer, E. Coutino-Gonzalez, M. B. J. Roeffaers, D. E. De Vos, J. Hofkens, T. Vosch and B. F. Sels, ChemPhysChem, 2010, 11, 1627 CrossRef CAS PubMed.
- R. Seifert, A. Kunzmann and G. Calzaferri, Angew. Chem., Int. Ed., 1998, 37, 1522 CrossRef CAS.
- R. Seifert, R. Rytz and G. Calzaferri, J. Phys. Chem. A, 2000, 104, 7473 CrossRef CAS.
- P. Sazama, H. Jirglova and J. Dedecek, Mater. Lett., 2008, 62, 4239 CrossRef CAS.
- T. Sun and K. Seff, Chem. Rev., 1994, 94, 857 CrossRef CAS.
- V. V. Terskikh, C. I. Ratcliffe, J. A. Ripmeester, C. J. Reinhold, P. A. Anderson and P. P. Edwards, J. Am. Chem. Soc., 2004, 126, 11350 CrossRef CAS PubMed.
- S. Jirak, V. Bosacek, S. Vratislav, H. Herden, R. Schollner, W. J. Mortier, L. Gellens and J. B. Uytterhoeven, Zeolites, 1983, 3, 255 CrossRef.
- P. A. Anderson, D. Barr and P. P. Edwards, Angew. Chem., Int. Ed., 1991, 30, 1501 CrossRef.
- B. Xu and L. Kevan, J. Phys. Chem., 1992, 96, 2642 CrossRef CAS.
- V. V. Terskikh, I. L. Moudrakovski, C. I. Ratcliffe, J. A. Ripmeester, C. J. Reinhold, P. A. Anderson and P. P. Edwards, J. Am. Chem. Soc., 2001, 123, 2891 CrossRef CAS.
- N. D. Hudson, S. C. Zajic and R. T. Yang, Ind. Eng. Chem. Res., 2000, 39, 1775 CrossRef.
- H. Yahiro, K. Kurohagi, G. Okada, Y. Itagaki, M. Shiotani and A. Lund, Phys. Chem. Chem. Phys., 2002, 4, 4255 RSC.
- E. Coutino-Gonzalez, M. B. J. Roeffaers, B. Dieu, G. De Cremer, S. Leyre, P. Hanselaer, W. Fyen, B. F. Sels and J. Hofkens, J. Phys. Chem. C, 2013, 117, 6998 CAS.
- M. Maus, M. Cotlet, J. Hofkens, T. Gensch, F. C. De Schryver, J. Schaffer and C. A. M. Seidel, Anal. Chem., 2001, 73, 2078 CrossRef CAS.
- Y. Kim and K. Seff, J. Am. Chem. Soc., 1977, 99, 7055 CrossRef CAS.
- P. A. Jacobs and J. B. Uytterhoeven, J. Chem. Soc., Faraday Trans., 1979, 75, 56 RSC.
- L. R. Gellens, W. J. Mortier, R. A. Schoonheydt and J. B. Uytterhoeven, J. Phys. Chem., 1981, 85, 2783 CrossRef CAS.
- P. J. Grobet and R. A. Schoonheydt, Surf. Sci., 1985, 156, 893 CrossRef CAS.
- T. Wasowicz and J. Michalik, Radiat. Phys. Chem., 1991, 37, 427 CrossRef CAS.
- A. Mayoral, T. Carey, P. A. Anderson, A. Lubk and I. Diaz, Angew. Chem., Int. Ed., 2011, 50, 11230 CrossRef CAS PubMed.
- N. T. Cuong, H. M. T. Nguyen and M. T. Nguyen, Phys. Chem. Chem. Phys., 2013, 15, 15404 RSC.
- H. S. Sherry, in Handbook of Zeolite Sciences, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker, New York, 2003, ch. 21, p. 1007 Search PubMed.
- I. Diez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldmann, A. H. E. Muller, O. Ikkala and R. H. A. Ras, Angew. Chem., Int. Ed., 2009, 48, 2122 CrossRef CAS PubMed.
- T. Miyanaga, H. Hoshino, H. Endo and H. Sakane, J. Synchrotron Radiat., 1999, 6, 442 CrossRef CAS PubMed.
- T. Miyanaga, Y. Suzuki, N. Matsumoto, S. Narita, T. Ainai and H. Hoshino, Microporous Mesoporous Mater., 2013, 168, 213 CrossRef CAS.
- V. Crupi, F. Longo, D. Majolino and V. Venuti, J. Phys.: Condens. Matter, 2006, 18, 3563 CrossRef CAS.
- V. Crupi, F. Longo, D. Majolino and V. Venuti, J. Chem. Phys., 2005, 123, 154702 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, TGA results, description of the home-made heating cell, FT-IR spectra, DRS spectra, time-resolved luminescence fitting information, and curve fitting parameters obtained from the EXAFS refinements. See DOI: 10.1039/c5tc02723c |
| This journal is © The Royal Society of Chemistry 2015 |