
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photophysical characterization of the 9,10-disubstituted anthracene chromophore and its applications in triplet–triplet annihilation photon upconversion†
Victor
Gray
,
Damir
Dzebo
,
Angelica
Lundin
,
Jonathan
Alborzpour
,
Maria
Abrahamsson
,
Bo
Albinsson
and
Kasper
Moth-Poulsen
*
Chalmers University of Technology, Gothenburg, Sweden. E-mail: kasper.moth-poulsen@chalmers.se
First published on 21st September 2015
Abstract
Molecules based on anthracene are commonly used in applications such as OLEDs and triplet–triplet annihilation upconversion. In future design of blue emitting materials it is useful to know which part of the molecule can be altered in order to obtain new physical properties without losing the inherent optical properties. We have studied the effect of substitution of 9,10-substituted anthracenes. Eight anthracenes with aromatic phenyl and thiophene substituents were synthesised, containing both electron donating and accepting groups. The substitutions were found to affect the UV/Vis absorption only to a small extent, however the fluorescence properties were more affected with the thiophene substituents that decreased the fluorescence quantum yield from unity to <10%. DFT calculations confirm the minor change in absorption and indicate that the first and second triplet state energies are also unaffected. Finally the three most fluorescent derivatives 4-(10-phenylanthracene-9-yl)pyridine, 9-phenyl-10-(4-(trifluoromethyl)phenyl)anthracene and 4-(10-phenylanthracene-9-yl)benzonitrile were successfully utilized as annihilators in a triplet–triplet annihilation upconversion (TTA-UC) system employing platinum octaethylporphyrin as the sensitizer. The observed upconversion quantum yields, ϕUC, slightly exceeded that of the benchmark annihilator 9,10-diphenylanthracene (DPA).
1 Introduction
Anthracene and its derivatives have played an important role as organic chromophores since its discovery in 1832 by Jean B. A. Dumas and Auguste Laurent.1,2 Many dyes are based on the anthracene structure and applications for these blue emitting chromophores are plentiful in a variety of fields, from organic light emitting diodes (OLEDs)3–5 and fluorescent probes6–8 to organic scintillators9 and more recently photon-upconversion through triplet–triplet annihilation.10–21Unsubstituted anthracene has a fluorescence quantum yield of about 30%, a consequence of the high intersystem crossing rate and a triplet yield of approximately 70%.22 Substituting anthracene at the 9- and 10-positions can drastically alter the probability of these transitions, e.g. 9,10-dimethylanthracene has a fluorescence quantum yield of about 70%23 and has been successfully used in triplet–triplet annihilation systems previously by Parker24 and more recently by McCusker and Castellano.25 For many applications it is desirable to have a high emission yield and therefore 9,10-substituted anthracenes are potential candidates. As the substitutions also influence other important factors, such as solubility, crystal structure, exciplex formation, surface affinity and spectral characteristics, there can be multiple purposes for the choice of substituents. Bulky substituents, such as phenyl groups, are also known to hamper the [4+4] photocycloaddition that anthracene and 9,10-dimethylanthracene undergo at high concentrations when irradiated.23,24,26
Photon upconversion is the process of generating high energy photons from two or more low energy photons. Through triplet–triplet annihilation (TTA) this can be achieved with incoherent low intensity light such as sunlight.10,13,27–32 Thus, TTA photon upconversion has gained a lot of interest as a way to improve the efficiency of solar energy applications33,34 such as photovoltaics,35–42 photoelectrochemical15,43 and solar driven photochemical reactions26,44 by utilizing sub-bandgap photons. TTA photon upconversion, schematically described in Fig. 1, generally occurs in a bimolecular system, consisting of a triplet sensitizer (S), typically metalloporphyrins, and an annihilator species (A), commonly polyaromatic molecules. The process was first described in the 1960s by Parker and Hatchard45–47 and consists of a series of events. First a low energy photon is absorbed by a sensitizer in its ground state (1S) which readily undergoes intersystem crossing (ISC) to its first excited triplet state (3S*). Subsequently the triplet state of the annihilator (3A*) is populated through a triplet-energy transfer (TET) process from the sensitizer. Two triplet-excited annihilators can then interact and undergo triplet–triplet annihilation (TTA), which can result in the formation of one singlet excited annihilator (1A*) and one annihilator in the ground state (1A). The excited singlet can subsequently relax to the ground state through fluorescence, emitting a photon of higher frequency than the ones initially absorbed. In reality, one TTA event does not necessarily form one singlet state if triplet and quintet states are energetically accessible, and spin-statistics was first assumed to limit the TTA process to efficiencies below 1/9. However experimental data have disproven this limit with observed efficiencies well above 1/9.15,48
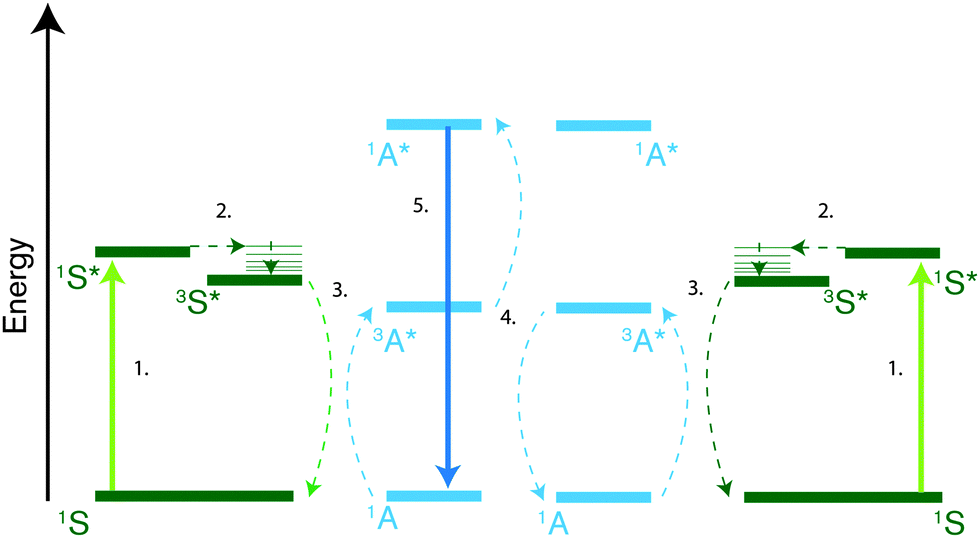 | ||
| Fig. 1 Jablonski diagram explaining the triplet–triplet annihilation upconversion process. First a sensitizer absorbs a low energy photon (1) and rapidly populates its first triplet excited state after intersystem crossing (ISC) from the singlet state (2). The triplet energy can then be transferred to an annihilator species through a triplet energy transfer (TET) process (3) generating one triplet excited annihilator molecule. When two triplet excited annihilator molecules come together they can undergo triplet–triplet annihilation (4) generating one singlet excited annihilator which can deactivate to its ground state by emitting a photon (5). | ||
One of the most efficient upconversion sensitizer–annihilator pairs is DPA, 1 (Fig. 2) and Pd or Pt octaethylporphyrin, with an upconversion quantum yield of up to 18%.15 Lately some promising attempts to modify 1 have been made in order to facilitate and achieve TTA-UC in different matrices.17,20,49,50 However no detailed study of how these modifications alter the photophysical properties of chromophore 1 has been done. With the aim of identifying the structure/property relationships for 9,10-substituted anthracenes and revealing the design parameters for future TTA-UC materials herein we present the synthesis and study of photophysical properties of eight 9,10-disubstituted anthracene derivatives (Fig. 2). To complement the experimental measurements DFT calculations of the position of singlet and triplet excited states were performed in order to determine the S–T energy level spacing. Both electron withdrawing and electron donating substituents have been chosen. We further demonstrate the use of the most promising derivatives as annihilators in the triplet–triplet annihilation based upconversion, using platinum octaethylporphyrin (PtOEP) as the sensitizer. The derivatives are compared to the well-known 9,10-diphenylanthracene (1) and 9,10-dimethylanthracene (2) chromophores.
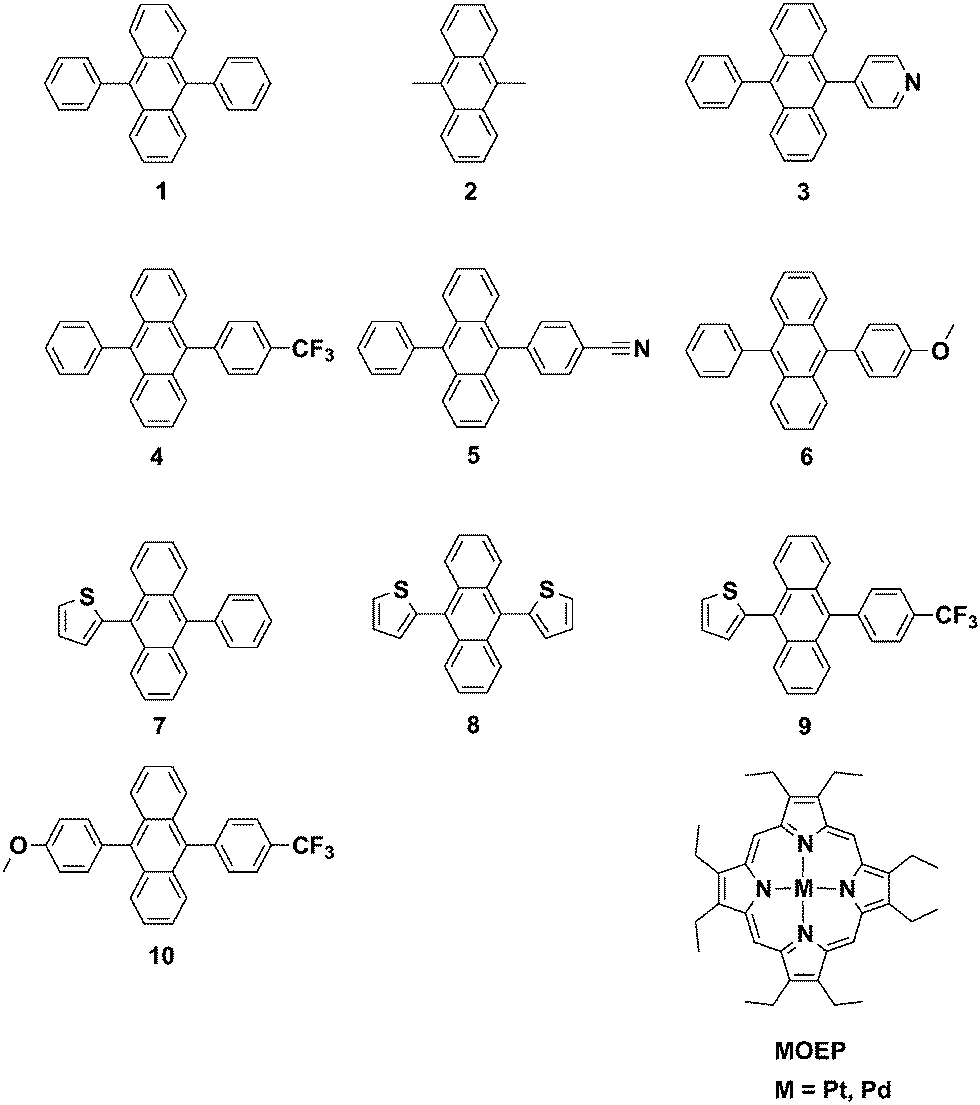 | ||
| Fig. 2 Structures of investigated 9,10-substituted anthracenes and the triplet sensitizers platinum and palladium octaethylporphyrin. | ||
2 Experimental method
2.1 Instrumentation and optical measurements
Steady-state absorption spectra were recorded on a Cary 4000 spectrophotometer and steady state fluorescence measurements were carried out on a Spex Fluorolog 3 spectrofluorimeter (JY Horiba). Fluorescence lifetimes were determined on a time correlated single photon counting (TCSPC) setup using PicoQuant laser diodes (377 nm) and a PMT detector (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts, 4096 channels). Nanosecond transient absorption measurements were performed on a home-built system using a Surelite Continuum Nd:YAG laser equipped with an OPO generating a ∼10 ns pump beam. A quartz-halogen lamp with a monochromator was used as the probe light and a monochromator together with a 5-stage PMT coupled to an oscilloscope was used for recording the transient. All photophysical measurements were carried out in toluene using quartz cuvettes except for samples used in the upconversion intensity study which were permanently sealed in Pyrex test-tubes after degassing following a freeze–pump–thaw procedure described in the ESI.† Samples for Stern–Volmer quenching studies and nanosecond transient absorption studies were degassed by stirring in a glovebox from Innovative Technologies (<0.1 ppm oxygen level) under a nitrogen atmosphere for at least 48 h.
000 counts, 4096 channels). Nanosecond transient absorption measurements were performed on a home-built system using a Surelite Continuum Nd:YAG laser equipped with an OPO generating a ∼10 ns pump beam. A quartz-halogen lamp with a monochromator was used as the probe light and a monochromator together with a 5-stage PMT coupled to an oscilloscope was used for recording the transient. All photophysical measurements were carried out in toluene using quartz cuvettes except for samples used in the upconversion intensity study which were permanently sealed in Pyrex test-tubes after degassing following a freeze–pump–thaw procedure described in the ESI.† Samples for Stern–Volmer quenching studies and nanosecond transient absorption studies were degassed by stirring in a glovebox from Innovative Technologies (<0.1 ppm oxygen level) under a nitrogen atmosphere for at least 48 h.
Fluorescence quantum yields were determined by relative actinometry employing 1 as the standard using deaerated dilute solutions, and comparative spectra were corrected for absorbance. Upconversion quantum yields were determined relative to zinc octaethyl porphyrin51 using a 532 nm green laser-pointer (33.8 mW, 0.0573 cm2) as the light source and a graduated neutral density filter to vary the intensity. Solubility experiments were carried out by saturating a solution of toluene with each derivative and then filtering the supernant with a syringe filter before recording the absorption spectra to monitor the amount of dissolved species.
1 was purchased from Sigma-Aldrich, PtOEP and PdOEP were purchased from PorphyChem and all were used as received. 2 was a kind gift from the late Prof. Hans-Dieter Becker and its purity was confirmed by H1-NMR spectroscopy prior to use. Spectroscopy and GC grade toluene was used for all measurements. Degassed and dry toluene was obtained from a M-Braun solvent drying system. NMR was run on a 400 MHz Varian NMR and IR was run either neat or in KBr pellets using a Perkin Elmer ATR-FTIR or Perkin Elmer FTIR respectively. Column chromatography was carried out using a Biotage Flash Column Chromatography system with Biotage prepacked SNAP columns if not stated otherwise. Melting points were determined using an automatic Mettler Toledo MP70 melting point apparatus.
2.2 Synthesis
Phenyl substitutents were coupled to the anthracene core by Suzuki–Miyaura cross-coupling procedures (Fig. 3) and thiophene-substitutents by Stille cross-coupling procedures (Fig. 3). 9-Bromo-10-phenylanthracene was synthesized according to the literature.52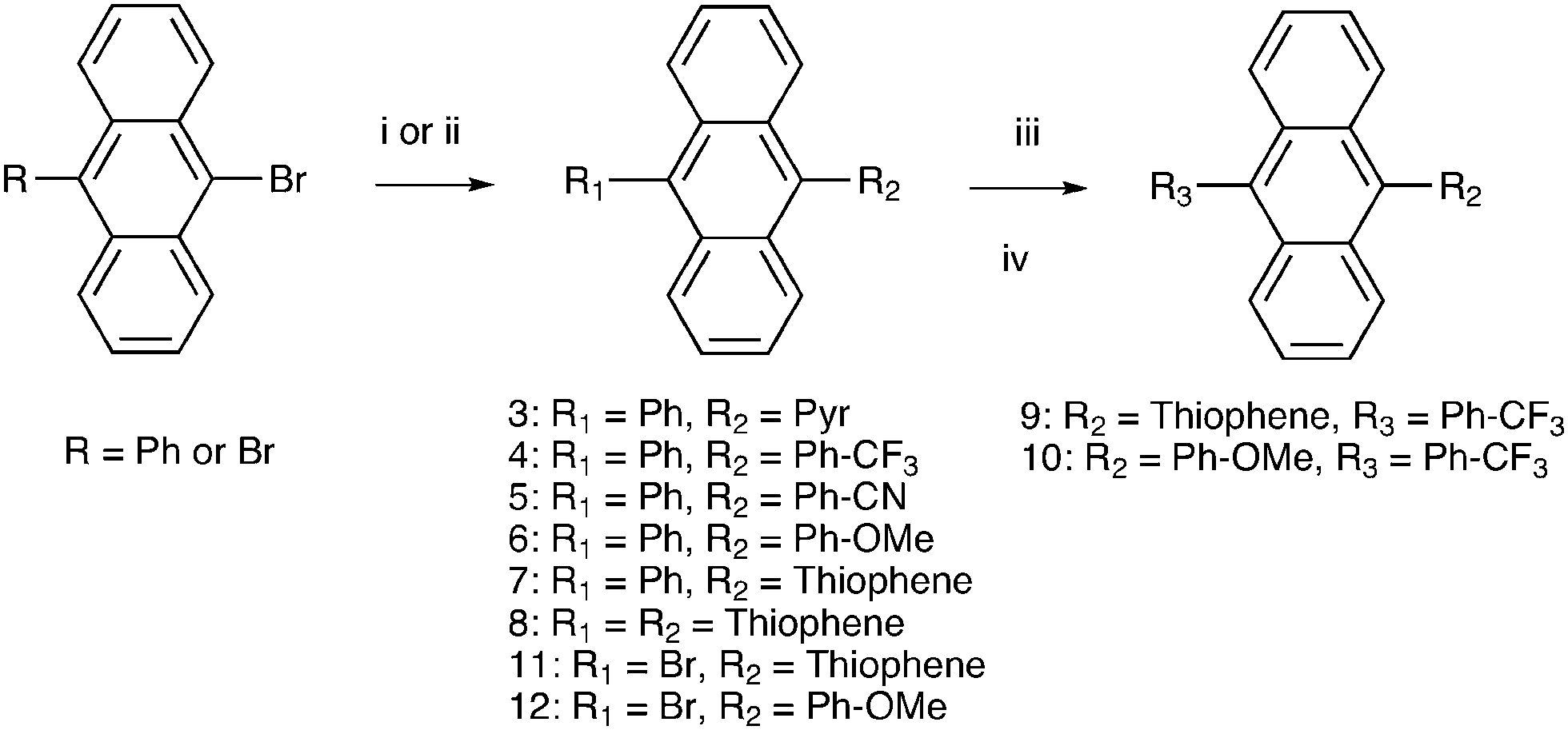 | ||
| Fig. 3 Synthesis of 9,10-disubstituted anthracenes. (i) Suzuki coupling; arylboronic acid, Pd(PPh3)4, Na2CO3 (2 M aq), THF, toluene, reflux, (ii) Stille coupling; 2-Bu3Sn-thiophene, Pd2(dba)3, tri-o-tolylphosphine, THF, reflux, (iii) CF3-Ph-B(OR)2, Pd(PPh3)4, and (iv) toluene, THF, Na2CO3 (2 M, aq), reflux, 18 h. | ||
2.3 Calculations
The popular hybrid functional B3LYP55,56 was used in connection with a triple-zeta basis set, 6-311+G(d,p)57–59 as implemented in Gaussian 09.60 All structures were fully optimized and consequently confirmed as local minima by calculation of second derivatives. Excited state calculations were performed using the time-dependent DFT method.61,623 Results and discussion
3.1 Synthesis
The commercially available 9,10-dibromoanthracene and 9-phenylanthracene were used as starting materials for Suzuki–Miyaura (S–M) and Stille cross-coupling reactions. The Stille cross-coupling procedure in dry THF was preferred for the thiophene–anthracene coupling as it proceeded smoothly and resulted in higher yields (46–80%) than the corresponding S–M procedures first attempted (<10%). The phenyl-substituents were successfully coupled using S–M cross-coupling procedures (58–84%). A system of THF/toluene/Na2CO3(aq)53 was generally used, except for the nitrogen containing phenyl groups which gave higher yields when carried out in a THF/K2CO3(aq) mixture. It was also observed that reducing the amount of solvent to half or less in the S–M procedure compared to what was initially used53 resulted in higher yields (from 35% to 70%).3.2 Photophysical characterization
Photophysical properties of the studied derivatives are presented in Table 1. Absorption spectra and molar absorptivities of the five representative anthracene derivatives are presented in Fig. 4 (for clarity, the remaining compounds were omitted and can be found in Fig. S1, ESI†). The absorption spectra are all similar to 1, with the characteristic vibronic peaks of 1 and anthracene. Only minor (<10 nm) red shifts (Fig. 4) are observed for derivatives with thiophene substituents. These results are in well agreement with calculations which predict only small changes in the S0–S1 gap, vide infra.| Compound | Solubility (mM) | Absmaxa (ε × 10−4) (wavelength (nm)) | Φ f | τ f (ns) | τ t (ms) | k TET (×109 M−1 s−1) | k TTA (×109 M−1 s−1) |
|---|---|---|---|---|---|---|---|
| a Reported values are the average of two independent measurements. b PtOEP as the sensitizer. | |||||||
| 1 | 93 | 1.21(395), 1.25(375), 0.76(356)68 | 1.064 | 6.97 | 8.61 | 2.15 | 2.51 |
| 2 | 115 | 0.92(401), 0.955(380), 0.56(360) | ∼0.723 | — | — | 3.97 | — |
| 3 | 39 | 1.21(395), 1.28(374), 0.79(356) | 0.96 ± 0.020 | 6.93 | 7.73 | 1.93 | 2.31 |
| 4 | 8 | 1.22(395), 1.29(374), 0.795(356) | 1.0 ± 0.010 | 6.84 | 9.55 | 1.92 | 2.25 |
| 5 | 83 | 1.24(395), 1.31(375), 0.80(357) | 0.99 ± 0.003 | 5.54 | 1.73 | 1.81 | 1.98 |
| 6 | 47 | 1.245(396), 1.32(375), 0.815(357) | 0.84 ± 0.065 | 5.50 | 18.95 | 2.25 | — |
| 7 | 256 | 1.255(397), 1.315(377), 0.8(358) | 0.09 ± 0.002 | — | 0.043 | 2.07 | — |
| 8 | 61 | 1.35(400), 1.365(379), 0.815(360) | 0.02 ± 0.000 | — | 0.005 | 2.52 | — |
| 9 | 76 | 1.28(397), 1.32(376), 0.80(358) | 0.026 ± 0.006 | — | 0.043 | 2.17 | — |
| 10 | 107 | 1.23(396), 1.30(375), 0.80(357) | 0.77 ± 0.016 | 4.69 | 8.50 | 1.90 | 1.77 |
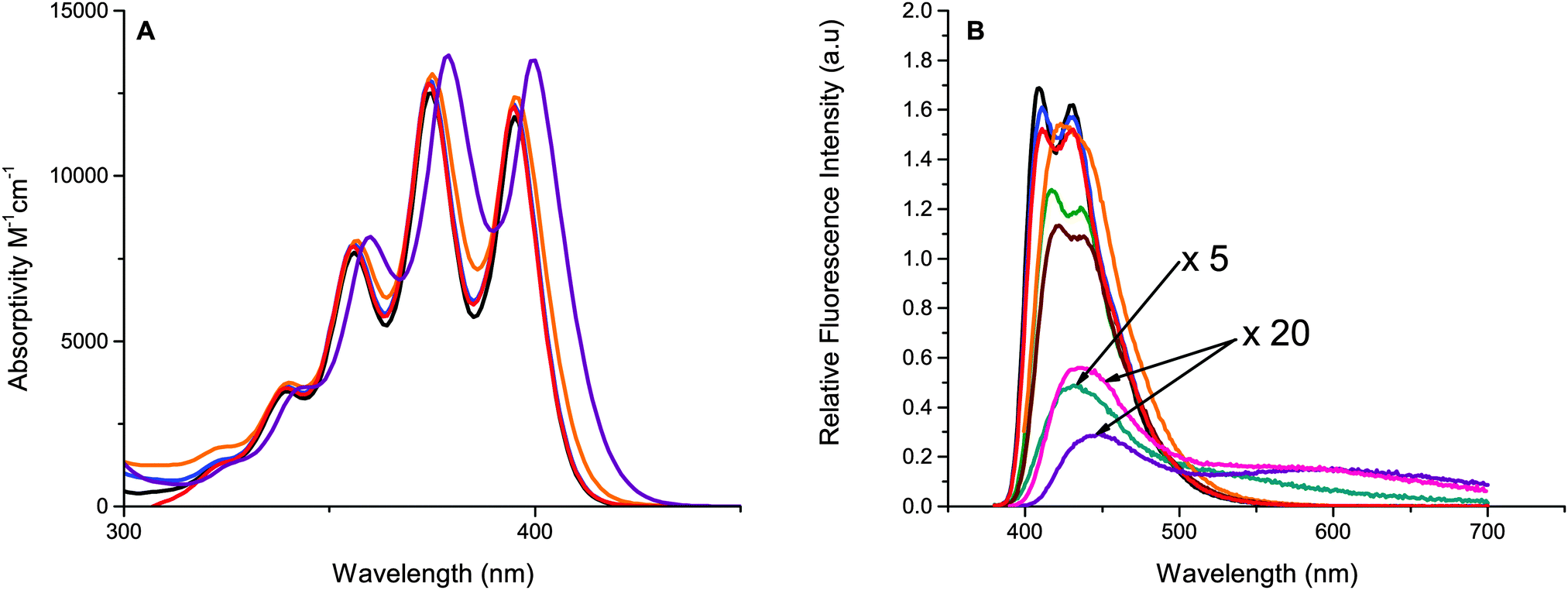 | ||
Fig. 4 (A) Molar absorptivity of the representative compounds 1 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), 3 ( ), 3 ( ), 4 ( ), 4 ( ), 5 ( ), 5 ( ), and 8 ( ), and 8 ( ) and (B) relative fluorescence intensity of synthesized anthracene derivatives compared to 1 (), 3 ( ) and (B) relative fluorescence intensity of synthesized anthracene derivatives compared to 1 (), 3 ( ), 4 ( ), 4 ( ), 5 ( ), 5 ( ), 6 ( ), 6 ( ), 7 ( ), 7 ( ), 8 ( ), 8 ( ), 9 ( ), 9 ( ) and 10 ( ) and 10 ( ); spectra are corrected for differences in absorbance at excitation wavelength. ); spectra are corrected for differences in absorbance at excitation wavelength. | ||
The fluorescence spectra of the phenyl-substituted anthracenes 3–6 and 10 are similar to that of 1, Fig. 4, and have high fluorescence quantum yields (Table 1). However, compounds containing thiophenes show a considerable decrease in their fluorescence quantum yield compared to 1, with quantum yields as low as 2% for 8. The tail of the emission of these derivatives (7–9) stretches further into red, up to 750 nm compared to the other derivatives which end closer to 600 nm, indicating that excitation is more delocalized as the smaller thiophene moiety can conform to a more planar structure compared to the phenyl substituents.63 The low fluorescence quantum yield is most probably explained by new non-radiative decay pathways that become possible as the rotation around the anthracene–thiophene bond is easier than the anthracene–phenyl bond which is substantially more sterically hindered. The lack of an efficient fluorescence state makes these derivatives less useful for applications requiring emissive materials, such as OLEDs or photon-upconversion. The phenyl containing derivatives, on the other hand, have high fluorescence quantum yields, many close to unity as observed for 1.64 Also listed in Table 1 are excited state lifetimes of the emissive compounds, these are in general similar or slightly shorter than the lifetime of 1.
Nanosecond transient absorption was used in order to determine the triplet lifetime of the derivatives (Table 1). The triplet absorption was monitored at 452 nm. No triplet states were detected for the derivatives when directly excited at 355 nm. Therefore samples of 1–10 (∼770 μM) were prepared with 1 μM PtOEP and were pumped at 532 nm using a fraction of the frequency doubled fundamental pump beam. All samples were degassed in a glove-box prior to measurement. The triplet decays were fit to eqn (1) which takes into account the contribution of triplet–triplet annihilation which can occur at high triplet state concentrations48,65,66 (Fig. S12–S20, ESI†):
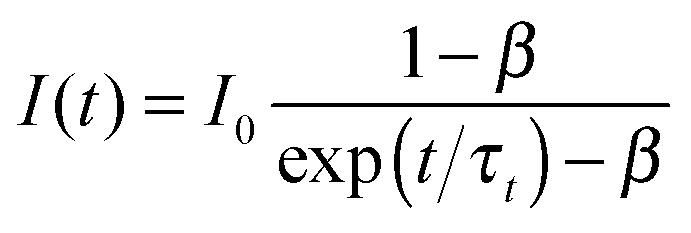 | (1) |
Long triplet state lifetimes (>1 ms) were observed for the diphenyl substituted derivatives (Table 1), which are beneficial in a TTA-UC system.67 As was the case in the first excited singlet state, thiophene containing derivatives also displayed shorter triplet state lifetimes.
The solubility in toluene was investigated and is also presented in Table 1. All derivatives are soluble above 40 mM in toluene, except for trifluoro-substituted 4 which is about an order of magnitude less soluble. The absorption spectra of the saturated solutions were recorded between two glass slides and showed no significant change compared to the dilute spectra (S2–S11, ESI†). Also the fluorescence of the saturated solutions were recorded. For aryl substituted anthracenes the only changes observed could be explained by reabsorption due to the overlap of absorption and emission, indicating that little or no aggregation occurred. 2 showed a new red-shifted emission which is characteristic of its exciplex and has been reported previously.24
3.3 Stern–Volmer quenching analysis
For an efficient triplet energy transfer from a triplet sensitizer to an acceptor it requires that the triplet state of the acceptor is lower in energy than that of the sensitizer. This is the case for the sensitizer–acceptor pairs platinum octaethylporphyrin (PtOEP) and 1 as well as palladium octaethylporphyrin (PdOEP) and 1. The energy transfer efficiency can be monitored by the quenching of the sensitizer phosphorescence as described by the Stern–Volmer relationship (eqn (2)) which relates the emission quenching to the concentration of the quencher:69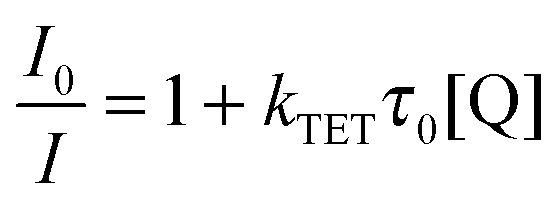 | (2) |
The quenching of PdOEP and PtOEP by the substituted anthracenes was studied. Stern–Volmer plots of the quenching dynamics of PtOEP are shown in Fig. 5. Similar graphs were obtained for the quenching of PdOEP (Fig. S26, ESI†). The obtained triplet energy transfer rate constants, kTET, (Table 1) are similar for all derivatives and 1, and all are diffusion limited as expected; they also agree with values reported for 1 in similar systems.27,70 This indicates that the first triplet state energies (T1) of 9,10-substituted anthracenes are all similar or lower than the triplet state of PdOEP, 1.86 eV,70 which is in agreement with that reported for 1, 1.77 eV.71 In this context all derivatives are good candidates for sensitized triplet–triplet annihilation photon upconversion utilizing PdOEP or PtOEP as a sensitizer and the upconverted emission was observed for all derivatives with >10% fluorescence quantum yields.
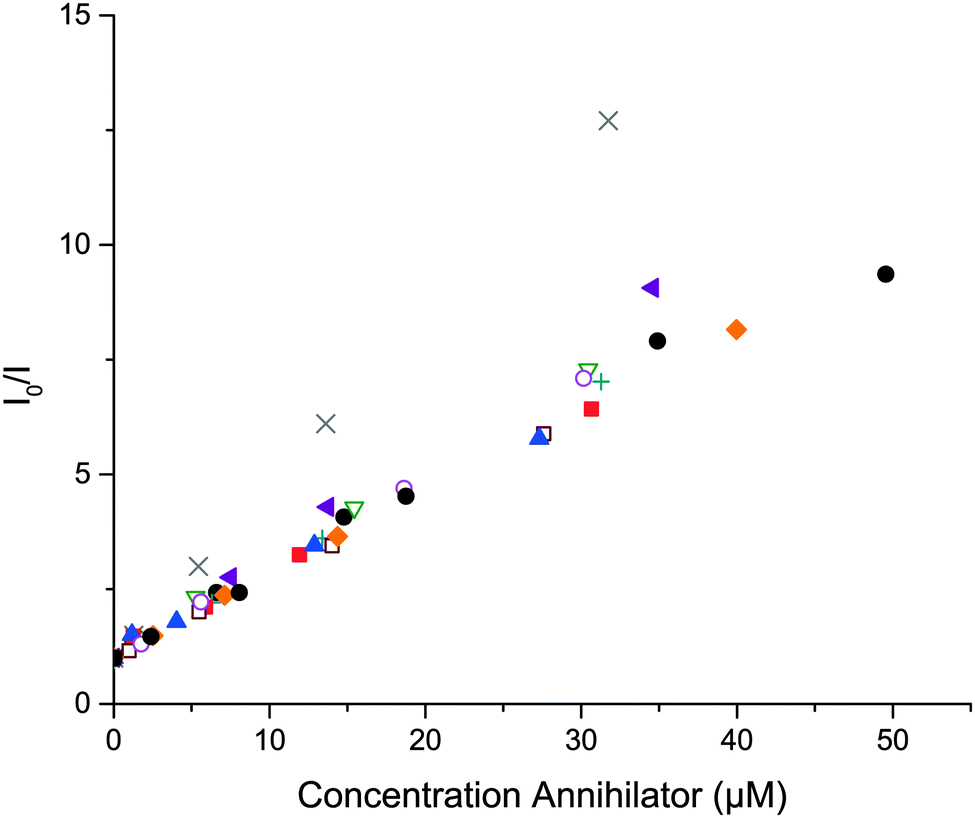 | ||
Fig. 5 Stern–Volmer plot of PtOEP (0.5 μM) phosphorescence quenching of 1 (●), 2 ( ), 3 ( ), 3 ( ), 4 ( ), 4 ( ), 5 ( ), 5 ( ), 6 ( ), 6 ( ), 7 ( ), 7 ( ), 8 ( ), 8 ( ), 9 ( ), 9 ( ) and 10 ( ) and 10 ( ). ). | ||
3.4 Theoretical calculations
In order to reveal the changes in electronic levels due to substitution patterns of 9,10-disubstituted anthracene DFT calculations were performed. Table 2 lists the calculated energy levels and the difference between two times the first triplet energy level (2 × T1) and the first singlet energy level (S1), which ideally is positive but close to zero for a good annihilator in a triplet–triplet annihilation upconversion system.33| Compound | S0–S1 | S0–T1 | S0–T2 | 2 × T1–S1 |
|---|---|---|---|---|
| a All energies are reported in eV. | ||||
| 1 | 3.10 | 1.72 | 3.23 | 0.34 |
| 2 | 3.03 | 1.67 | 3.24 | 0.31 |
| 3 | 3.10 | 1.73 | 3.23 | 0.36 |
| 4 | 3.10 | 1.73 | 3.23 | 0.36 |
| 5 | 3.10 | 1.73 | 3.23 | 0.36 |
| 6 | 3.10 | 1.73 | 3.23 | 0.36 |
| 7 | 3.08 | 1.71 | 3.22 | 0.34 |
| 8 | 3.05 | 1.69 | 3.20 | 0.33 |
| 9 | 3.07 | 1.71 | 3.22 | 0.35 |
| 10 | 3.10 | 1.73 | 3.23 | 0.36 |
The rotation around the single bond between the anthracene and the ligand group was explored and a global minima at 90 degrees was always found in good agreement to that calculated for 9-phenylanthracene and close to 85° as determined by X-ray diffraction of substituted diphenylanthracenes.72 This explains the minor effect of the substitution on the energy levels as the conjugation between the perpendicular aryl-substituent and the anthracene core is minimal compared to the coplanar orientation. The coplanar orientation is unlikely, due to the sterical clash between the hydrogen of substituents and the hydrogens of 1,4,5,8-anthracene, resulting in a high rotational barrier for the phenyl groups.72 The smaller thiophene substituent is expected to have a less hindered rotation and is consistent with the observed red-shifted emission, vide supra.
The substitution effect on the calculated energies is minimal (Table 2), as confirmed by optical absorption experiments. For applications in TTA upconversion this is a promising finding as the spacing of the energy levels is of crucial importance for an efficient system and 1, being a benchmark system, could be modified with substituents to obtain desired properties without affecting the energetics of the chromophore.
3.5 Upconversion study
A long triplet lifetime and an efficient triplet–triplet annihilation rate constant are key features of an upconverting system.67 Triplet–triplet annihilation rate constants were determined for the derivatives 3 (1 mM), 4 (1 mM), 5 (0.7 mM) and 10 (1 mM) as well as for the reference 1 (1 mM) with PtOEP (3.4 μM) as the sensitizer. Samples were monitored at 410 nm as well as at 650 nm after excitation at 532 nm. Fig. 6 displays the transient decays of 4/PtOEP at 410 nm and 650 nm (inset in Fig. 6 shows the first 5 μs) and the best fit to the data. Decays of 1, 3, 4, 5 and 10 with the corresponding fits and the determined fitting parameters can be found in Fig. S21–S25 (ESI†). As is seen in Fig. 6 there is an initial positive feature at 410 nm, which corresponds to the absorption of the annihilator triplet state and the sensitizer triplet, after about 100 ns delayed fluorescence of the annihilator starts to take part, resulting in a negative feature in the transient. Whereas at 650 nm only the phosphorescence of the sensitizer is observed. Both long (1 ms) and short (5 or 50 μs, inset Fig. 6) time windows were recorded at 410 nm. The three transients (1 ms and 5 μs or 50 μs at 410 nm and 5 μs at 650 nm) were fitted globally to the rate equations governing the system, as presented in the ESI,† using MATLAB and the built-in differential equation solver ode23s. The fitting parameters were the triplet–triplet annihilation rate constant (kTTA), the triplet energy transfer rate constant (kTET), the annihilator triplet absorptivity (ΔεET), the sensitizer triplet absorptivity (ΔεST), as well as two scaling factors for the relative magnitude of phosphorescence (aphos) and upconverted emission (afl). The initial triplet concentration of the sensitizer was estimated from the initial ground state bleach at 540 nm.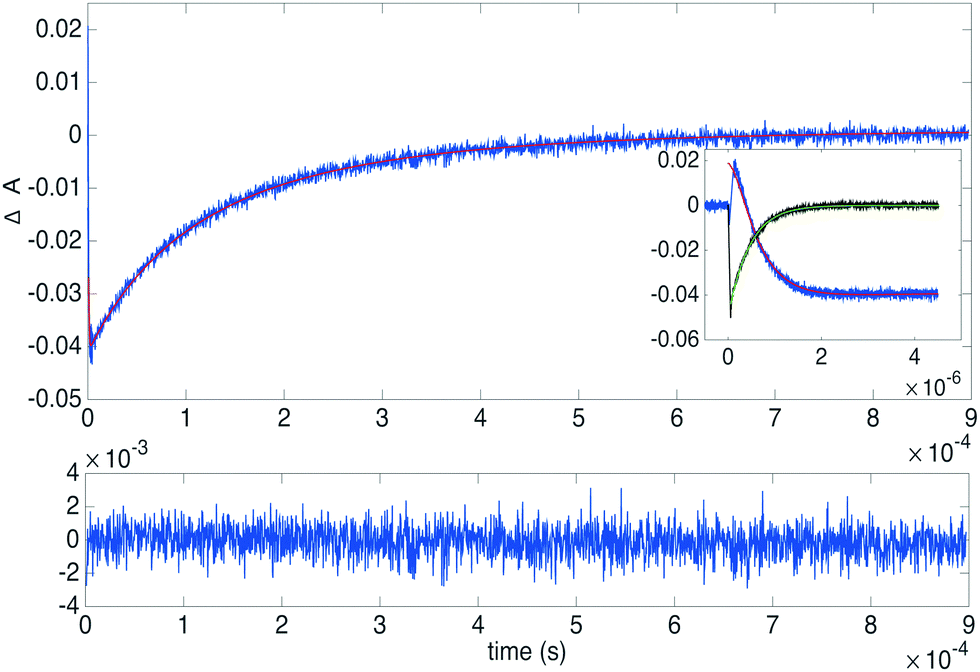 | ||
| Fig. 6 Transient absorption measurements of 4 (1 mM) and PtOEP (3.4 μM) at 410 nm (annihilator decay, blue) and 650 nm (sensitizer decay, black) and the respective fits. The inset shows the first 5 μs. The bottom panel shows the residual of the fitted annihilator decay. | ||
Derivatives 3 and 4 exhibit similar annihilation rate constants but slightly lower than 1, and compounds 5 and 10 display the lowest values of the series. The fitting also yielded triplet energy transfer rate constants kTET close to those determined in the Stern–Volmer analysis as well as triplet absorptivities close to half of that reported at ∼450 nm for 1, which is consistent with the triplet–triplet absorption spectra of 1.71,73 The obtained values can be found in the ESI† together with the transient decays (Fig. S21–S25, ESI†).
The quantum yield of upconversion, the number of emitted high-energy photons compared to the number of absorbed low energy photons, is challenging to determine and to compare with reported values for which one requires to know the exact experimental conditions, such as the sensitizer and annihilator concentrations, light intensity and oxygen concentration.34 The upconversion quantum yield is the product of the quantum yields for each step required to produce upconverted photons (eqn (3)):
| ΦUC = ΦISC × ΦTET × ΦTTA × Φf | (3) |
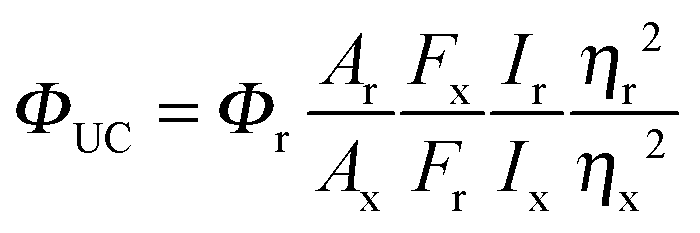 | (4) |
Characteristics of the upconverted emission show a quadratic dependence on the excitation intensity at low intensities, which shifts to a linear dependence at higher intensities as the annihilator triplet concentration no longer limits the annihilation process.65 Consequently the upconversion quantum yield will increase with the excitation intensity until the linear regime is reached. The intensity where the dependence shifts from quadratic to linear is referred to as the threshold intensity Ith and is an important parameter as it can be compared to the light intensity provided from the sun at the specific wavelength. For an efficient and practical system Ith should be equal or lower than the intensity of the sun.
As described in eqn (3), ΦUC scales with the fluorescence quantum yield of the annihilator, an efficient system thus requires an annihilator with a fluorescence quantum yield close to unity. Therefore only the three derivatives with the highest quantum yields, namely 3, 4 and 5, were chosen for the upconversion study. As mentioned previously compounds 6 and 10 with intermediate fluorescence quantum yields also displayed upconverted emission.
The excitation power density dependence on the upconverted emission is presented in Fig. 7. All three annihilators perform similar to 1 where 3 and 4 perform within error the same as 1 with Ith = 18 mW cm−2, whereas 5 has a slightly higher Ith = 40 mW cm−2. In Fig. 7ΦUC is seen to level out at higher excitation intensities as expected. Consistent with the observed annihilation rate constants, 3 and 4 exhibit yields similar to 1, but slightly higher; 7.9% and 8.7%, respectively, compared to 7.7% for DPA (1). Again 5 performs slightly worse with a ΦUC of 6.9%, this minor decrease of ΦUC could be explained by the higher molar absorptivity and a more red-shifted absorption onset of 5 which would result in an increased reabsorption at high concentrations used for the UC samples, but could also be a consequence of the shorter triplet lifetime and the less efficient triplet–triplet annihilation process. Overall 3, 4 and 5 are efficient annihilators for TTA-UC employing PtOEP as a sensitizer.
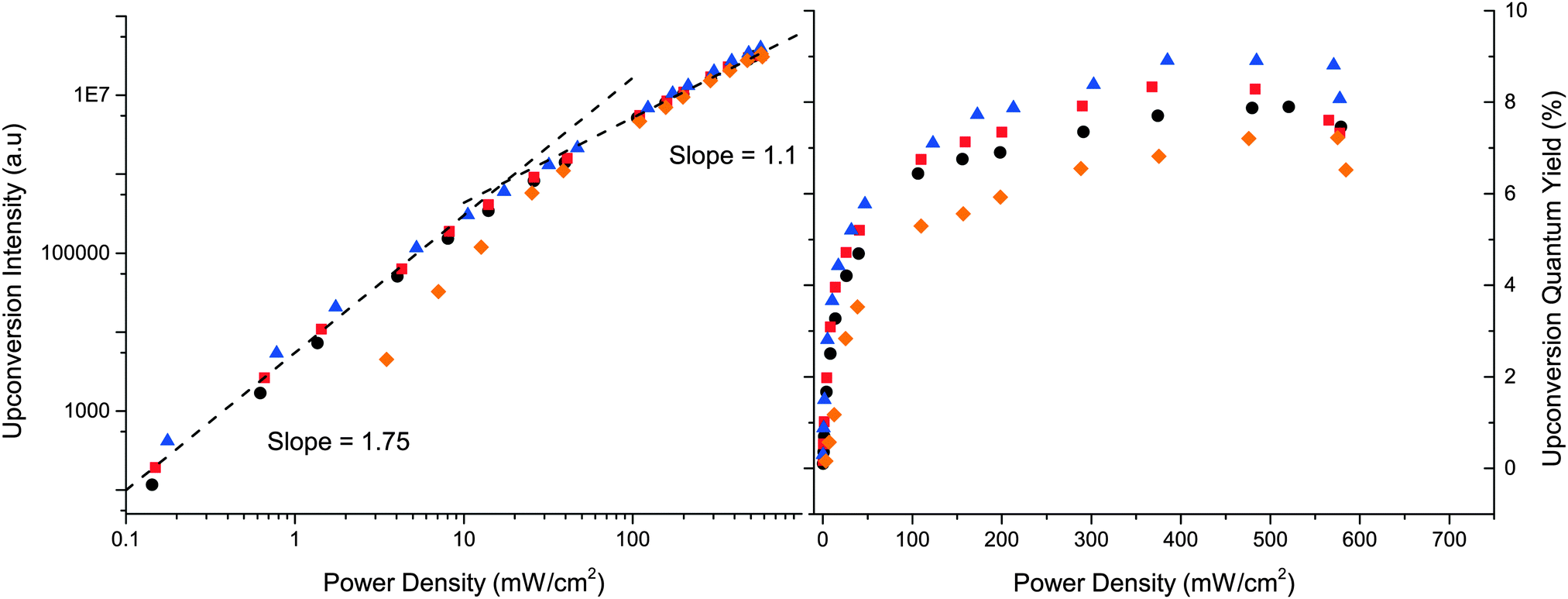 | ||
Fig. 7 Left: A double logarithmic plot of integrated upconverted emission dependence on excitation power density (532 nm) of solutions containing PtOEP, 15 μM and 0.5 mM annihilator. Right: Upconverted emission quantum yield (ΦUC) as a function of excitation power density of the same solutions. 1 (●), 3 ( ), 4 ( ), 4 ( ) and 5 ( ) and 5 ( ). ). | ||
4 Conclusion
Eight 9,10-substituted anthracenes containing either electron donating, electron withdrawing or both types of groups have been synthesized and their photophysical properties have been studied and compared to the two previously known 9,10-substituted anthracenes DPA and DMA (1 and 2 respectively). The type of substitution at the 9,10-position was shown to have only a minor influence on the absorption spectrum, where phenyl-substituents were slightly more blue-shifted compared to thiophene and sp3-C substituents. However the fluorescence quantum yield decreased considerably for thiophene containing derivatives, most probably a result of a larger non-radiative decay rate. Thus for applications requiring blue emissive chromophores phenyl-substituted 9,10-anthracenes are more suitable. DFT calculations are in well agreement with experimental measurements indicating a minimal change of the energy levels upon substitution.The three most fluorescent compounds 3, 4 and 5 were successfully used as annihilators for triplet–triplet annihilation upconversion with PtOEP as the sensitizer and quantum yields slightly exceeding that of DPA were observed. The main challenge for TTA upconversion is to achieve highly efficient systems in the solid state and some attempts to achieve this have been reported. Mainly two methods have been employed in designing such materials, capturing annihilators and sensitizers in a polymeric or gel matrix,29,74–79 or developing supra-molecular or polymeric structures containing annihilators and/or sensitizers.80,81
Aiding the future design of efficient blue-emitting materials based on 9,10-substituted anthracene we demonstrate that the well-known chromophore 1 may be modified in the para-positions with both electron withdrawing and donating substituents without changing the absorption and emission properties. However 9,10-substituents that introduce new non-radiative relaxation pathways should be avoided. The design parameters discussed here would be helpful in future synthesis of 9,10-substituted anthracenes as modifying 1 in this way can be practical and in some cases necessary for materials design. For example, one can envision improving the affinity of 1 for the matrix or the sensitizer by introducing suitable side groups without affecting the useful properties inherent to chromophore 1 and recently two examples of this were reported by Kimizuka and co-workers.20,50
Acknowledgements
The authors acknowledge funding from the Swedish Energy Agency (36436-1), Chalmers Materials Area of Advance, the Swedish Research Council (B0361301), Swedish Strategic Research Council (SSF) and Knut and Alice Wallenberg Foundation. Also computer time at Chalmers Centre for Computational Science and Engineering (C3SE) provided by the Swedish National Infrastructure for Computing (SNIC) is acknowledged.References
- M. O. Forster, Nature, 1921, 108, 108–109 CrossRef PubMed
.
-
H.-G. Franck and J. Stadelhofer, Industrial Aromatic Chemistry, Springer-Verlag, Berlin, Heidelberg, 1st edn, 1988 Search PubMed
- M. H. Ho, B. Balaganesan and C. H. Chen, Isr. J. Chem., 2012, 52, 484–495 CrossRef CAS PubMed
- J. Huang, J.-H. Su and H. Tian, J. Mater. Chem., 2012, 22, 10977 RSC
- T. Serevičius, R. Komskis, P. Adomėnas, O. Adomėnienė, V. Jankauskas, A. Gruodis, K. Kazlauskas and S. Juršėnas, Phys. Chem. Chem. Phys., 2014, 16, 7089–7101 RSC
- H. Ihmels, A. Meiswinkel and C. J. Mohrschladt, Org. Lett., 2000, 2, 2865–2867 CrossRef CAS PubMed
- B. Valeur, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS
- H. Lu, B. Xu, Y. Dong, F. Chen, Y. Li, Z. Li, J. He, H. Li and W. Tian, Langmuir, 2010, 26, 6838–6844 CrossRef CAS PubMed
-
W. Leo, Techniques for nuclear and particle physics experiments: a how-to approach, Springer-Verlag, Berlin, Heidelberg, 2nd edn, 1994 Search PubMed
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 7–9 CrossRef
- T. N. Singh-Rachford, R. R. Islangulov and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3906–3910 CrossRef CAS PubMed
- H.-C. Chen, C.-Y. Hung, K.-H. Wang, H.-L. Chen, W. S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.-P. Hsu and S.-S. Sun, Chem. Commun., 2009, 4064–4066 RSC
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626–1629 CrossRef CAS PubMed
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834–7841 RSC
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209 RSC
- F. Deng, J. Blumhoff and F. N. Castellano, J. Phys. Chem. A, 2013, 117, 4412–4419 CrossRef CAS PubMed
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef CAS PubMed
- X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 2334–2338 CrossRef CAS
- S. H. C. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS PubMed
- A. R. Horrocks and F. Wilkinson, Proc. R. Soc. A, 1968, 306, 257–273 CrossRef CAS
- R. L. Barnes and J. B. Birks, Proc. R. Soc. A, 1966, 291, 570–582 CrossRef CAS
- C. A. Parker and T. A. Joyce, Chem. Commun., 1967, 744–745 RSC
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2015, 54, 6035–6042 CrossRef CAS PubMed
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957–5959 CrossRef CAS PubMed
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC
- S. Baluschev, V. Yakutkin, G. Wegner, T. Miteva, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, Appl. Phys. Lett., 2007, 90, 1–4 CrossRef PubMed
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed
- S. Baluschev, V. Yakutkin, T. Miteva, G. Wegner, T. Roberts, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, New J. Phys., 2008, 10, 013007 CrossRef
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912–5917 CrossRef CAS PubMed
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 CAS
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 4117–4122 CrossRef CAS PubMed
- N. J. Ekins-Daukes and T. W. Schmidt, Appl. Phys. Lett., 2008, 93, 063507 CrossRef PubMed
- J. de Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. van Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835 CAS
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953 CAS
- T. F. Schulze, J. Czolk, Y.-Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, M. J. Crossley, B. Stannowski, K. Lips, U. Lemmer, A. Colsmann and T. W. Schmidt, J. Phys. Chem. C, 2012, 116, 22794–22801 CAS
- T. F. Schulze, Y. Y. Cheng, B. Fckel, R. W. MacQueen, A. Danos, N. J. L. K. Davis, M. J. Y. Tayebjee, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Aust. J. Chem., 2012, 65, 480–485 CrossRef CAS
- J. A. Briggs, A. C. Atre and J. A. Dionne, J. Appl. Phys., 2013, 113, 124509 CrossRef PubMed
- A. Nattestad, Y. Cheng, R. Macqueen, T. Schulze, F. Thompson, A. Mozer, B. Fu, T. Khoury, M. Crossley, K. Lips, G. Wallace and T. W. Schmidt, J. Phys. Chem. Lett., 2013, 4, 2073–2078 CrossRef CAS PubMed
- J.-H. Kim and J.-H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef CAS PubMed
- K. Börjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. A, 2013, 1, 8521–8524 Search PubMed
- C. A. Parker and C. G. Hatchard, Proc. R. Soc. A, 1962, 269, 574–584 CrossRef
- C. A. Parker and C. G. Hatchard, Proc. Chem. Soc., 1962, 1962, 386–387 Search PubMed
- C. A. Parker, Proc. R. Soc. A, 1963, 276, 125–135 CrossRef CAS
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795–1799 CrossRef CAS
- R. Vadrucci, C. Weder and Y. C. Simon, J. Mater. Chem. C, 2014, 2, 2837–2841 RSC
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed
- X. Liu, E. K. L. Yeow, S. Velate and R. P. Steer, Phys. Chem. Chem. Phys., 2006, 8, 1298–1309 RSC
- K. Börjesson, M. Gilbert, D. Dzebo, B. Albinsson and K. Moth-Poulsen, RSC Adv., 2014, 4, 19846 RSC
- S. Kotha, A. K. Ghosh and K. D. Deodhar, Synthesis, 2004, 549–557 CrossRef CAS
- J. Y. Hu, Y. J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064–2071 CrossRef CAS PubMed
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS PubMed
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Rucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS
- E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef PubMed
- J.-H. Ho, Y.-H. Chen, L.-T. Chou, P.-W. Lai and P.-S. Chen, Tetrahedron Lett., 2014, 55, 5727–5731 CrossRef CAS PubMed
- G. Heinrich, S. Schoof and H. Gusten, J. Photochem., 1974, 3, 315–320 CrossRef CAS
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef CAS
- S. Bachilo and R. Weisman, J. Phys. Chem. A, 2000, 104, 7711–7714 CrossRef CAS
- A. Danos, R. W. MacQueen, Y. Y. Cheng, M. Dvorak, T. A. Darwish, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 3061–3066 CrossRef CAS PubMed
-
E. Gazis, UV Atlas of organic compounds, Butterworths, London, vol. III, 1967 Search PubMed
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science, New York, 3rd edn, 2006 Search PubMed
- M. Penconi, F. Ortica, F. Elisei and P. L. Gentili, J. Lumin., 2013, 135, 265–270 CrossRef CAS PubMed
- S. Chattopadhyay, C. V. Kumar and P. Das, Chem. Phys. Lett., 1983, 98, 250–254 CrossRef CAS
- K. Nikitin, H. Müller-Bunz, Y. Ortin, J. Muldoon and M. J. McGlinchey, Org. Lett., 2011, 13, 256–259 CrossRef CAS PubMed
- T. J. Kemp and J. P. Roberts, Trans. Faraday Soc., 1969, 65, 725–731 RSC
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817 RSC
- J. H. Kim, F. Deng, F. N. Castellano and J. H. Kim, Chem. Mater., 2012, 24, 2250–2252 CrossRef CAS
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed
- K. Sripathy, R. W. MacQueen, J. R. Peterson, Y. Y. Cheng, M. Dvorák, D. R. McCamey, N. D. Treat, N. Stingelin and T. W. Schmidt, J. Mater. Chem. C, 2015, 3, 616–622 RSC
- R. Vadrucci, C. Weder and Y. C. Simon, Mater. Horiz., 2015, 2, 120–124 RSC
- A. J. Tilley, M. J. Kim, M. Chen and K. P. Ghiggino, Polymer, 2013, 54, 2865–2872 CrossRef CAS PubMed
- A. J. Tilley, B. E. Robotham, R. P. Steer and K. P. Ghiggino, Chem. Phys. Lett., 2015, 618, 198–202 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra, transient absorption spectra, the Stern–Volmer plot for PdOEP and NMR spectra. See DOI: 10.1039/c5tc02626a |
| This journal is © The Royal Society of Chemistry 2015 |