
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High brightness deep blue/violet fluorescent polymer light-emitting diodes (PLEDs)†
Javan H.
Cook
a,
José
Santos
b,
Hameed A.
Al-Attar
a,
Martin R.
Bryce
*b and
Andrew P.
Monkman
*a
aDepartment of Physics, Durham University, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 2nd September 2015
Abstract
Two new deep blue/violet emitting alternating co-polymers, comprising readily-available carbazole (C) and fluorene (F) monomer units, have been synthesised and shown to produce extremely bright solution-processed polymer light-emitting diodes (PLEDs) with the structure ITO/PEDOT:PSS/polymer/TPBi/LiF/Al. The para-conjugated polymer, CF1, gave PLED devices with external quantum efficiency (EQE) values of ηext,max 1.4%, Lmax of 565 cd m−2 with CIEx,y (0.16, 0.07). The EQE was raised to ηext,max 2.1%, after the addition of a TAPC hole injection layer. For the isomeric meta-conjugated polymer, CF2, values of ηext,max 0.35%, Lmax of 16 cd m−2 with CIEx,y (0.18, 0.12) were obtained. The λELmax was 409 nm for both the CF1 and CF2 devices. The CF1 devices also possess low turn-on and low operating voltages for devices of such high brightness. Moreover, the CF1 emission is very stable from 10 cd m−2 up to peak brightness, with only a negligible shift in CIE coordinates. The combination of a simple co-polymer structure synthesised using readily-available monomer units, and high brightness and good colour stability from a simple device architecture, makes CF1 suitable for a wide range of applications requiring deep blue/violet emission.
1. Introduction
Organic light emitting devices (OLEDs) are a rapidly advancing technology due to their applications in full-colour flat-panel displays1–4 and energy-saving solid-state lighting,5–7 with numerous potential benefits over their inorganic competitors. These benefits include lower power consumption, faster response times, larger viewing angles, smaller sizes and greater ranges of colour and contrast, along with being cheap and quick to produce. OLEDs typically comprise multilayer structures which can give very bright and efficient devices when an emitter layer in sandwiched between additional layers that facilitate hole-transport and electron-transport. However, these multilayer devices have practical limitations as their fabrication is complicated by the need for sequential layer-by-layer deposition procedures. Furthermore, exciplex formation can occur at the organic–organic interfaces,8,9 which can be detrimental for efficient and stable emission – especially in the blue spectral region.10,11OLEDs which emit from the deep blue/violet region of the spectrum, especially with Commission Internationale de l'Eclairage (CIE) coordinates that match the National Television System Committee (NTSC) standard blue CIEx,y (0.14, 0.08) have been the subject of increased investigation to meet the demands of high quality displays.12–21 Maximum external quantum efficiencies (EQEs) as high as 3–6% for emission peaks in the range 400–480 nm have been achieved for a range of small molecule based devices through rational molecular design.12,21 However, efficient deep blue polymeric emitters in simple device architectures remain under-developed.11,21 Polymeric systems which possess good solubility in organic solvents offer the advantages of rapid industrial-scale polymer LED (PLED) fabrication by solution processing techniques, such as ink-jet printing, spin-coating and roll-to-roll processing.22 The key challenge in the molecular design of saturated blue emitting polymers is that backbone conjugation should be restricted to short (oligomer) segments, while the charge carrier transport capabilities of the polymer must be retained.23
Poly(9,9-dialkylfluorene-2,7-diyl) derivatives are widely used as blue/deep blue emitting polymers because of their high charge-carrier mobilities, good thermal and electrochemical stability, high photoluminescence quantum yields (PLQY), and versatile chemical modification.24 Specific examples include: poly(3,6-silafluorene-co-2,7-fluorene),25 poly(fluorene-co-thiophene) host–guest systems26 and spiro-polyfluorenes.27 Incorporation of tetrafluoro-p-phenylene units into a polyfluorene backbone affords PLEDs with EQEs as high as 5% with CIE coordinates of (0.16, 0.05).28 It has recently been shown that the attachment of bulky side chains to a polyfluorene backbone increases intrachain torsion angles and isolates minimal conjugated segments.29–31 In an alternative approach, 9,9-diphenylfluorene units serve to disrupt conjugation in the polymer backbone leading to PLEDs with EQE values of ηext,max 3.9%, Lmax of 274 cd m−2 with CIEx,y coordinates (0.17, 0.07).32 It is also established that meta-linked aromatic units in the backbone can blue shift emission by reducing the effective conjugation length.33–35 Sergent et al. have recently synthesised regiorandom fluorene–carbazole copolymers [with emphasis on different length alkyl chains at the fluorene C(9) position]. Very preliminary electroluminescence data showed blue emission (EQE 1.32%; CIEx,y 0.16; 0.11).36
The aim of the present work is to study new readily-available polymers which yield efficient deep blue/violet PLEDs. This has been achieved in two new high triplet energy poly(carbazole-alt-fluorene) co-polymers CF1 and CF2 which function as both the charge carrier transporters and the fluorescent emitters simultaneously. In both polymers the carbazole unit (C) is linked through 3,6-positions, while the linkage to the fluorene unit (F) is varied (2,7 in CF1 – para-conjugated; 3,6 in CF2 – meta-conjugated) in order to modulate the conjugation along the backbone. The photophysics and PLED applications of these co-polymers are reported. In particular, CF1 devices emit deep blue/violet light with remarkably high brightness (Lmax of 565 cd m−2) when the luminosity function is taken into account. The luminosity function shows the perception of brightness to different wavelengths of light, as observed by the human eye and is the function by which OLED emission is normalised. Because the luminosity function peaks at 555 nm, blue and red emission is reduced in comparison to green emission, so more emission is required for these colours to achieve the same brightness. This becomes more of a problem the further the emission is from 555 nm, making the high brightness of CF1 at 409 nm especially notable.
2. Results and discussion
2.1. Polymer synthesis and characterisation
The synthesis of the alternating copolymers CF1 and CF2 (Fig. 1) was carried out under standard Suzuki-Miyaura co-polymerisation conditions37 followed by end-capping, as described in the ESI.† The co-polymers were characterised by GPC-UV-Vis data using polystyrene standards: CF1Mw 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, Mn 10800; CF2Mw 21000 Da, Mn 6051 Da. The thermal properties of the polymers were characterised by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. The decomposition temperature (Td), which corresponds to a weight loss of 5% at a heating rate of 10 °C min−1, was 433 °C and 422 °C for CF1 and CF2, respectively, demonstrating that the polymers have very good thermal stability. Glass transition temperatures (Tg) of 134 °C and 130 °C for CF1 and CF2 were determined by DSC during the second heating cycle. In addition, no exothermic crystallisation or endothermic melting peaks were observed over the range 40–200 °C, implying that the polymers should exhibit high morphological stability in a device. These data are typical of copolymers of 9,9-dialkylfluorenes38 and represent increased thermal stability compared to poly(9,9-dialkylfluorene)s, due to the reduced number of alkyl chains in CF1 and CF2.39 In particular Tg is raised significantly compared to poly(9,9-dihexylfluorene) (Tg 83 °C)40 showing that the N-hexylcarbazole units effectively suppress the dense packing and crystallisation of the polymer chains.41
000 Da, Mn 10800; CF2Mw 21000 Da, Mn 6051 Da. The thermal properties of the polymers were characterised by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. The decomposition temperature (Td), which corresponds to a weight loss of 5% at a heating rate of 10 °C min−1, was 433 °C and 422 °C for CF1 and CF2, respectively, demonstrating that the polymers have very good thermal stability. Glass transition temperatures (Tg) of 134 °C and 130 °C for CF1 and CF2 were determined by DSC during the second heating cycle. In addition, no exothermic crystallisation or endothermic melting peaks were observed over the range 40–200 °C, implying that the polymers should exhibit high morphological stability in a device. These data are typical of copolymers of 9,9-dialkylfluorenes38 and represent increased thermal stability compared to poly(9,9-dialkylfluorene)s, due to the reduced number of alkyl chains in CF1 and CF2.39 In particular Tg is raised significantly compared to poly(9,9-dihexylfluorene) (Tg 83 °C)40 showing that the N-hexylcarbazole units effectively suppress the dense packing and crystallisation of the polymer chains.41
 | ||
| Fig. 1 Structures of the new deep blue/violet emitting carbazole-alt-fluorene copolymers studied in this work. | ||
2.2. Optical properties
The spectroscopic data for polymers CF1 and CF2 is summarised in Table 1. Their absorption spectra are shown in the ESI† and their photoluminescence spectra are shown in Fig. 2. The polymers are deep blue/violet emitters (λPLmax in thin films: 421 and 401 nm, respectively). They exhibit very little solvatochromism, in agreement with previous work on fluorene co-polymers;29,30,32 the small changes observed can be explained by the different refractive index of the solvents.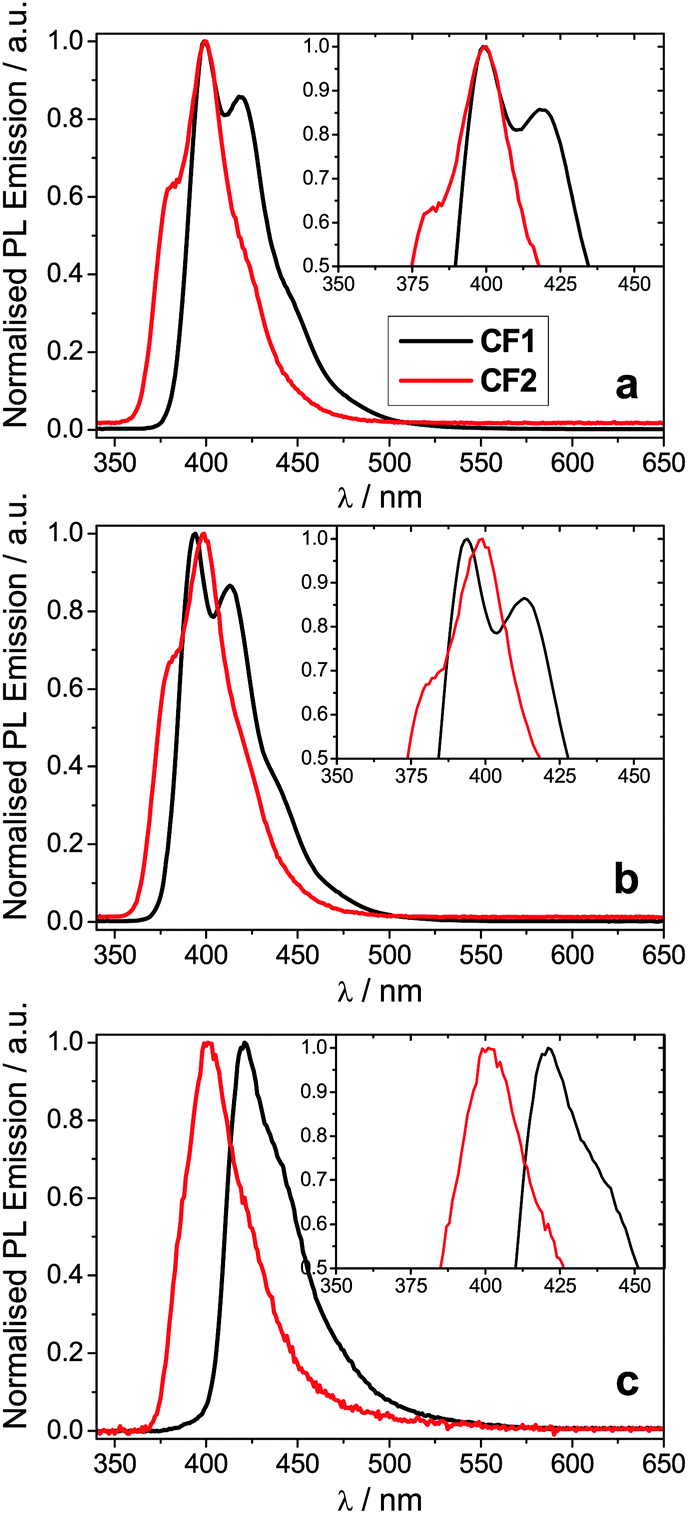 | ||
| Fig. 2 Normalised PL emission spectra for polymers CF1 and CF2 (a) in ethyl acetate; (b) in cyclohexane; (c) in thin film. The insets show a magnification of the λmax region. | ||
| Polymer | Solvent/film | λ absmax/nm | λ PLmax/nm | PLQY, ΦPLa |
E
onsetTb/eV |
|---|---|---|---|---|---|
| a PLQY is the photoluminescence quantum yield. b E T is the triplet energy determined for solid state samples at 16 K. | |||||
| CF1 | Ethyl acetate | 348 | 399 | ||
| Cyclohexane | 344 | 394 | |||
| Film | 350 | 421 | 0.20 | 2.34 | |
| CF2 | Ethyl acetate | 300 | 400 | ||
| Cyclohexane | 304 | 399 | |||
| Film | 287 | 401 | 0.17 | 2.34 | |
From the photophysical data in Table 2 it can be observed that both CF1 and CF2 emissions peak at ca. 400 nm in solution with negligible solvent polarity effect (Table 2, Fig. 2a and b). Nevertheless, PL from thin films shows a different trend: while CF1 (para-conjugated) shows the typical aggregation induced red-shift (20 nm) in its emission, CF2's (meta-conjugated) λPLmax remains basically unaltered (Table 2 and Fig. 2c).
| V on /V | Brt/cd m−2 | EQE/% | Dev eff/cd A−1 | Brtmax/cd m−2 | EQEmax/% | Dev effmax/cd A−1 | Lummax/lm W−1 | CIEx,ye | CIEx,yf | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Device architecture: ITO/PEDOT:PSS/polymer/TPBi/LiF/Al. b V on is the turn-on voltage, defined here as the voltage at which the device reaches a brightness of 10 cd m−2. c A comparison current density of 10 mA cm−2 was selected. d Data in brackets are for devices with an additional TAPC layer between PEDOT:PSS and CF1 (see text for details). e CIE coordinates at the turn-on voltage (10 cd m−2). f CIE coordinates at the maximum brightness. CIE diagrams are shown in the ESI. | ||||||||||
| CF1 | 3.40 | 232c | 1.10c | 0.51c | 565 (492)d | 1.4 (2.1)d | 0.65 | 0.63 | 0.16, 0.07 | 0.16, 0.07 |
| CF2 | 4.50 | 16c | 0.05c | 0.04c | 16 | 0.35 | 0.25 | 0.99 | 0.18, 0.12 | 0.20, 0.15 |
No significant difference is observed in the film PLQY for polymers CF1 and CF2, with values of ΦPL 0.20 and 0.17, respectively. The observed triplet level for CF1 and CF2 (EonsetT 2.34 eV) corresponds well to the reported values for poly(9,9-dialkylfluorene)42 and poly(N-alkylcarbazole) derivatives43 (ca. 2.3 eV) and related polymers with limited backbone π-conjugation.29,30,32
2.3. PLED device fabrication and properties
Hybrid devices were fabricated with CF1 or CF2 as the spin-coated light-emitting polymer (LEP) layer with the structure: glass|ITO (150 nm)|PEDOT:PSS 1.5 (70 nm)|LEP (CF1 or CF2 20 nm)|TPBi (20 nm)|LiF (1 nm)|Al (100 nm). For all of the devices, no other dopants or transporters were added to the LEP layer. An evaporated layer of 1,3,5-tris(N-phenylbenzimidazole-2-yl)benzene (TPBi) was incorporated as a standard electron-injecting material to improve device performance (efficiency, brightness and turn-on voltage).44 The thickness of the LEP layer was optimised for maximum device performance.The device results are shown in Fig. 3 and 4. Devices containing CF1 display deep blue/violet EL with λmax of 409 nm and maximum EQE of 1.4% and CIEx,y coordinates [turn on (0.16, 0.07) and peak (0.16, 0.07)] exhibiting excellent stability under device operation. CF2 also showed deep blue/violet emission, again with λmax 409 nm, but with a substantially reduced EQE of 0.35% and less impressive colour coordinates [turn on (0.18, 0.12) and peak (0.20, 0.15)] with reduced stability. The CIE coordinates for CF1 are considerably deeper in the blue than data recently reported for other fluorene–carbazole co-polymer PLEDs [(0.16, 0.11) and (0.18, 0.14)].36 This excellent colour stability of the CF1 emission is in contrast to most other deep blue polymers32 for which there is a shift to less blue emission at maximum brightness (as seen here for CF2). CIE diagrams can be found in the ESI.† The results for CF1 compare very favourably with pure PFO devices,28 having a deeper blue λmax (409 nm compared to 420 nm), superior CIE coordinates ((0.16, 0.07) compared to (0.16, 0.11)) and higher EQE and device efficiencies (1.10% and 0.51 cd A−1 compared to 0.56% and 0.20 cd A−1). The emission is also deeper blue than reported recently in very preliminary EL data for regiorandom fluorene–carbazole copolymers (CIEx,y 0.16; 0.11),36 demonstrating the benefits of the well-defined alternating structure of CF1.
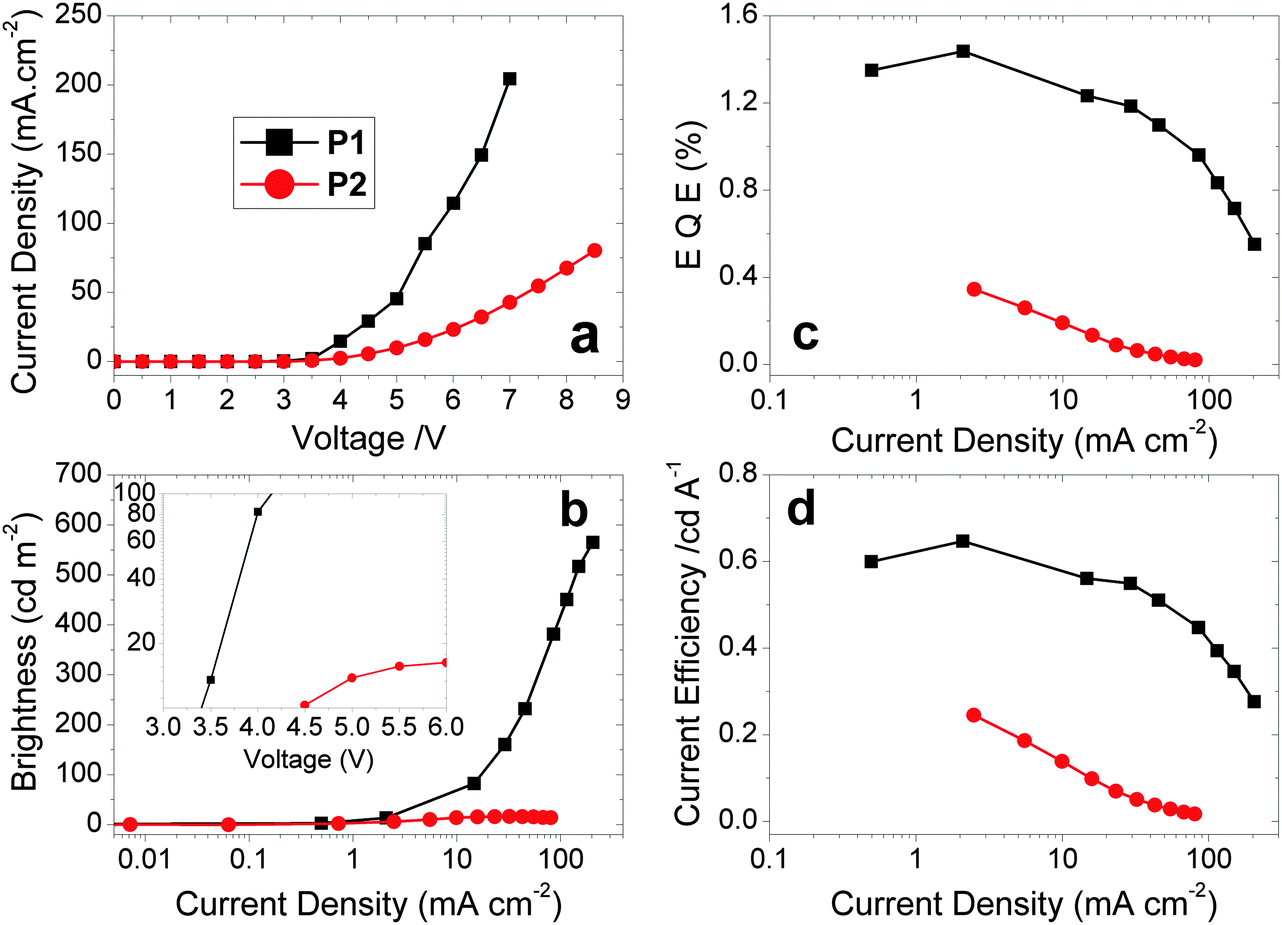 | ||
| Fig. 3 Plots of (a) J–V curves. (b) Luminance vs. J. (c) EQE vs. J. (d) Device efficiency vs. J for the polymers CF1 and CF2. Inset to (b) shows the low turn-on voltages for the two devices in a plot of luminance vs. V. The device structure is stated in Table 2, footnote a. | ||
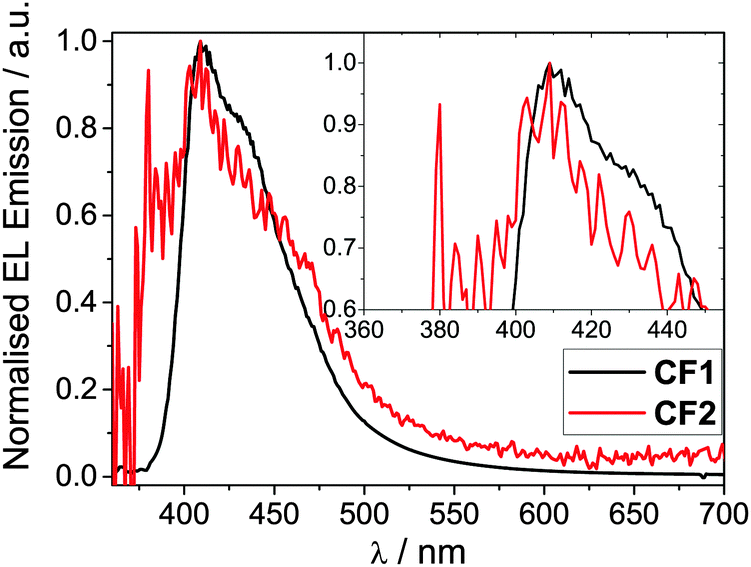 | ||
| Fig. 4 Normalised EL emission spectra for polymers CF1 and CF2 devices. Inset shows a magnification of the λmax region. The device structure is stated in Table 2, footnote a. | ||
Polymer CF1 produced an excellent maximum brightness of 565 cd m−2 for a device with these CIE coordinates. As stated in the introduction, this deep blue spectral region is heavily affected by the luminosity function (the eye sensitivity here is very low) making such a high value particularly notable. The low turn-on voltage (3.40 V) and low potential operating voltage makes this polymer well suited for a wide range of practical applications, including displays1–4 and medical devices: for the latter “blue light therapy” has a range of beneficial effects.45CF2 has a substantially reduced maximum brightness of 16 cd m−2. Also, despite the emission showing a bluer onset compared to CF1 and the same λmax of 409 nm for both devices, the CIE coordinates of CF2 are less blue than CF1 due to broadened emission into the green region which skews the coordinates (Fig. 4).
The meta-linkage through the 3,6-positions of fluorene in CF2 limits the backbone conjugation; this in turn reduces the conductivity which explains the decrease in the current density passing through the CF2 device, compared to CF1. This reduces both the maximum brightness and EQE of the CF2 device, whilst causing no substantial blue shift in EL emission.
To further investigate CF1, devices were fabricated with an additional hole-injection and electron-blocking layer of 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC).46 The primary purpose of this layer was to boost device efficiency by preventing electrons from reaching the anode and trapping them in the emissive layer, utilising TAPC's poor electron mobility. Two TAPC thickness were trialled to give the structure: glass|ITO (150 nm)|PEDOT:PSS HIL 1.5 (70 nm)|TAPC (60 nm or 90 nm)|LEP (20 nm)|TPBi (20 nm)|LiF (1 nm)|Al (100 nm). Whilst these TAPC thicknesses are both high relative to the LEP thickness, it was assumed that a portion would be washed away during deposition of the LEP layer leaving a more reasonable thickness behind that could be assessed by ellipsometry. The EQE was raised to ηext,max 2.1%. There was, however, a 25% increase in turn-on voltage and a 13% decrease in the maximum brightness. These were initially believed to be the result of increased device thickness and potential absorption by the TAPC layer, respectively. However, ellipsometry of the multilayer structures returns a combined thickness for the two layers between 19–20 nm. This indicates that the majority of the TAPC layer is removed when the LEP layer is deposited. Despite this, the observed changes in device properties fit with those expected for the addition of an electron blocking layer and so further investigation would be required to determine whether either a very thin film of TAPC remains, or whether the two layers have blended.
4. Conclusions
This work has addressed the synthesis, photophysical characterisation and device properties of two well-defined fluorene-alt-carbazole co-polymers CF1 and CF2 which are deep blue/violet emitters, with λELmax 409 nm. The para-conjugated polymer, (CF1), with 2,7-linked fluorene units, is the most efficient emitter, giving PLED devices with external quantum efficiency (EQE) values of ηext,max 1.4%, Lmax of 565 cd m−2 with CIEx,y (0.16, 0.07). The meta-conjugated polymer, CF2, with 3,6-linked fluorene units, gives analogous devices with ηext,max 0.35%, Lmax of 16 cd m−2 with CIEx,y (0.18, 0.12). The emission of CF1 devices shows excellent colour stability from 10 cd m−2 up to peak brightness, with essentially no shift in the CIE coordinates. The unique combination of a simple co-polymer structure that is based on cheap, readily-available monomer units, low turn-on voltage, excellent colour stability and high brightness in a simple PLED architecture makes CF1 a very promising material for a wide range of applications requiring deep blue/violet emission.5. Experimental
5.1. Synthesis of polymers CF1 and CF2
The syntheses followed standard routes as reported in the ESI.†5.2. Optical characterisation
Absorption spectra for solution and solid state samples were obtained using a Shimadzu UV-vis-NIR spectrophotometer whilst emission spectra were acquired using a Jobin-Yvon fluoromax spectrofluorimeter. The PLQY data was also obtained using the Jobin-Yvon fluoromax spectrofluorimeter, along with a small integrating sphere and a neutral density filter (optical density = 2).47 The excitation wavelengths were determined from the maximum absorbance of the polymers as obtained from their absorption spectra. The triplet energies of solid state samples at 16 K were calculated using a gated luminescent measurement of the phosphorescence. Solutions were produced in ethyl acetate or cyclohexane, and the OD kept below 1.0. Solid state samples were drop-cast from a 1:1 mixture of 175 mg mL−1 zeonex and 0.5 mg mL−1 of the polymer, both in chlorobenzene, and had a maximum absorbance of 2.0 OD.
5.3. Device fabrication and characterisation
These devices featured an ITO anode (150 nm, 16 Ω −1) commercially pre-coated on a glass substrate (24 mm × 24 mm). The substrates were cleaned with acetone, isopropanol and acetone sequentially in a sonic bath for a period of 9 min each. They were then exposed to low pressure plasma for a period of 3 min and treated with UV-ozone for a further 4 min. A hole-injection layer (HIL) of PEDOT:PSS of thickness 70 nm was deposited by spin coating and then baked on a hotplate at 200 °C for 3 min to remove any remaining moisture. The PEDOT:PSS used was the commercially available HIL 1.5 from Heraeus Precious Metals, Germany. Active layers of polymers CF1 and CF2 were prepared in a solution of chlorobenzene, with the concentration varied to produce layers of 60 nm thickness, and then spun on top of the PEDOT:PSS. The device was then annealed at 120 °C on a hotplate for 10 min to remove residual chlorobenzene. An electron injection layer (EIL) consisting of a 20 nm layer of 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) was thermally evaporated directly on top of the polymer layer. This was followed by a 1 nm thick lithium fluoride (LiF) cathode, which was thermally evaporated using a shadow mask to produce parallel strips perpendicular to the ITO anodes, forming four individually addressable pixels per substrate each of 5 mm × 4 mm area. The LiF was capped with a 100 nm thick layer of aluminium to protect it from oxidation. An evaporation pressure of the order of 10−6 mbar and a rate of approximately 0.1 nm s−1 was used for all of the evaporated produced layers. The devices were then encapsulated with DELO UV curable epoxy (Katiobond) and a 12 × 12 mm glass cover slide.The devices were characterised in a calibrated Labsphere LMS-100 integrating sphere, connected to a USB 4000 CCD spectrometer supplied by a 30 μm UV/Vis fibre optic cable, under steady state conditions. Layer thicknesses were measured using a J. A. Woolam VASE Ellipsometer after having been spin coated onto Si/SiO2 substrates. The non-uniformity of the organic layer thicknesses across the samples leads to a 5–10% error in device efficiencies: all measurements were averages from at least four devices.
Acknowledgements
The authors acknowledge the financial support of the Durham Energy Institute and EPSRC.References
- R. Mertens, The OLED Handbook. A Guide to OLED Technology, Industry and Market, 2014, http://www.oled-info.com/handbook Search PubMed.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- K. Müllen and U. Scherf, Organic Light-Emitting Devices, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- Z. Li and H. Meng, Organic Light-Emitting Materials and Devices, CRC Press, Boca Raton, Florida, 2006 Search PubMed.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- S. Beaupré, P. L. T. Boudrealt and M. Leclerc, Adv. Mater., 2010, 22, E6–E27 CrossRef PubMed.
- L. Ying, C.-L. Ho, H. Wu, Y. Cao and W.-Y. Wong, Adv. Mater., 2014, 26, 2459–2473 CrossRef CAS PubMed.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- M. Castellani and D. Berner, J. Appl. Phys., 2007, 102, 024509 CrossRef PubMed.
- M. Carvelli, A. van Reenen, R. A. J. Janssen, H. P. Loebl and R. Coehoorn, Org. Electron., 2012, 13, 2605–2614 CrossRef CAS PubMed.
- V. Jankus, C.-J. Chiang, F. Dias and A. P. Monkman, Adv. Mater., 2013, 25, 1455–1459 CrossRef CAS PubMed.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- A. L. Fisher, K. E. Linton, K. T. Kamtekar, C. Pearson, M. R. Bryce and M. C. Petty, Chem. Mater., 2011, 23, 1640–1642 CrossRef CAS.
- X. J. Feng, S. F. Chen, Y. Ni, M. S. Wong, M. M. K. Lam, K. W. Cheah and G. Q. Lai, Org. Electron., 2014, 15, 57–64 CrossRef CAS PubMed.
- S. H. Jeong and J. Y. Lee, J. Mater. Chem., 2011, 21, 14604–14609 RSC.
- S. Nau, N. Schulte, S. Winkler, J. Frisch, A. Vollmer, N. Koch, S. Sax and E. J. W. List, Adv. Mater., 2013, 25, 4420–4424 CrossRef CAS PubMed.
- Y.-H. Chung, L. Sheng, X. Xing, L. Zheng, M. Bian, Z. Chen, L. Xiao and Q. Gong, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- Y. Yang, P. Cohn, A. L. Dyer, S.-H. Eom, J. R. Reynolds, R. K. Castellano and J. Xue, Chem. Mater., 2010, 22, 3580–3582 CrossRef CAS.
- J. Ye, Z. Chen, M.-K. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C.-S. Lee, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- X. Zhan, N. Sun, Z. Wu, J. Tu, L. Yuan, X. Tang, Y. Xie, Q. Peng, Y. Dong, Q. Li, D. Ma and Z. Li, Chem. Mater., 2015, 27, 1847–1854 CrossRef CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- D. Y. Kim, H. N. Cho and C. Y. Kim, Prog. Polym. Sci., 2000, 25, 1089–1139 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- E. G. Wang, C. Li, Y. Q. Mo, Y. Zhang, G. Ma, W. Shi, J. B. Peng, W. Yang and Y. Cao, J. Mater. Chem., 2006, 16, 4133–4140 RSC.
- G. Vamvounis, G. L. Schulz and S. Holdcroft, Macromolecules, 2004, 37, 8897–8902 CrossRef CAS.
- C.-W. Huang, C.-L. Tsai, C.-Y. Liu, T.-H. Jen, N.-J. Yang and S.-A. Chen, Macromolecules, 2012, 45, 1281–1287 CrossRef CAS.
- U. Giovanella, C. Botta, F. Galeotti, B. Vercelli, S. Battiato and M. Pasinbi, J. Mater. Chem. C, 2013, 1, 5322–5329 RSC.
- K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481–4488 CrossRef CAS.
- J. H. Cook, J. Santos, H. Li, H. A. Al-Attar, M. R. Bryce and A. P. Monkman, J. Mater. Chem. C, 2014, 2, 5587–5592 RSC.
- J. Liu, S. Hu, W. Zhao, Q. Zou, W. Luo, W. Yang, J. Peng and Y. Cao, Macromol. Rapid Commun., 2010, 31, 496–501 CrossRef CAS PubMed.
- J. Santos, J. H. Cook, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, J. Mater. Chem. C, 2015, 3, 2479–2483 RSC.
- J. Ritchie, J. A. Crayston, J. P. J. Markham and I. D. W. Samuel, J. Mater. Chem., 2006, 16, 1651–1656 RSC.
- P. Taranekar, M. Abdulbaki, R. Krishnamoorti, S. Phanichphant, P. Waenkaew, D. Patton, T. Fulghum and R. Advincula, Macromolecules, 2006, 39, 3848–3854 CrossRef CAS.
- Z. L. Wu, Y. Xiong, J. H. Zou, L. Wang, J. C. Liu, Q. L. Chen, W. Yang, J. B. Peng and Y. Cao, Adv. Mater., 2008, 20, 2359–2364 CrossRef CAS PubMed.
- A. Sergent, G. Zucchi, R. B. Pansu, M. Chaigneau, B. Geffroy, D. Tondelier and M. Ephritikhine, J. Mater. Chem. C, 2013, 1, 3207–3216 RSC . In Scheme 1 in this article, the structures are drawn with the carbazole units linked into the polymer chain through the 3,7-positions. Presumably this is an error as it is stated that the monomer used in the synthesis is 3,6-disubstituted carbazole.
- (a) J. Murage, J. W. Eddy, J. R. Zimbalist, T. B. McIntyre, Z. R. Wagner and F. E. Goodson, Macromolecules, 2008, 41, 7330–7338 CrossRef CAS; (b) J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schluter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS PubMed.
- L. Kinder, J. Kanicki, J. Swensen and P. Petroff, Proceedings of SPIE Organic Field Effect Transistors II, ed. C. D. Dimitrakopoulos and A. Dodabalapur, SPIE, Bellingham, WA, 2003, vol. 5217, pp. 35–42 Search PubMed.
- S. Jegadesan, S. Sindhu, R. C. Advincula and S. Valiyaveettil, Langmuir, 2006, 22, 780–786 CrossRef CAS PubMed.
- K. L. Chan, S. E. Watkins, C. S. K. Mak, M. J. McKiernan, C. R. Towns, S. I. Pascu and A. B. Holmes, Chem. Commun., 2005, 5766–5768 RSC.
- Y. Zhu, K. M. Gibbons, A. P. Kulkarni and S. A. Jenekhe, Macromolecules, 2007, 40, 804–813 CrossRef CAS.
- A. P. Monkman, H. D. Burrows, L. J. Hartwell, L. E. Horsburgh, I. Hamblett and S. Navaratnam, Phys. Rev. Lett., 2001, 86, 1358–1361 CrossRef CAS.
- K. Zhang, Y. Tao, C. Yang, H. You, Y. Zou, J. Qin and D. Ma, Chem. Mater., 2008, 20, 7324–7331 CrossRef CAS.
- J.-H. Jou, W.-B. Wang, S.-M. Shen, S. Kumar, I.-M. Lai, J.-J. Shyue, S. Lengvinaite, R. Zostautiene, J. V. Grazulevivius, S. Grigalevicius, S.-Z. Chen and C.-C. Wu, J. Mater. Chem., 2011, 21, 9546–9552 RSC.
- (a) S. Vandersee, M. Beyer, J. Lademann and M. E. Darvin, Oxid. Med. Cell. Longevity, 2015, 2015, 579675 Search PubMed; (b) Y. Zhang, Y. Zhu, A. Gupta, Y. Huang, C. K. Murray, M. S. Vrahas, M. E. Sherwood, D. G. Baer, M. R. Hamblin and T. Dai, J. Infect. Dis., 2014, 209, 1963–1971 CrossRef CAS PubMed.
- G. Liaptsis and K. Meerholz, Adv. Funct. Mater., 2013, 23, 359–365 CrossRef CAS PubMed.
- L.-O. Palsson and A. P. Monkman, Adv. Mater., 2002, 14, 757–758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of CF1 and CF2; absorption spectra; CIE diagrams. See DOI: 10.1039/c5tc02162f |
| This journal is © The Royal Society of Chemistry 2015 |