
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphotropic azobenzene derivatives with oligooxyethylene and glycerol based polar groups†
Xiaoping
Tan‡
a,
Ruilin
Zhang‡
ab,
Chunxiang
Guo
a,
Xiaohong
Cheng
*a,
Hongfei
Gao
a,
Feng
Liu
*c,
Johanna R.
Bruckner
d,
Frank
Giesselmann
*d,
Marko
Prehm
e and
Carsten
Tschierske
*e
aKey Laboratory of Medicinal Chemistry for Natural Resources, Chemistry Department, Yunnan University, Kunming, Yunnan 650091, P. R. China. E-mail: xhcheng@ynu.edu.cn; Fax: +86 871 5032905
bForensic Medicine of Kunming Medical University, Kunming 650500, P. R. China
cState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an, 710049, P. R. China. E-mail: feng.liu@mail.xjtu.edu.cn; Fax: +86 29 82663453
dUniversity of Stuttgart, Institute of Physical Chemistry, Pfaffenwaldring 55, D-075069, Stuttgart, Germany. E-mail: f.giesselmann@ipc.uni-stuttgart.de; Fax: +49 711-685-62569
eInstitute of Chemistry, Organic Chemistry, Martin Luther University Halle-Wittenberg, Kurt-Mothes Str. 2, 06120 Halle/Saale, Germany. E-mail: carsten.tschierske@chemie.uni-halle.de; Fax: +49 345 55 27346
First published on 29th September 2015
Abstract
A series of amphiphilic azobenzenes with one to three lipophilic alkyl chains at one end and polar groups with oligooxyethylene (EO) and racemic 3-glyceryl units at the opposite end was synthesized and their thermotropic and lyotropic liquid crystalline self-assemblies were studied by POM, DSC and XRD. Tilted and non-tilted lamellar phases with interdigitated double layer structures (SmCd and SmAd, respectively) were found for the compounds with a single alkyl chain, whereas hexagonal columnar phases were formed by the compounds with two or three alkyl chains. The effect of protic solvents, like formamide, ethylene glycol and water, was investigated for representative examples. For the compounds with the single chain, induction and stabilization of SmA phases were observed, though broad regions of lyotropic SmC phases were retained in most cases. Depending on the structure of the polar group, the hexagonal columnar phases were either removed or drastically stabilized by the solvents. Photoisomerisation of an azobenzene chromophore was also studied.
1. Introduction
Design of molecules which can self-assemble into ordered nanostructures is of current interest for the general understanding of the processes of molecular self-assembly.1 Amphiphilic and anisometric compounds have aroused special interest as they can organize into ordered liquid crystalline (LC) superstructures with surprising complexity and great interest for materials science as well as life science.2 By combining an anisometric molecular shape and an amphiphilic structure, both representing structural concepts supporting LC self-assembly, new amphotropic molecules can be obtained.3–5 Such molecules can display thermotropic and lyotropic LC phases and form well defined monolayers at interfaces.12 Typical examples are amphotropic LCs involving biphenyl,6–8 phenylpyrimidine9,10 and other rigid cores11,12 terminated with alkyl chains at one end and oligooxyethylene (EO) or glycerol-based groups at the other end.5 Amphotropic LCs involving EO chains and glycerol units are potentially useful, for example, for design of nanostructured 1D, 2D or 3D ion conducting materials in combination with ionic liquids.13 On the other hand, LC azobenzene derivatives have attracted attention because of their photoresponsive properties caused by the trans–cis photoisomerization of the azobenzene units.14 Therefore, a wide diversity of azobenzene derivative LCs have been designed to introduce light-modulated functionalities, for example for application as organic light-driven actuators.15,16 Photoisomerizable amphiphiles can find applications in photoswitchable surfactants,17 for drug delivery,18 switchable vesicles19 and controlled gelation.20 Considering the vast variety of useful properties and potential applications, photoswitchable azobenzene based amphiphiles21–27 and especially photoswitchable amphotropic LCs28–30 have received comparatively little attention to date. Herein we report the design, synthesis and thermotropic as well as lyotropic liquid crystalline self-assembly of new amphiphilic azobenzenes, consisting of EO31 or glycerol based polar groups or combinations of both involving one or two hydroxyl groups in the polar groups, and having one to three lipophilic n-alkyl chains at the opposite end.In the notation of the compounds 1–3 (1nEOmOH to 3nEOmOH and 1nEOm(OH)2 to 3nEOm(OH)2, see Scheme 1), the first number indicates the number of terminal n-alkyl chains and the superscript n represents the length of these chains. Note that in compounds of type 1 the single alkoxy chain is directly attached to the azobenzene core whereas for compounds of types 2 and 3 with more than only one alkoxy chain these chains are attached via an additional benzylether unit to the azobenzene core. EO indicates the presence of an EO chain with a number m of EO units in this chain. At the end OH denotes the presence of a single terminal hydroxyl group, whereas (OH)2 indicates a racemic 3-glyceryl unit. Compounds 1n(OH)2–3n(OH)2 have the 3-glyceryl units directly connected to the azobenzene core by an ether linkage.
 | ||
| Scheme 1 Synthesis of compounds 1–3. Reagents and conditions: (i) KOH, H2O, 195–200 °C, 5 h; (ii) TsCl, CH2Cl2, Et3N, 0–25 °C; (iii) K2CO3, DMF, 90 °C, 12 h; (iv) CH3CN, K2CO3, 70 °C, 24 h; (v) allyl bromide, NaH, THF, 70 °C, 12 h; (vi) OsO4, NMMNO, H2O, acetone, 25 °C, 24 h. | ||
2. Results and discussion
2.1. Synthesis
The synthesis of the compounds under discussion is shown in Scheme 1. The key intermediate is azobenzene-4,4′-diol C, which was prepared based on the method of Willstätter and Benz from 4-nitrophenol32 and monoalkylated with appropriate n-bromoalkanes or monobenzylated with alkoxysubstituted benzyl chlorides, leading to the 4′-alkoxysubstituted azobenzene-4-ols 1nOH and the 4′-benzyloxy-azobenzene-4-ols 214OH and 3nOH, respectively. Compounds 1nOH and 3nOH were alkylated using tri- and tetraethylene glycol monotosylates EOmTs yielding compounds 1nEOmOH and 3nEOmOH. These compounds and the azobenzene-4-ols 1nOH–3nOH were etherified with allylbromide, followed by dihydroxylation of the double bonds using OsO4/N-methylmorpholine-N-oxide (NMMNO).33 The obtained target compounds, the alcohols 1nEOmOH and 3nEOmOH and the glycerol ethers 214(OH)2, 3n(OH)2 and 3nEOm(OH)2 were purified by column chromatography or crystallization from ethyl acetate. Detailed synthetic procedures and analytical data are given in the ESI.†2.2 Thermotropic self-assembly of the single chain compounds 1 in SmA and SmC phases
The phase transition temperatures and enthalpies of the azobenzene derivatives with a single alkyl chain (compounds 1) are listed in Tables 1 and 2. No LC phases were found for the EO ethers with only one OH group and comparatively short hexyl chains (compounds 16EOmOH with m = 3, 4). Elongation of the alkyl chain leads to SmC phases. All compounds with terminal glycerol groups show smectic phases with enhanced stability compared to the compounds with only a single OH group. Among them, compounds 1n(OH)2 without EO units have enhanced mesophase stability compared to the related compounds 1nEOm(OH)2 with additional EO units. SmC phases were found in all cases and for most compounds with glycerol groups, additional SmA phases are formed above the SmC phases. The SmA–SmC transitions have comparatively large enthalpy values (ΔH = 1.3–2.6 kJ mol−1) for this kind of transition and thus this phase transitions appear to be first order. Fig. 1 shows representative DSC traces for compound 112(OH)2 with a SmC–SmA sequence. Interestingly, the Sm–Iso transition enthalpy values of the glycerol derivatives, having two terminal OH groups, are only about one half of those of the amphiphiles with only one hydroxyl group (compare Tables 1 and 2), which is discussed later.|
|
|||
|---|---|---|---|
| Comp | R1 | m | Phase transitions |
| a Transition temperature and enthalpy changes (in square brackets) were determined by DSC (peak temperature, first heating scan, 10 °C min−1); abbreviations: Cr = crystalline solid; SmCd = tilted smectic phase with a double layer structure; Iso = isotropic liquid state. b Supercooling was possible to 95 °C. | |||
| 16EO3OH | C6H13O | 3 | Cr 102 [51.5] Iso |
| 16EO4OH | C6H13O | 4 | Cr 104 [49.5] Isob |
| 112EO3OH | C12H25O | 3 | Cr 97 [35.6] SmCd 106 [12.3] Iso |
| 112EO4OH | C12H25O | 4 | Cr 96 [42.6] SmCd 113 [16.5] Iso |
| 118EO3OH | C18H37O | 3 | Cr 106 [52.1] SmCd 114 [16.1] Iso |
|
|
|||
|---|---|---|---|
| Comp | R1 | m | Phase transitions |
| a SmAd = non-tilted smectic phase with a double layer structure; for conditions and other abbreviations, see Table 1. | |||
| 112(OH)2 | C12H25O | 0 | Cr 156 [30.8] SmCd 168 [1.3] SmAd 176 [5.1] Iso |
| 114(OH)2 | C14H29O | 0 | Cr 144 [31.1] SmCd 166 [2.6] Iso |
| 112EO3(OH)2 | C12H25O | 3 | Cr 94 [18.6] SmCd 129 [2.6] SmAd 131 [6.0] Iso |
| 114EO3(OH)2 | C14H29O | 3 | Cr 95 [41.9] SmCd 121 [2.0] SmAd 126 [7.4] Iso |

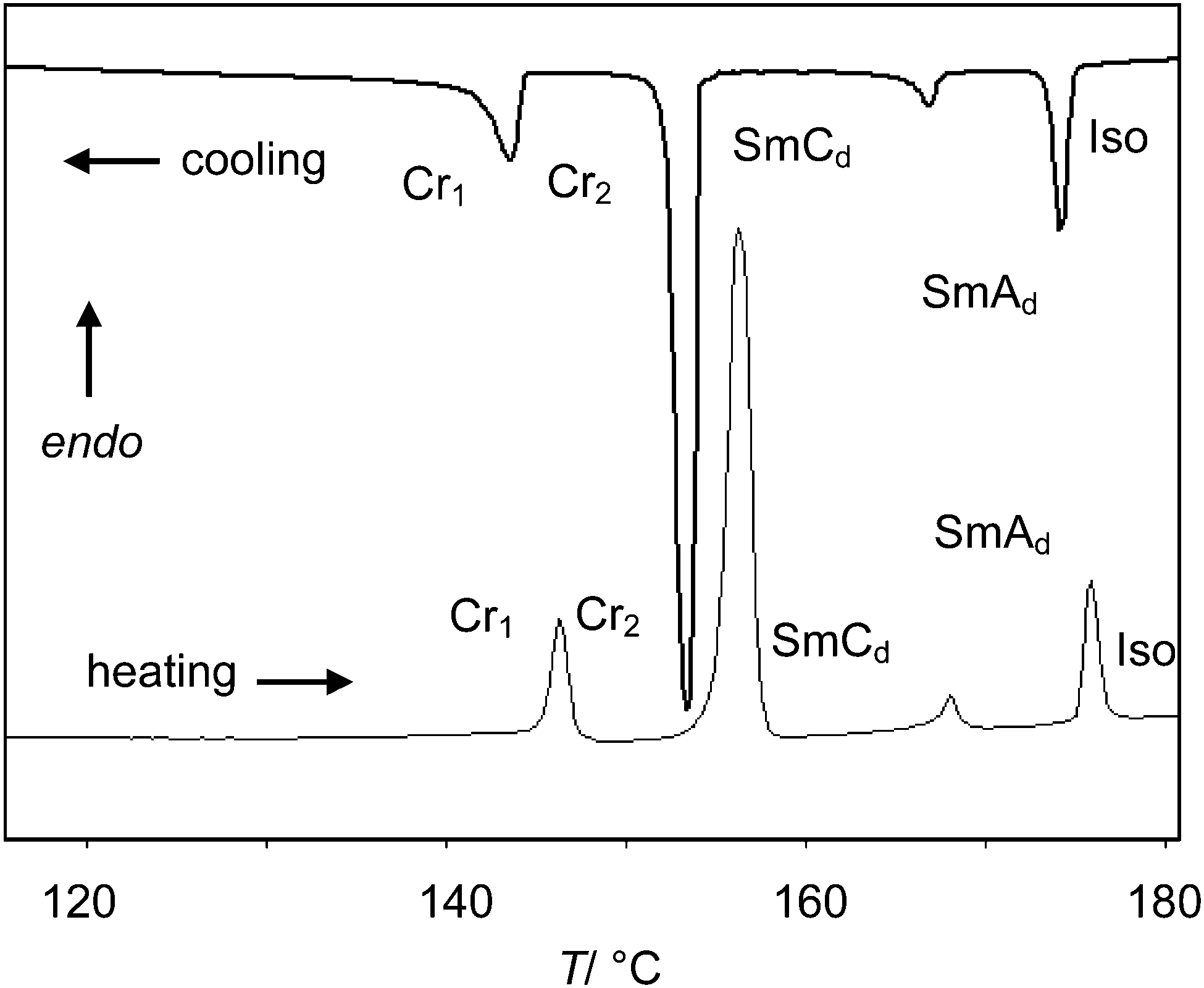 | ||
| Fig. 1 DSC traces of 112(OH)2 (10 K min−1). | ||
By microscopic investigation between non-treated glass plates through crossed polarizers, the smectic C phases of these compounds occur with typical broken fan and SmC schlieren textures34–36 (see Fig. 2a) and the SmA phases are characterized by focal-conic fan-like textures and pseudoisotropic regions (Fig. 2b), often separated by oily streaks.
 | ||
| Fig. 2 Textures of 112(OH)2 in the (a) SmCd phase at T = 167 °C and (b) in the SmAd phase at T = 169 °C as observed upon heating; at the right homeotropic alignment and at the left predominately planar alignment with fan texture. | ||
Compounds 112EO3OH and 112(OH)2 were investigated as representative examples by X-ray diffraction (XRD; see Fig. 3). Diffuse wide angle scattering with maxima at d = 0.45–0.47 nm, corresponding to the mean lateral distance between the molecules, indicate fluid smectic phases without in-plane order for all investigated compounds. This excludes the presence of other low temperature smectic mesophases37 and thus assignment to SmA and SmC was confirmed by XRD. The sharp fundamental and second-order pseudo-Bragg peaks in the small-angle regime originate from the long-range lamellar order along a single direction. For compound 112EO3OH the layer reflection has a tilt angle of θ = 29° with respect to the maxima of the diffuse wide angle scattering, indicating the presence of a synclinic SmC phase with an average tilt angle of 29° (see Fig. 3c). The layer spacing is d = 5.57 nm while the length of the molecule is only Lmax = 3.7 nm (calculated by the MM2 method38 and assuming a molecule in the most stretched conformation with all-trans conformation for the alkyl chains and EO chains39). Thus the layer thickness is significantly larger than the molecular length (d/L = 1.5). Considering the tilt, the effective molecular length is reduced to cos 29° Lmol = 3.24 nm, leading to the effective d/L ratio of 1.72. This means that the layer distance is quite a bit smaller than twice the effective molecular length, indicating a bilayer structure with a partial intercalation of the alkyl chains as shown in Fig. 3d (the SmCd phase). The degree of alkyl chain intercalation could become smaller if there is strong disorder of the alkyl chains and especially of the EO chains.
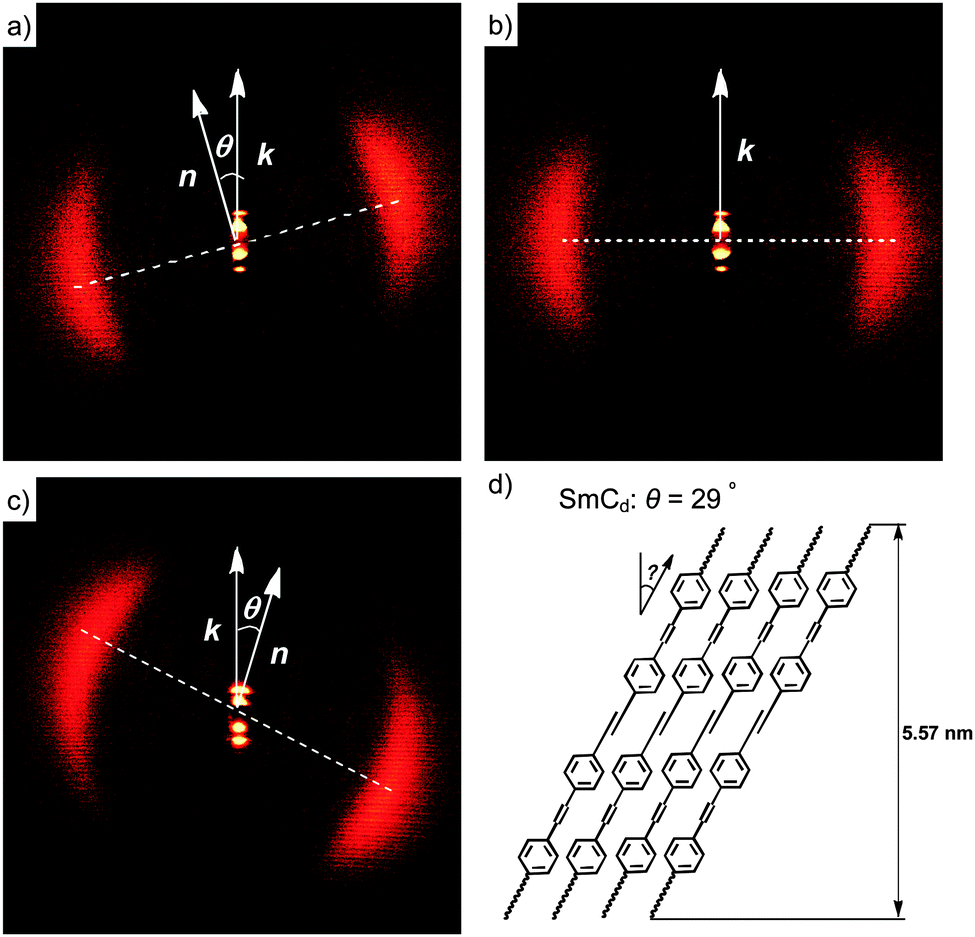 | ||
| Fig. 3 XRD patterns of the smectic phases after subtraction of the scattering in the isotropic liquid states: (a) 2D-XRD pattern of a surface aligned sample of compound of 112(OH)2 in the SmCd phase at T = 167 °C and (b) in the SmAd phase at T = 169 °C and (c) 2D pattern of a surface aligned sample of the SmC phase of 112EO3OH at T = 105 °C; (d) shows a model of the molecular organization in one layer of the double layer SmC phase with partial intercalation of the alkyl chains; complete intercalation would lead to d ∼ 5.2 nm. For additional crystallographic data, see Tables S1–S3 (ESI†). | ||
In the SmC phase of the glycerol derivative 112(OH)2 with the same alkyl chain length, but without EO units the thickness of the smectic layers is d = 5.16 nm at 167 °C and the tilt angle is only 12° (Fig. 3a). The layer thickness decreases to 5.05 nm at the transition from the SmA phase to the SmC phase at 167 °C, in line with the emerging uniform tilt angle of only 12°. The ratio d/L = 1.56 (Lmax = 3.3 nm) also indicates an intercalated bilayer structure for the SmC and SmA phases (SmCd and SmAd phases) with a degree of intercalation being very similar to that found in the SmCd phase of compound 112EO3OH (Table 3).
| Comp. | Phase | d/nm (T/°C) | L/nm |
|---|---|---|---|
| 112(OH)2 | SmAd | 5.16 (169) | 3.3 |
| SmCd | 5.05 (167) | 3.3 | |
| 112EO3OH | SmCd | 5.57 (105) | 3.7 |
XRD investigation of the Iso phases occurring immediately above the smectic–Iso phase transition of compounds 112EO3(OH)2 and 112EO3OH, as shown in Fig. 4, indicates a higher intensity of the diffuse small angle scattering in the Iso phase of the glycerol derivative 112EO3(OH)2 compared to 112EO3OH having only one OH group. This indicates the formation of clusters in the Iso phase of 112EO3(OH)2 which is much weaker for compound 112EO3OH with only one terminal OH group. This could contribute to the observation that the enthalpy values of the Sm–Iso transition of the glycerol substituted compounds are only one half or even less than those measured for the related compounds with a single OH group (see Tables 1 and 2). It appears that for 112EO3(OH)2 significant cybotactic clusters are already formed in the Iso phase which then fuse to infinite layers at the Iso–Sm phase transition, whereas for 112EO3OH the clusters are much smaller and just at the Iso–Sm transition layer formation starts with much smaller clusters, thus leading to a larger enthalpy for the phase transition.
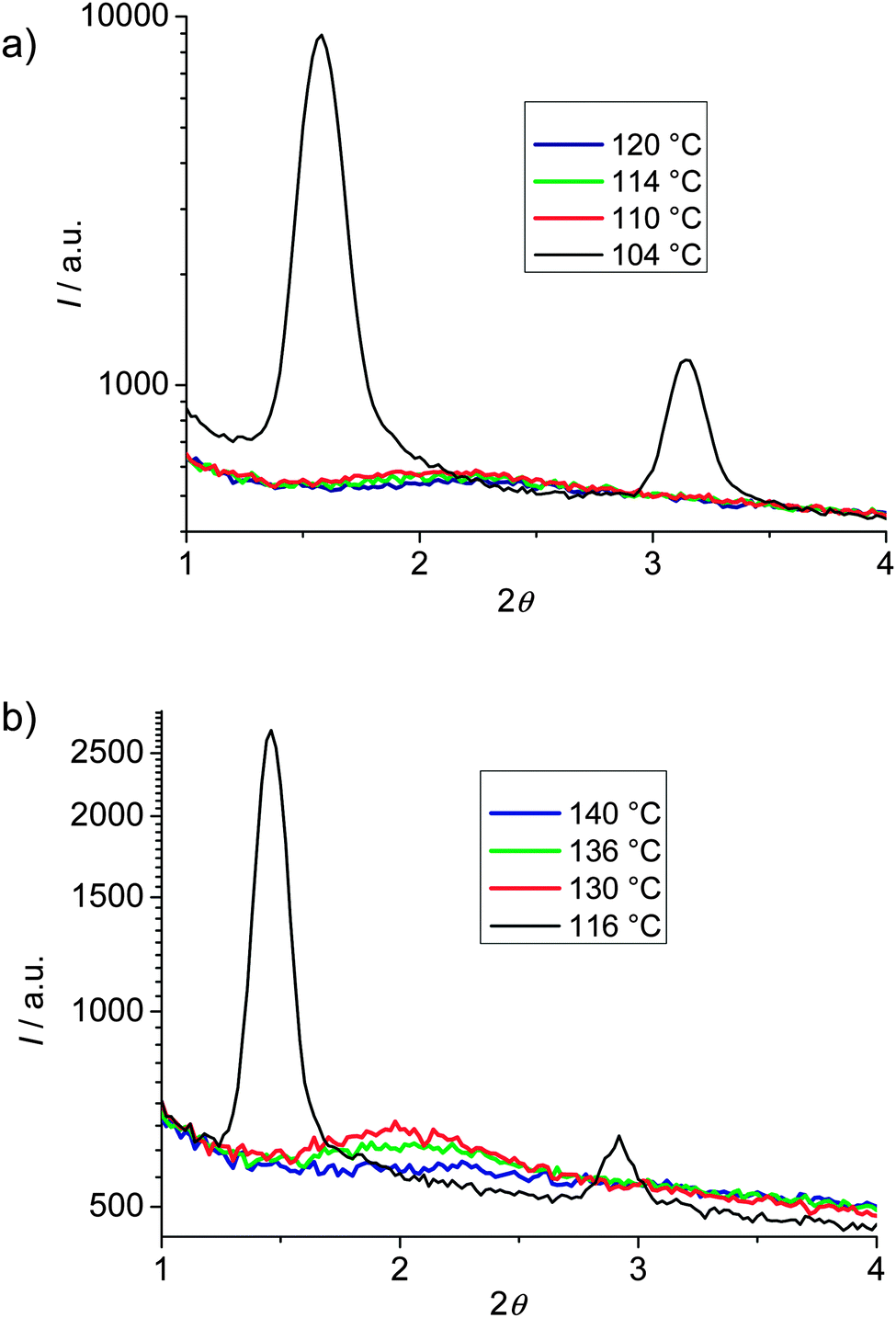 | ||
| Fig. 4 Small angle XRD patterns of compounds (a) 112EO3OH and (b) 112EO3(OH)2 at the indicated temperatures in the smectic phase (black lines) and at different temperatures in the Iso phase in proximity to the SmAd–Iso transition (colored lines), showing a significant cybotaxis for 112EO3(OH)2 being much weaker for 112EO3OH. | ||
2.3 Thermotropic self-assembly of compounds 2 and 3 with two and three alkyl chains in columnar LC phases
Double and triple chain benzylether 2 and 3 are collated in Table 4. Except for compound 314EO3OH, other compounds show exclusively hexagonal columnar phases (Colhex). In general the stability of LC phases increases with increasing number of OH groups. The Colhex–Iso transition enthalpies are in the range between ΔH = 0.4 and 1.5 kJ mol−1, which is about one order of magnitude smaller than those for the Sm–Iso transitions of compounds 1, and there is obviously no dependence of the Col–Iso transition enthalpy on the number of OH groups.|
|
||||||
|---|---|---|---|---|---|---|
| Comp | R1 | R2, R3 | m | Phase transitions | a/nm (T/°C) | n cell |
| a Abbreviations: Colhex = hexagonal columnar phase with p6mm symmetry; for conditions and other explanations see Table 1. b No LC phase was observed upon cooling before crystallization at T = 62 °C. | ||||||
| 214(OH)2 | H | OC14H29 | 0 | Cr 115 [25.1] Colhex 165 [1.5] Iso | 6.65 (140) | 13.0 |
| 312(OH)2 | OC12H25 | OC12H25 | 0 | Cr 59 [34.4] Colhex 119 [0.7] Iso | 6.20 (90) | 9.6 |
| 314(OH)2 | OC14H29 | OC14H29 | 0 | Cr 64 [36.7] Colhex 115 [0.4] Iso | ||
| 312EO3OH | OC12H25 | OC12H25 | 3 | Cr 74 [57.9] (Colhex 70 [0.4]) Iso | ||
| 314EO3OH | OC14H29 | OC14H29 | 3 | Cr 78 [55.3] Isob | ||
| 312EO3(OH)2 | OC12H25 | OC12H25 | 3 | Cr 66 [49.6] Colhex 115 [0.9] Iso | 7.17 (90) | 11.3 |
| 314EO3(OH)2 | OC14H29 | OC14H29 | 3 | Cr 71 [39.5] Colhex 116 [0.5] Iso | ||

There is a strong effect of the number of alkyl chains, reducing the mesophase stability with growing chain number (compare compounds 214(OH)2 and 314(OH)2 in Table 4). In contrast, the effects of the EO unit between the azobenzene core and the glycerol unit on melting temperatures and mesophase stability are only marginal (compare compounds 3nEO3(OH)2 and 3n(OH)2). The textures of the columnar mesophases, as observed between crossed polarizers, are characterized by ‘spherulitic’ domains (see Fig. 5a, Fig. S1a and c, ESI†), combined with regions appearing completely dark, where the optic axis is normal to the LC film (homeotropic regions), see Fig. S1b (ESI†). These textures indicate an optically uniaxial columnar LC phase The orientation of the π-conjugated aromatic cores is deduced from the colour of the fans in the micrograph taken with a λ-retarder plate (Fig. 5b and Fig. S1c, ESI†). The directions of yellow and blue fans with respect to the indicatrix of the retarder confirm the orientation of the high-index axis as radial to the fans. As the columns are tangential and the high-index axis is parallel to the π-conjugated aromatic cores. This means that in the columns the preferred direction of the intra-molecular π-conjugation pathway (i.e. the long axis of the azobenzene cores) is on average perpendicular to the column long axis, in line with the proposed mode of self-assembly (see below).
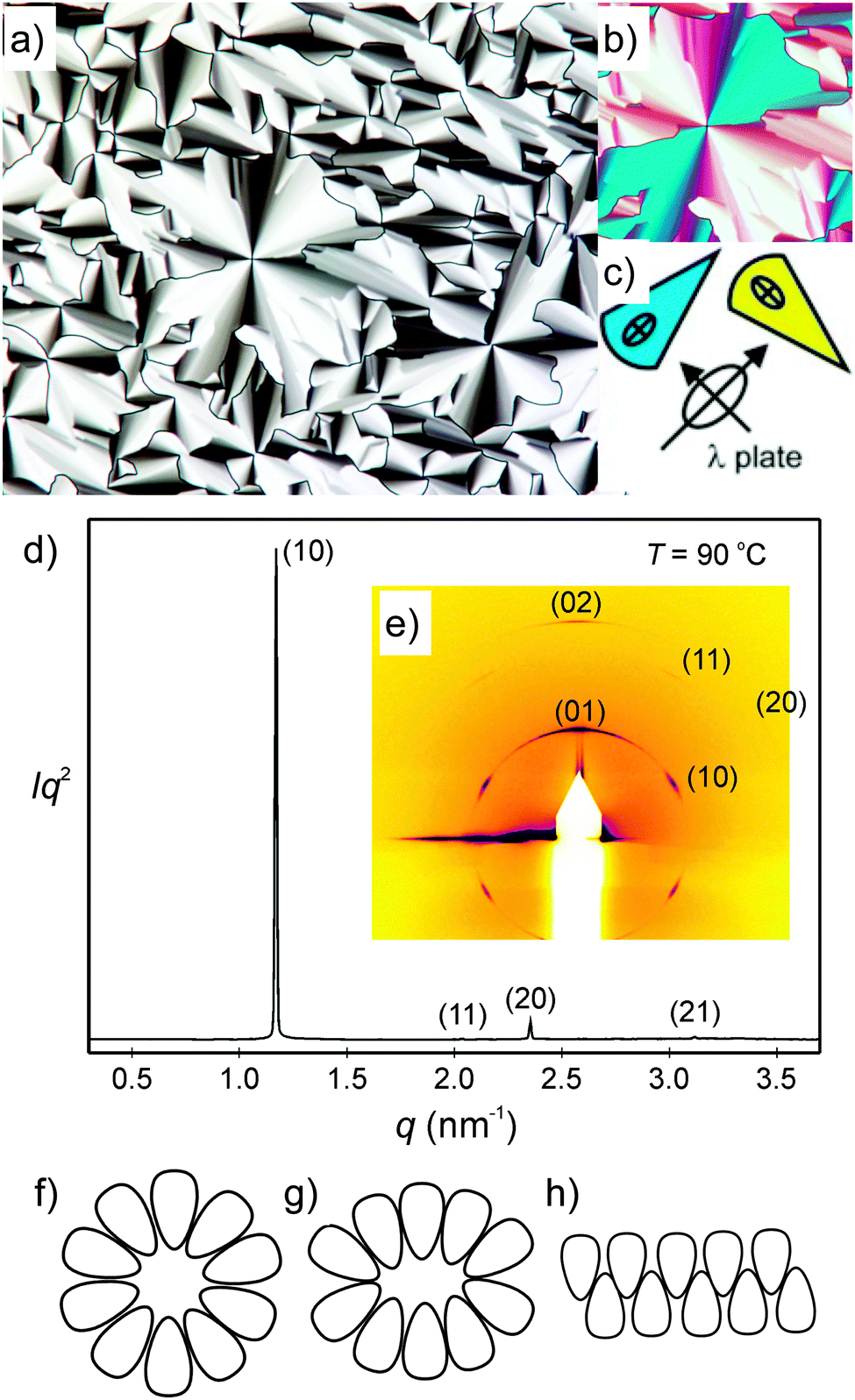 | ||
| Fig. 5 Investigation of compound 312(OH)2 (a) texture of the columnar phase at T = 90 °C as observed between crossed polarizers; (b) shows a section of the texture with an additional λ-retarder plate and the orientation of the indicatrix of the λ-plate is shown in (c); (d) powder X-ray diffraction pattern at T = 90 °C and (e) GISAXS pattern of a surface-aligned sample at T = 90 °C; (f–h) models showing the organization of the amphiphilic molecules, (f) in circular columns, (g) in elliptical columns and (h) in ribbons; for more details, see Tables S4–S7 and Fig. S2 (ESI†). | ||
The columnar phases of the double chain glycerol substituted compound 214(OH)2, the three chain compound 312(OH)2 and the related compound with additional (EO)3 unit 312EO3(OH)2 were investigated as representative examples by small-angle X-ray scattering (SAXS), and grazing-incidence small-angle X-ray scattering (GISAXS) on thin surface-aligned films on silicon using a synchrotron source (see Fig. 5d, e and Fig. S2, ESI†). All columnar phases show three or four small angle reflections with a ratio of their reciprocal spacing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31/2:2:(71/2) which can be indexed to the 10, 11, 20 (and 21) reflections of a hexagonal lattice with p6mm symmetry (Fig. 5d and Fig. S2c, ESI†). The 2D pattern of an aligned sample of compound 312(OH)2 also confirms this phase assignment (Fig. 5e and Fig. S2b, ESI†). Although SAXS was not carried out on all of the columnar phases, based on textural similarities (Fig. S1, ESI†), it is very likely that the columnar phases of the other compounds represent the hexagonal columnar phases too. The number of molecules organized in a slice of the columns with a height of h = 0.45 nm (a typical value for the maximum of the diffuse wide angle scattering) ncell was estimated using eqn (1) and assuming a density of ρ = 1 g cm−3 (NA = Avogadro constant; M = molecular mass, see Table 4). The number decreases from ncell = 13 for the two-chain compound 214(OH)2 to ncell = 10 for the three-chain compound 312(OH)2. Introduction of the (EO)3 unit at constant alkyl chain volume leads to a slight increase of the number of molecules in the column cross section from ncell = 10 to 11 for compound 312EO3(OH)2.
:31/2:2:(71/2) which can be indexed to the 10, 11, 20 (and 21) reflections of a hexagonal lattice with p6mm symmetry (Fig. 5d and Fig. S2c, ESI†). The 2D pattern of an aligned sample of compound 312(OH)2 also confirms this phase assignment (Fig. 5e and Fig. S2b, ESI†). Although SAXS was not carried out on all of the columnar phases, based on textural similarities (Fig. S1, ESI†), it is very likely that the columnar phases of the other compounds represent the hexagonal columnar phases too. The number of molecules organized in a slice of the columns with a height of h = 0.45 nm (a typical value for the maximum of the diffuse wide angle scattering) ncell was estimated using eqn (1) and assuming a density of ρ = 1 g cm−3 (NA = Avogadro constant; M = molecular mass, see Table 4). The number decreases from ncell = 13 for the two-chain compound 214(OH)2 to ncell = 10 for the three-chain compound 312(OH)2. Introduction of the (EO)3 unit at constant alkyl chain volume leads to a slight increase of the number of molecules in the column cross section from ncell = 10 to 11 for compound 312EO3(OH)2.
 | (1) |
These two- and triple-alkyl chain amphotropic azobenzene derivatives can be either considered as taper shaped molecules,40,41 providing a radial organization of the molecules in circular columnar aggregates, thus, directly leading to a packing of the circular columns on a hexagonal lattice (see the model in Fig. 5f), or as polycatenar (three- and tetracatenar, respectively) mesogens, which can organize into ribbons of antiparallel organized molecules, resembling the organization in the smectic phases of the single chain compounds (see the model in Fig. 5h).42,43 In the latter case the time and space averaging of the ribbon local orientation would lead to the globally averaged hexagonal lattice. Probably the truth is somewhere in the middle, i.e. the columns have an elliptical shape which retains some ribbon-like organization in the middle and simultaneously the contact between polar groups and aliphatic chains in the periphery is minimized by fusing the aromatic regions to elliptical shells covering the polar regions (Fig. 5g). These elliptical columns are then arranged on a time and space averaged hexagonal lattice. This model is in line with the reconstructed electron density maps (see Fig. 6a and c) where the column centers with high electron density (purple) are surrounded by medium electron density shells (blue/green/yellow). The medium electron density shells might result from the rotational disorder of the elliptical columns leading to a space and time averaging of the high electron density polar cores and the low electron density alkyl chains in the continuum around the columns. In the column cores only the aromatic/polar cores of the elliptical columns overlap on the space and time average, leading to high electron density. Independent of the precise shape of the columns the rigid aromatics, the polar groups (polyether and glycerol unit) and the non-polar alky chains segregate into distinct domains. The polar units form the column centers, which are surrounded by the azobenzene cores and assembled on a hexagonal lattice within the lipophilic continuum mainly formed by the alkyl chains. The three alkyl chains of 312(OH)2 provide a stronger interface curvature leading to a 30% decrease of the number of molecules organized in the cross section of the columns (ncell = 10) compared to the double chain compound 214(OH)2 (ncell = 13). Enlargement of the polar group by the (EO)3 chain increases the polar volume and this reduces the interface curvature, thus leading to a slight increase in the number of molecules organized in the cross section of the columns (ncell = 11 for 312EO3(OH)2).
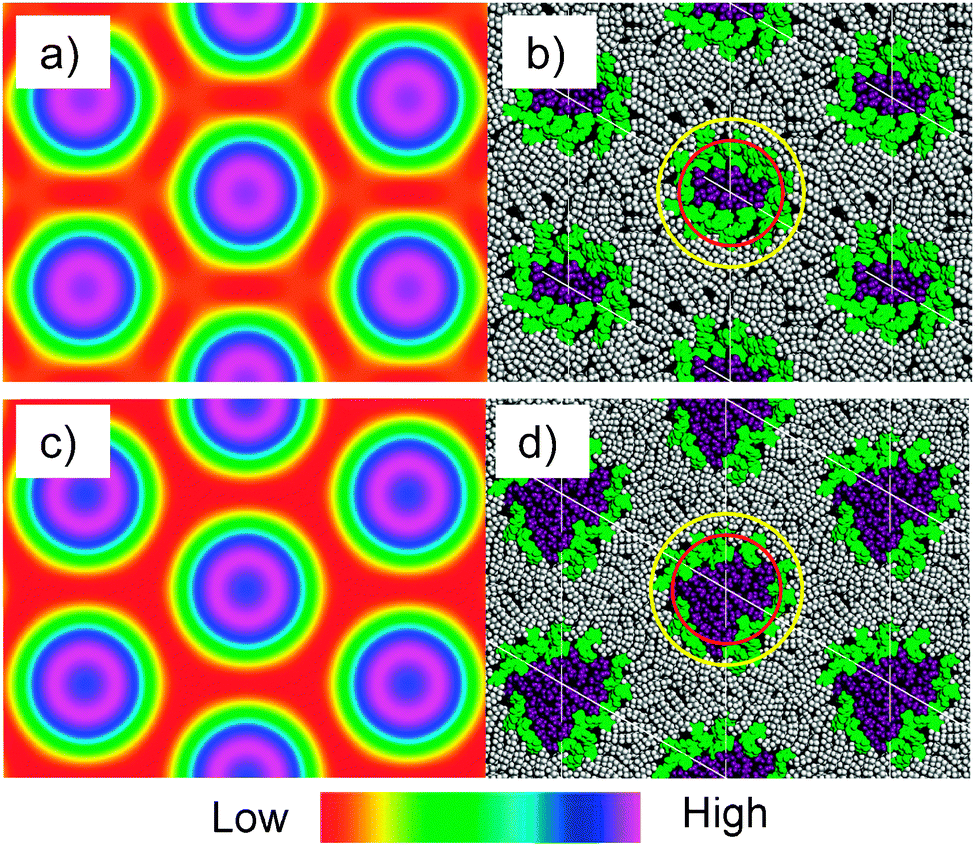 | ||
| Fig. 6 Electron density maps of the Colhex phases of (a) compound 312(OH)2 and (c) compound 312EO4(OH)2 as reconstructed from the synchrotron powder diffraction patterns (color code: purple/blue = high, green/yellow = medium and red = low electron density; for details of the reconstruction, see the ESI†); (b) snapshot of the structure of the Colhex phase of 312(OH)2 and (d) of 312EO3(OH)2 after MD annealing (polar groups are colored purple and the aromatic cores green, alkyl chains gray); red and yellow circles represent the boundaries between high and medium and medium and low electron density regions in the electron density maps, respectively. | ||
In Fig. 6 the reconstructed electron density maps of the Colhex phases of compounds 312(OH)2 and 312EO3(OH)2 (left) are compared with the molecular dynamics (MD) annealed models of the columnar phases (right). The MD annealed model of the columnar phase of 312EO3(OH)2 (Fig. 6d) indicates that the polar chains form the majority of the column cores and the azobenzenes form a kind of shell separating the polar core from the aliphatic continuum, in which the alkyl chains are completely disordered and partly interdigitated. The formation of this kind of aromatic shell is favored by the possibility of hydrogen bonding of the OH groups to the azo groups, to the electron rich aromatic π-systems and to the ether oxygens at the aromatics. The MD annealed model of the columnar phase of 312(OH)2 (Fig. 6b) without EO units shows better defined shells of the aromatic cores with a larger degree of parallel alignment of the cores around the smaller core regions involving the polar groups (structure corresponding to the model in Fig. 5g).
2.4 Effects of solvents and lyotropic self-assembly
The effect of protic solvents on the phase behavior of 112EO3OH as a representative example was investigated by POM and XRD. By adding ethylene glycol, the SmC phase of this compound is first stabilized, but already at nEO ∼ 1 (nEO = n(EO)/n(112EO3OH)) the stability of the SmC phase decreases and a SmA phase occurs (Fig. 7). With further increase in solvent concentration, the SmC–SmA transition temperature decreases whereas the SmA phase stability increases. The SmC phase exhibits a typical schlieren texture as observed for the dry compound and the SmA phase shows oily streaks and homeotropic areas as characteristic of lyotropic SmA phases (Fig. 8). At nEO ∼ 4 the maximum of the phase stability is reached and with a further increase in solvent concentration the transition temperatures slightly decrease up to nEO ∼ 11 which represents the maximum uptake of the solvent (Fig. 7). After reaching this concentration the system remained heterogeneous with excess solvent coexisting with the lyotropic LC system. | ||
| Fig. 7 Phase diagram of the system 112EO3OH/ethylene glycol (EO). | ||
 | ||
| Fig. 8 Texture of the solvent induced lyotropic SmA phase of 112EO3OH at T = 105 °C in a mixture with ethylene glycol (EO) at nEO = 10. | ||
The system was in addition investigated by XRD for the concentrations nEO ∼ 1 (∼10 wt% EO), nEO ∼ 3 (∼30 wt% EO) and nEO ∼ 10 (∼50 wt% EO). The XRD patterns showed the typical diffraction patterns of smectic phases. There is a significant increase of the d-spacings with increasing solvent content, in line with a swelling of the polar layers by the added solvent molecules. Remarkably, the layer shrinkage at the SmA–SmC transition and in the temperature range of the SmC phase increases with increasing solvent content. In contrast, the layer spacing d remains nearly constant in the SmC phase region of the solvent-free sample (see Fig. 9). This could have different reasons, for example, the solvation of the head groups could change, i.e. the number of molecules separating the head groups laterally increases and simultaneously the number of solvent molecules arranged between the layers decreases with decreasing temperature. This could further lead to a stronger intercalation of the alkyl chains and an increase of the uniform (SmC) or average tilt (SmA) of the aromatic at lower temperature. The combined contribution of these effects is thought to lead to the observed effect.
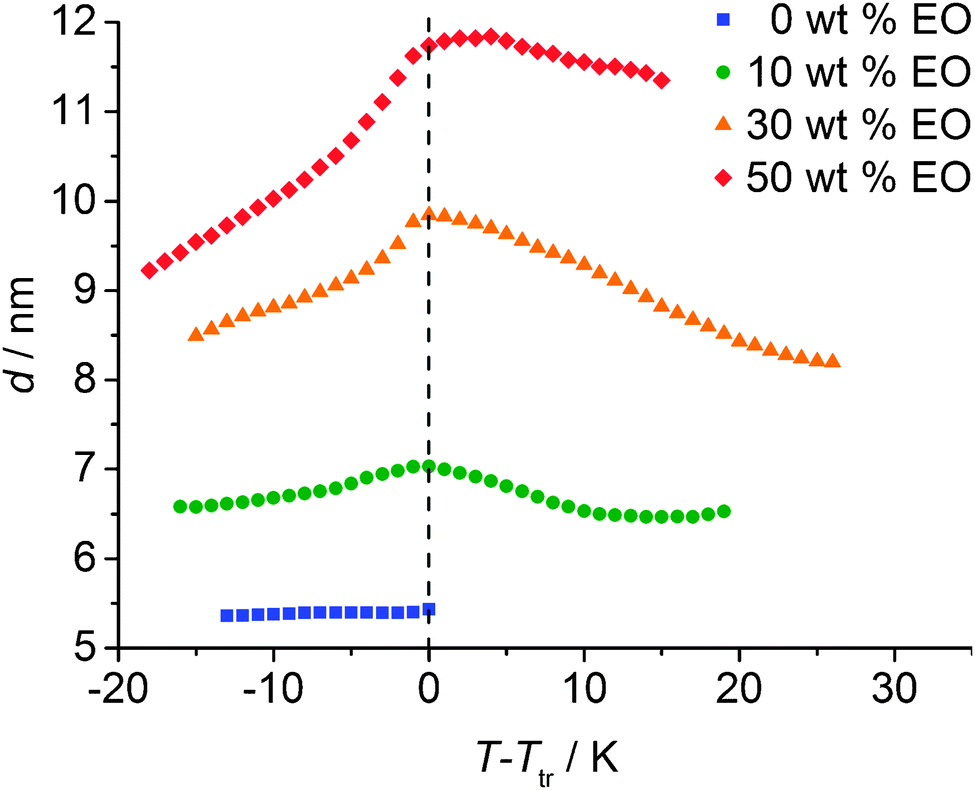 | ||
| Fig. 9 Temperature dependence of the layer spacing depending on the EO content of the system 112EO3OH/ethylene glycol (EO). | ||
A similar behaviour to that with EO was also observed using water and formamide, which were investigated by contact preparation. For this purpose the amphiphile and the solvent were placed side-by-side between two glass plates and the phases developing in the contact region due to the developing concentration gradient were observed under a polarizing microscope. In all cases a SmA phase was induced and an SmC–SmA–Iso phase sequence was found for the solvent enriched samples. For formamide a bit higher transition temperatures were observed for the solvent saturated sample (Cr 77 °C SmC 90 °C SmA 135 °C Iso), compared with EO, whereas for water the observed transition temperatures are lower (Cr 78 °C SmC 80 °C SmA 97 °C Iso for the water saturated sample). This is a bit surprising, as usually water provides the most efficient polar protic solvent, leading to the highest lyotropic phase stabilities.5
Additional compounds were investigated using formamide as the protic solvent. In the case of 112EO3(OH)2 with an additional glycerol group there is an even stronger stabilization of the SmA phase, reaching about 180 °C for the formamide saturated sample. This formamide saturated sample crystallizes at 68 °C without formation of a SmC phase, i.e. there is a stronger destabilization of the tilted SmC phase. Compound 312(OH)2 with a glycerol unit but without the EO segments behaves similar, the SmA phase being stabilized to >200 °C, but the formamide saturated sample rapidly crystallizes already at 123 °C before the transition of the SmA to the SmC phase can take place.
The lyotropic behavior of the three chain compounds is very different and for these compounds a strong dependence of the phase sequence on the type of polar group is observed. Compounds 312EO3(OH)2, 312EO3OH and 312(OH)2 having identical alkyl chain lengths but different types of polar groups were investigated as representative examples using formamide as the polar protic solvent (contact preparations). The columnar phases of 312(OH)2 and 312EO3OH, having either the glycerol group or the triethyleneglycol chain, were completely removed in the contact region and only crystallization was observed at T = 50 °C for both compounds. In contrast, for compound 312EO3(OH)2, combining the EO chain with the glycerol group, a dramatic stabilization of the Colhex phase was observed (see Fig. 10), it is retained up to the boiling point of this solvent at 210 °C. It appears that the molecular organization in columns with a polar interior restricts the effect of the solvent on the self assembly. In the case of relatively small polar groups the volume added by the solvent molecules to the polar groups distorts the formation of columnar aggregates, but it appears to be too small for a transition to a lamellar organization; therefore order is lost and an isotropic liquid is formed. For compound 312EO3(OH)2 with the largest polar group, involving a strong hydrogen bond donor group (the glycerol units) and having several additional hydrogen bonding acceptor sites along the EO chains, a small number of formamide molecules might be sufficient to dramatically increase the number of attractive hydrogen bonds, thus stabilizing the columnar aggregates without requiring too much additional space, and thus retaining the interface curvature. Moreover, as shown in Fig. 6d, the major part of the columns is formed by the polar groups, i.e. polar–apolar segregation is dominating the behaviour of 312EO3(OH)2. The azobenzenes being strongly tilted and some of them organized nearly parallel to the column surface form a relatively thin shell. This might reduce the sensitivity to steric distortions of columnar self assembly and could lead to the very special behavior of this compound.
 | ||
| Fig. 10 Texture of the solvent stabilized lyotropic Colhex phase of 312EO3(OH)2 (right) at T = 170 °C in the contact region with formamide (left) as observed upon cooling, the inset shows the texture with the additional λ-plate. | ||
Investigation of aqueous systems was more difficult due to easy water evaporation, but in principle the behaviour can be considered as similar to formamide. So the Colhex phase of 312EO3(OH)2 is retained in the contact region in the whole temperature region available for investigation, whereas for 312EO3OH and 312(OH)2 the columnar phases are removed for the water saturated samples. It appears that for 312(OH)2 an additional lamellar phase is induced in the water saturated sample with a clearing temperature of 71 °C, but this is not completely certain due to the above mentioned difficulties in handling these amphiphile water systems.
2.5 Photoisomerization behavior
All compounds 1–3 exhibit the expected reversible photoresponsive behavior (see Fig. 11). All azobenzene derivatives in the E-form show a strongly absorbing band in the UV region (∼360 nm) which is attributed to the π–π* transition, and a weakly absorbing band in the visible region (∼450 nm) due to the n–π* transition. The E-form is generally more stable than the Z-form, but each isomer can be converted into the other by light-irradiation caused a decrease of the absorption at around 355 nm and an increase of the absorption at around 450 nm. For example, dark incubation of a dichloromethane solution (1 × 10−5 M) of 112EO3OH have a maximum absorbance at 358 nm corresponding to the E-azobenzene chromophore (π–π* transition). Upon irradiation of this solution with 365 nm light resulted in photoisomerization from the E-azobenzene to the Z-azobenzene chromophore, as evidenced by a decrease in absorbance at 358 nm and an increase in absorbance at 450 nm (n–π* transition). Also the absorbance at 313 nm (π–π* transition) increased. The E–Z transition was complete in 35 s (see Fig. 11a). Under visible light, the reverse E–Z transition process was achieved within 6 min (see Fig. 11b).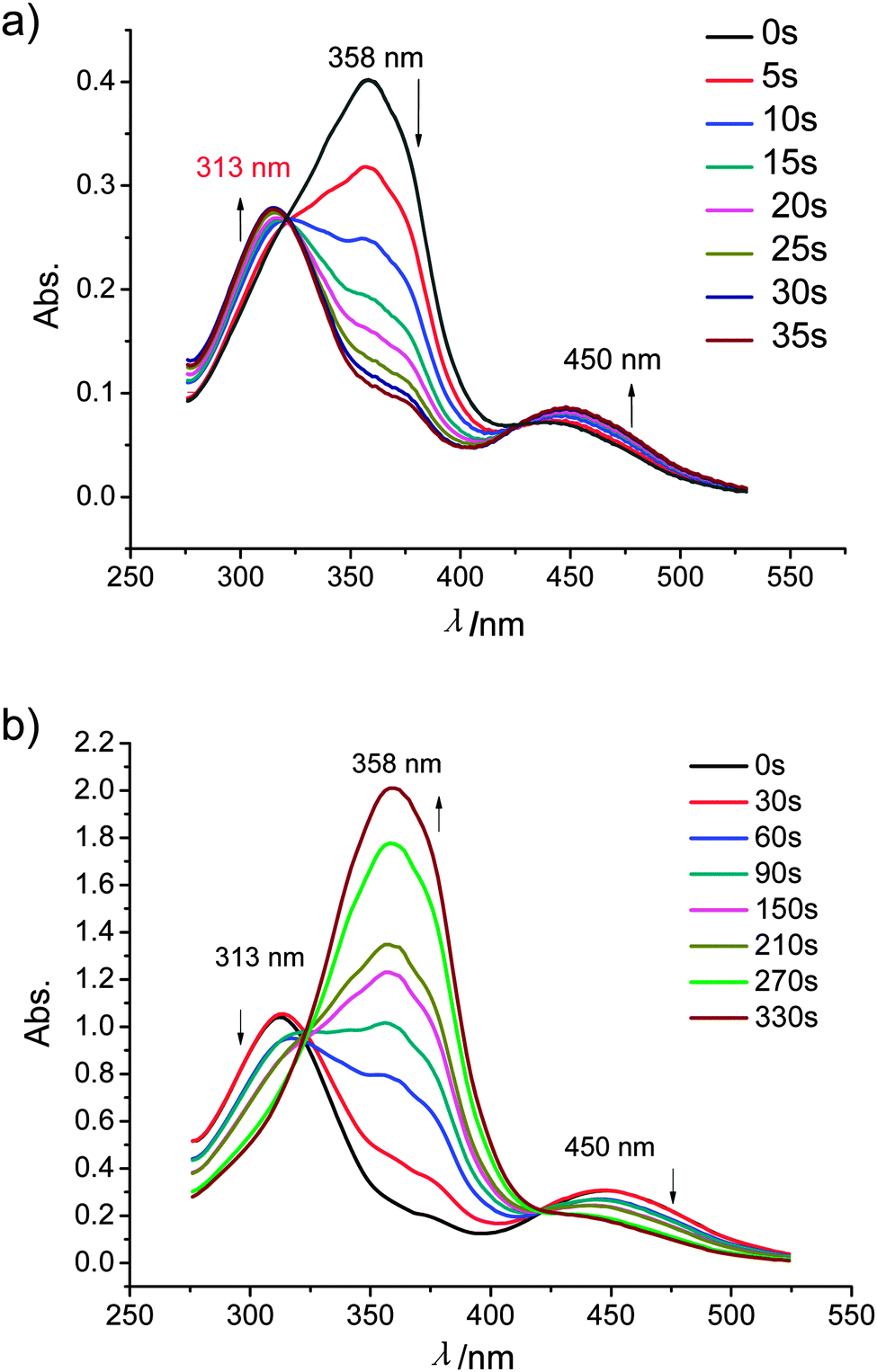 | ||
| Fig. 11 UV spectra of 112EO3OH in CH2Cl2 solution (c = 10−5 mol L−1), changing with time: (a) from E- to Z-azobenzene under irradiation with 365 nm light; (b) from Z- to E-azobenzene under irradiation with visible light; for additional examples, see Fig. S3 and S4 (ESI†). | ||
The effect of E-to-Z photoisomerization caused by the UV irradiation on the thermotropic LC phases was investigated using compound 112EO3OH as an example. The sample was placed between two thin quartz glass plates to form a thin film and placed on a Linkam hot stage that can control the sample temperature within 0.1 °C. The sample was cooled from the isotropic liquid state to 98 °C in the range of the SmCd phase (see Fig. S6a, ESI†). After annealing for 5 minutes the sample was exposed to UV irradiation at 365 nm (5 mW cm−2) for 60 s. After ∼20 s exposure time the schlieren texture of the SmCd phase has changed to a plain fan texture separated by homeotropic areas, indicating that the SmCd phase is replaced by a SmA phase (most probably SmAd; see Fig. S6b and c, ESI†). After thermal annealing at the same temperature of 98 °C without irradiation a slow change in the texture back to the SmCd phase took place, indicating a thermal Z–E isomerization (see Fig. S6d and e, ESI†). So the E–Z photoisomerization allows a reversible switching between SmC and SmA at a fixed temperature. This is due to a change in the phase transition temperatures. For example, a sample exposed for 30 s has the phase sequence Cr 86 °C SmC 94 °C SmA 99 °C Iso. This means that photoisomerization induces a SmA phase and reduces the Sm–Iso transition temperature (compare with Table 1). It appears that the isomerization process in the LC state is significantly slower than in the isotropic solution. Nevertheless, in the LC state the photoinduced E-to-Z transitions is still sufficiently fast and requires only a few seconds to induce a SmA phase, whereas the thermal relaxation to the SmC phases is much slower, requiring about 1 hour at the used temperature. Such photoisomerizations could be useful for light sensitive displays or for data storage applications.
3. Conclusion
A series of amphotropic azobenzene derivatives with a different number of alkyl chains and a distinct structure of the polar groups was synthesized and investigated with respect to thermotropic and lyotropic self-assembly in LC phases. Most compounds with only one n-alkyl chain form fluid tilted smectic phases with a double layer structure (SmCd). Compounds with a single hydroxyl group form exclusively the SmCd phase whereas a SmCd/SmAd dimorphism was observed for most of the related diols. SmA phases were induced and stabilized by addition of protic solvents. This means that increased head group size favors SmA phases, but nevertheless broad regions of lyotropic SmC phases were retained. Such SmC phases are rarely observed in lyotropic LC phases and these materials are of interest as lyotropic ferroelectrics after introduction of chirality.10b The SmC–SmA transition can be considered as mainly resulting from the randomization of the tilt by the lateral separation of the molecules by the solvent molecules located around the polar groups. The increased effective polar group volume also reduces the tilt correlation between the layers.Compounds with two or three alkyl chains form exclusively Colhex phases and there is a strong effect of protic solvents, stabilizing Colhex phases of compounds combining EO chains with glycerol units and removing columnar phases for compounds having only one of them, either EO chains or glycerol units. All amphiphiles show photoresponsive behavior, which makes these compounds interesting for potential applications as photoresponsive amphiphiles and LCs.
Acknowledgements
This work was supported by NSFC (No. 21364017, 21274119, 21374086), YNSF (2013FA007), YED (ZD2015001), DFG FOR 1145 (Ts 39/21-2). MP acknowledges support from the Cluster of Excellence “Nanostructured Materials” of the government of Saxony-Anhalt and JRB acknowledges financial support from the “Landesgraduiertenförderung” of the state Baden-Württemberg.References
- J. W. Steed and J. L. Atwood, Encyclopedia of Supramolecular Chemistry, Marcel Dekker, New York, 2004 Search PubMed; J. M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed.
- (a) C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828–8878 CrossRef CAS PubMed; (b) B. Donnio and D. Guillon, Adv. Polym. Sci., 2006, 201, 145–155 CrossRef; (c) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., 2006, 118, 44–74 CrossRef PubMed; (d) M. Lee, B.-K. Cho and W.-C. Zin, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS PubMed; (e) I. M. Saez and J. W. Goodby, J. Mater. Chem., 2005, 15, 26–40 RSC; (f) G. Ungar and X. Zeng, Soft Matter, 2005, 1, 95–106 RSC.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., 1988, 100, 117–162 CrossRef CAS PubMed.
- (a) R. Duran and P. Gramain, Makromol. Chem., 1987, 188, 2001–2009 CrossRef CAS PubMed; (b) R. Duran, D. Guillon, P. Gramain and A. Skoulios, J. Phys., 1988, 49, 1455–1466 CrossRef CAS; (c) H. R. Allcock and C. Kim, Macromolecules, 1989, 22, 2596–2602 CrossRef CAS; (d) V. Percec and C.-S. Hsu, Polym. Bull., 1990, 23, 463–470 CrossRef CAS; (e) V. Percec, D. Tomazos, J. Heck, H. Blackwell and G. Ungar, J. Chem. Soc., Perkin Trans. 2, 1994, 31–44 RSC.
- (a) C. Tschierske, Prog. Polym. Sci., 1996, 21, 775–852 CrossRef CAS; (b) M. Kölbel, T. Beyersdorff, C. Tschierske, S. Diele and J. Kain, Chem. – Eur. J., 2000, 6, 3821–3837 CrossRef; (c) C. Tschierske, Curr. Opin. Colloid Interface Sci., 2002, 7, 355–370 CrossRef CAS.
- (a) B. Neumann, C. Sauer, S. Diele and C. Tschierske, J. Mater. Chem., 1996, 6, 1087–1098 RSC; (b) C. Tschierske, J. A. Schröter, N. Lindner, C. Sauer, S. Diele, R. Festag, M. Wittenberg and J.-H. Wendorff, SPIE, 1998, 3319, 8–13 CrossRef CAS PubMed; (c) C. Sauer, S. Diele and C. Tschierske, Liq. Cryst., 1997, 23, 911–917 CrossRef CAS PubMed; (d) C. Sauer, S. Diele, N. Lindner and C. Tschierske, Liq. Cryst., 1998, 25, 109–116 CrossRef CAS PubMed.
- N. Lindner, M. Kölbel, C. Sauer, S. Diele, J. Jokiranta and C. Tschierske, J. Phys. Chem. B, 1998, 102, 5261–5273 CrossRef CAS.
- A. Bubnov, M. Kašpar, V. Hamplová, U. Dawin and F. Giesselmann, J. Org. Chem., 2013, 9, 425–436 CAS.
- N. Pietschmann, A. Lunow, G. Brezesinski, C. Tschierske, F. Kuschel and H. Zaschke, Colloid Polym. Sci., 1991, 269, 636–639 CAS.
- (a) J. R. Bruckner, D. Krueerke, J. H. Porada, S. Jagiella, D. Blunk and F. Giesselmann, J. Mater. Chem., 2012, 22, 18198–18203 RSC; (b) J. R. Bruckner, J. H. Porada, C. F. Dietrich, I. Dierking and F. Giesselmann, Angew. Chem., Int. Ed., 2013, 52, 8934–8937 CrossRef CAS PubMed.
- (a) C. Tschierske, A. Lunow, D. Joachimi, F. Hentrich, D. Girdziunaite, H. Zaschke, A. Mädicke, G. Brezesinski and F. Kuschel, Liq. Cryst., 1991, 9, 821–829 CrossRef CAS PubMed; (b) H. Müller and C. Tschierske, J. Chem. Soc., Chem. Commun., 1995, 645–646 RSC.
- (a) D. Joachimi, C. Tschierske, A. Öhlmann and W. Rettig, J. Mater. Chem., 1994, 4, 1021–1027 RSC; (b) D. Joachimi, A. Öhlmann, W. Rettig and C. Tschierske, J. Chem. Soc., Perkin Trans. 2, 1994, 2011–2019 RSC.
- T. Kato, Angew. Chem., Int. Ed., 2010, 49, 7847–7848 CrossRef CAS PubMed.
- (a) T. Ube and T. Ikeda, Angew. Chem., Int. Ed., 2014, 53, 10290–10299 CrossRef CAS PubMed; (b) H. M. Dhammika Bandarab and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- (a) T. Ikeda, J. Mamiya and Y. Yu, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS PubMed; (b) E. V. Fleischmann and R. Zentel, Angew. Chem., Int. Ed., 2013, 52, 8810–8827 ( Angew. Chem. , 2013 , 125 , 8972–8991 ) CrossRef CAS PubMed.
- (a) Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed; (b) Y. Yu, M. Nakano, A. Shishido, T. Shishido and T. Ikeda, Chem. Mater., 2004, 16, 1637–1643 CrossRef CAS; (c) M. Kondo, Y. Yu and T. Ikeda, Angew. Chem., Int. Ed., 2006, 45, 1378–1382 CrossRef CAS PubMed; (d) Y. Yu, T. Maeda, J. Mamiya and T. Ikeda, Angew. Chem., Int. Ed., 2007, 46, 881–883 CrossRef CAS PubMed; (e) Y. Zhang, J. Xu, F. Cheng, R. Yin, C.-C. Yen and Y. Yu, J. Mater. Chem., 2010, 20, 7123–7130 RSC.
- (a) R. F. Tabor, D. D. Tan, S. S. Han, S. A. Young, Z. L. E. Seeger, M. J. Pottage, C. J. Garvey and B. L. Wilkinson, Chem. – Eur. J., 2014, 20, 13881–13884 CrossRef CAS PubMed.
- X. M. Liu, B. Yang, Y. L. Wang and J. W. Wang, Chem. Mater., 2005, 17, 2792–2799 CrossRef CAS.
- (a) S. K. M. Nalluri and B. Jan Ravoo, Angew. Chem., Int. Ed., 2010, 49, 5371–5374 ( Angew. Chem. , 2010 , 122 , 5499–5502 ) CrossRef CAS PubMed; (b) F. P. Hubbard, G. Santonicola, E. W. Kaler and N. L. Abbott, Langmuir, 2005, 21, 6131–6136 CrossRef CAS PubMed.
- C. T. Jr. Lee, K. A. Smith and T. A. Hatton, Macromolecules, 2004, 37, 5397–5405 CrossRef.
- J. Eastoe and A. Vesperinas, Soft Matter, 2005, 1, 338–347 RSC.
- (a) N. Drillaud, E. Banaszak-Leonard, I. Pezron and C. Len, J. Org. Chem., 2012, 77, 9553–9561 CrossRef CAS PubMed; (b) R. Bianchini, G. Catelani, R. Cecconi, F. D'Andrea, E. Frino, J. Isaad and M. Rolla, Carbohydr. Res., 2008, 343, 2067–2074 CrossRef CAS PubMed.
- (a) J. M. Kuiper and J. B. F. N. Engberts, Langmuir, 2004, 20, 1152–1160 CrossRef CAS; (b) Y. Okuia and M. HanRational, Chem. Commun., 2012, 48, 11763–11765 RSC; (c) E. Chevallier, C. Monteux, F. Lequeux and C. Tribet, Langmuir, 2012, 28, 2308–2312 CrossRef CAS PubMed.
- (a) T. G. Shang, K. A. Smith and T. A. Hatton, Adv. Mater., 2014, 26, 1918–1922 CrossRef PubMed; (b) S. Aya, H. Obara, D. Pociecha, F. Araoka, K. Okano, K. Ishikawa, E. Gorecka, T. Yamashita and H. Takezoe, Adv. Mater., 2014, 26, 1918–1922 CrossRef CAS PubMed; (c) M. Badis, M. H. Guermouche, J.-P. Bayle, M. Rogalski and E. Rogalska, Langmuir, 2004, 20, 7991–7997 CrossRef CAS PubMed.
- C. Kördel, C. S. Popeney and R. Haag, Chem. Commun., 2011, 47, 6584–6586 RSC.
- Y. Y. Lin, Y. Qiao, C. Gao, P. F. Tang, Y. Liu, Z. B. Li, Y. Yan and J. B. Huang, Chem. Mater., 2010, 22, 6711–6717 CrossRef CAS.
- (a) D. G. Whitten, L. H. Chen, H. C. Geiger, J. Perlstein and X. D. Song, J. Phys. Chem. B, 1998, 102, 10098–10111 CrossRef CAS; (b) E. H. G. Backus, J. M. Kuiper, J. B. F. N. Engberts, B. Poolman and M. Bonn, J. Phys. Chem. B, 2011, 115, 2294–2302 CrossRef CAS PubMed.
- (a) D.-Y. Kim, S.-A. Lee, Y.-J. Choi, S.-H. Hwang, S.-W. Kuo, C. Nah, M.-H. Lee and K.-U. Jeong, Chem. – Eur. J., 2014, 20, 5689–5695 CrossRef CAS PubMed; (b) Z. H. Shi, D. Z. Chen, H. J. Lu, B. Wu, J. Ma, R. S. Cheng, J. L. Fang and X. F. Chen, Soft Matter, 2012, 8, 6174–6184 RSC.
- N. Laurent, N. Lafont, F. Dumoulin, P. Boullanger, G. Mackenzie, P. H. J. Kouwer and J. W. Goodby, J. Am. Chem. Soc., 2003, 125, 15499–15506 CrossRef CAS PubMed.
- B. Soberats, E. Uchida, M. Yoshio, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2014, 136, 9552–9555 CrossRef CAS PubMed.
- F. A. Khan, K. Parasuraman and B. Donnio, Tetrahedron, 2010, 66, 8745–8755 CrossRef CAS PubMed.
- (a) R. Willstätter and M. Benz, Chem. Ber., 1906, 339, 3492–3503 CrossRef PubMed; (b) W. H. Wei, T. Tomohiro, M. Kodaka and H. Okuno, J. Org. Chem., 2000, 65, 8979–8987 CrossRef CAS PubMed.
- V. Van Rheenen, D. Y. Cha and W. M. Hartley, Org. Synth., 1978, 58, 43–51 CrossRef CAS.
- T. P. Rieker, N. A. Clark, G. S. Smith, D. S. Parmar, E. B. Sirota and C. R. Safinya, Phys. Rev. Lett., 1987, 59, 2658–2661 CrossRef CAS.
- Y. Ouchi, H. Takano, H. Takezoe and A. Fukuda, Jpn. J. Appl. Phys., 1988, 27, 1–7 CrossRef CAS.
- N. A. Clark, T. P. Rieker and J. E. Mclennan, Ferroelectrics, 1988, 85, 79–97 CrossRef PubMed.
- G. W. Gray and J. W. Goodby, Smectic Liquid Crystals – Textures and Structures, Leonard Hill, sgoe and London, 1984 Search PubMed.
- CAChe 3.2, Oxford Molecular Ltd, Oxford, UK, 1999 Search PubMed.
- Identical values were obtained with CPK models.
- K. Borisch, S. Diele, P. Göring, H. Kresse and C. Tschierske, J. Mater. Chem., 1998, 8, 529–543 RSC.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- (a) H.-T. Nguyen, C. Destrade and J. Malthete, Adv. Mater., 1997, 9, 375–388 CrossRef CAS PubMed; (b) M. Gharbia, A. Gharbi, H. T. Nguyen and J. Malthete, Curr. Opin. Colloid Interface Sci., 2002, 7, 312–325 CrossRef CAS.
- N. G. Nagaveni, M. Gupta, A. Roy and V. Prasad, J. Mater. Chem., 2010, 20, 9089–9099 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc02583d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |