
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable plasmonic surfaces via colloid assembly†
T.
Honold
,
K.
Volk
,
A.
Rauh
,
J. P. S.
Fitzgerald
and
M.
Karg
*
Physical Chemistry I, University of Bayreuth, Universitaetsstr. 30, 95440 Bayreuth, Germany. E-mail: matthias.karg@uni-bayreuth.de; Web: http://www.karg.uni-bayreuth.de
First published on 6th October 2015
Abstract
Self-assembly of plasmonic colloids is a key challenge on the road to the cost-efficient fabrication of optically active surfaces. Here, we show that core–shell particles with plasmonic cores and hydrogel shells are ideal building blocks for coatings with engineered optical properties. The plasmonic properties of the colloids are tuned by selective overgrowth of the gold cores with either gold or silver shells of variable thicknesses. This library of particles is used for monolayer preparation by self-assembly at the air/water interface and subsequent transfer onto solid supports. These monolayers of hexagonally packed particles have very similar particle densities and center-to-center distances, independent of the metal core composition and size. Consequently, our simple and robust bottom-up approach allows us to precisely tailor the optical properties of solid surfaces.
Introduction
Optically functional coatings are critical to materials science with widespread applications in sensing,1–7 photovoltaics8–12 and metamaterials.13–18 Ideal building blocks for such coatings are noble metal nanoparticles due to their ability to support localised surface plasmon resonances (LSPRs).19–22 These LSPRs are collective oscillations of the conduction electrons, causing intense electromagnetic fields in close proximity to the nanoparticle.2,23 The plasmon resonance position, width and intensity depend on the particle size, shape and composition, as well as on the refractive index of the surrounding medium.24–26 Fortunately, the last several decades have seen tremendous progress in the synthesis of nanoparticles with precise control over these aspects,27–31 and a broad toolbox of plasmonic particles with tailored resonances is readily available.Several challenges limit the widespread use of colloidal plasmonic particles in optical devices: colloidal stability, structural control in assembly, processability, and availability. The first three are primarily the result of an uncontrolled surface chemistry, and the last is due to the low concentrations achievable in most metal nanoparticle synthesis protocols – usually in the nanomolar range. Furthermore, typical reduction protocols yield only particles of a single size and shape population.
A clever way to extend the range of optical properties available through wet-chemical synthesis of plasmonic colloids is the controlled overgrowth of preformed precursor particles. Such seeded-growth protocols allow further tuning of size and shape starting from seed particles.32–34 Through this approach, a library of colloidal building blocks covering a broad range of optical properties (resonance position, width and number of resonances) is accessible.
In this work, we propose core–shell colloids with small, spherical gold cores and semi-transparent hydrogel shells as a convenient precursor material for seeded-growth and the realisation of plasmonic surfaces with tailored optical properties. The hydrogel encapsulation was realised by seeded precipitation copolymerisation of N-isopropylacrylamide (NIPAM) and N,N-methylenebisacrylamide (BIS) according to a well-established protocol.35 The role of the hydrogel shell is threefold: (1) to ensure colloidal stability in aqueous dispersion and different organic solvents such as alcohols. (2) To control surface coverage upon assembly of the particles into, e.g., a monolayer.36 (3) To allow for diffusion-controlled overgrowth of the gold nanoparticle cores.37–39
We employed this strategy to generate a library of plasmonic core–shell particles with the same hydrogel shell and hydrodynamic diameter but with different plasmonic metal cores. Our synthetic protocol allows us to adjust precisely the absorbance and the LSPR position of our particles within a range of wavelengths as large as 100 nm. This is confirmed by simple Mie theory calculations. These particles were assembled at the air/water interface and subsequently transferred onto glass slides. The particle density and the hexagonal packing of the transferred particle arrays are found to be independent of the size and composition of the plasmonic cores. The particle packing is solely determined by the hydrogel shell. Consequently, monolayers with very similar particle densities but significantly different absorption/scattering ratios and LSPR peak wavelength were prepared. Such structures are easily fabricated in a fast, cost-efficient and reproducible manner on cm-scale substrates. Upscaling to larger sample sizes may become possible in a roll-to-roll production approach.40 Our plasmonic coatings are particularly interesting as light management structures in hybrid photovoltaic devices.8
Experimental section
Chemicals
Ascorbic acid (Aldrich, >99%), 3-butenylamine hydrochloride (Aldrich, 97%), cetyltrimethylammonium bromide (CTAB, Aldrich, >98%), cetyltrimethylammonium chloride (CTAC, Aldrich, 25 wt%, H2O), ethanol (abs. VWR), glycine (Merck, >99.7%), H2O2 (VWR, 30%), N-isopropylacrylamide (Aldrich, 97%), N,N′-methylenebis(acrylamide) (Aldrich, 99%), NH4OHaq (Applichem, 25%), potassium peroxodisulfate (Merck, 99%), silver nitrate (Fluka, >99.5%), sodium dodecylsulfate (SDS, SERVA, 99%) and tetrachloroauric(III) acid (Aldrich, 99.999%) were used as received. Milli-Q water with a resistivity of 18.2 MΩ cm was used in all synthesis and purification procedures.Synthesis
Monolayer preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5 mixture by volume of H2O2, NH4OHaq (25%) and H2O for 30 min. Afterwards, the glass slides were cleaned with water several times and dried using nitrogen gas.
:1:5 mixture by volume of H2O2, NH4OHaq (25%) and H2O for 30 min. Afterwards, the glass slides were cleaned with water several times and dried using nitrogen gas.
Characterisation
Results and discussion
Au–PNIPAM precursor particles
To address the challenges of colloidal stability and control of particle–particle separation, we encapsulated plasmonic gold nanoparticles with rather thick hydrogel shells. This shelling procedure is based on a seeded precipitation polymerisation protocol.35 The obtained core–shell precursor particles (Au–PNIPAM) have single gold nanoparticle cores of 20 nm in diameter as obtained by TEM and soft, cross-linked poly(N-isopropylacrylamide) (PNIPAM) shells. The overall hydrodynamic diameter of the core–shell particles dispersed in water is 325 nm as determined by DLS (25 °C, swollen state). Consequently, the swollen shell has a thickness on the order of 150 nm. The porous, highly swollen structure of the hydrogel allows diffusion of small molecules into the shell, so post-modification of the gold core is possible.Overgrowth of the cores of the precursor particles
In order to achieve a whole toolbox of core–shell particles with different plasmonic properties but the same overall particle dimensions and interactions, we applied successive overgrowth of the precursor cores by gold and silver. This way we changed the diameter and composition of the cores starting from only one precursor system (Au–PNIPAM).We used surfactant assisted seeded-growth protocols in combination with mild reducing conditions to guarantee homogeneous overgrowth of the gold precursor cores and suppress secondary nucleation. Previous studies have shown that the ratio of reactants, pH, temperature, concentration of the surfactant, and nature of the halide counter ion strongly influence the final nanoparticle morphology.49,50 The surfactant cetyltrimethylammonium chloride (CTAC) was chosen for the gold overgrowth because it helps to suppress the formation of non-spherical geometries. Due to the presence of the surfactant, the redox potential of the metal salt is changed through complexation. Consequently, complete reduction takes place only at the metal surface when a mild reducing agent is used.51 Control on the core size was achieved by adding increasing amounts of feed solution and the reducing agent ascorbic acid to the precursor particles. A similar protocol was used for the silver overgrowth, which was done under basic conditions (pH approx. 10) in order to increase the reducing strength of ascorbic acid.52 The synthetic protocol was adjusted in order to get similar core sizes as for the Au@Au–PNIPAM particles by variation of the amount of seed particles.
The core–shell particles upon successive overgrowth of the precursor cores will be denoted Aux@Au20–PNIPAM and Agy@Au20–PNIPAM throughout the rest of the manuscript. Here x (y) refers to the thickness of the gold (silver) shell, and Au20 refers to the initial gold core of 20 nm in diameter. For instance, Ag4@Au20–PNIPAM is a core–shell system with a 4 nm-thick silver shell, grown on the gold core with 20 nm in diameter. Therefore, the total core diameter of this example particle is 28 nm.
Fig. 1 shows schematic depictions of the structure of the different kinds of core–shell particles along with photographs of samples before overgrowth (Au–PNIPAM) and after progressive steps of gold growth (Au@Au–PNIPAM) as well as silver growth (Ag@Au–PNIPAM). The change in core size is clearly visible from the photographs. While the precursor particles appear as a slightly red and turbid dispersion, the overgrown samples exhibit much stronger colour: red for gold and yellow for silver. This increase in colour intensity is related to the increasing absorption of the cores with larger sizes. At the same time, the photographs of the particle dispersions show that the scattering of the samples increases with the core size. This is the result of the increased scattering cross-section for the larger cores.
 | ||
| Fig. 1 Controlled overgrowth: the gold core of the initial core–shell particles, Au–PNIPAM, with gold cores of 20 nm in diameter (left column, middle) can be selectively overgrown with silver (bottom) or with gold (top) leading to an increase in core size. The respective absorbance spectra (right column) illustrate the changes of the optical properties during the overgrowth with silver (bottom) and with gold (top). The spectra were normalized to the absorbance at 400 nm (gold overgrowth) and at 300 nm (silver overgrowth). | ||
The optical impression from the photographs of the samples is confirmed by UV-Vis absorbance spectra shown in the right column of Fig. 1. The spectrum in the middle of the right column represents the absorbance of the precursor particles, Au–PNIPAM. The spectrum reveals a weak LSPR peak, originating from the gold cores, at approximately 523 nm. The strong scattering of the relatively large hydrogel shell dominates the spectrum. This is visible as an increase in absorbance for shorter wavelengths.35
The changes of the optical properties for the overgrown samples are entirely attributed to the scattering and absorption of the plasmonic cores because the hydrogel shell dimensions will be nearly unaffected by the core growth (see hydrodynamic diameter Dh in Table 1). When the precursor particles are overgrown, the absorption to scattering ratio changes, and the plasmonic properties become more pronounced (Fig. 1, right column, top and bottom). For both growth materials, Au and Ag, the plasmon resonance intensifies with increasing core size. Furthermore, the position of the plasmon resonance is affected by the overgrowth of the metal cores: for gold as growth material, a continuous shift of the plasmon resonance to longer wavelengths is observed for increasing core dimensions. For growth with silver, the plasmon resonance initially shifts to shorter wavelengths as compared to the initial Au–PNIPAM precursor particles. This is caused by the higher restoring force from the nuclei in silver as compared to gold. Upon growth of the silver shell the plasmon resonance shifts to longer wavelengths as the thickness of the silver shell increases. In order to better understand this transition, we synthesised four different metal shell thicknesses using gold and silver growth each aiming at comparable shell thicknesses for the two metals.
| Sample | D core [nm] | λ LSPR(Exp) [nm] | λ LSPR(Mie) [nm] | D h [nm] | ζ [mV] |
|---|---|---|---|---|---|
| Au–PNIPAM | 20 ± 2 | 523 | 530 | 325 | −17.3 |
| Au5@Au20–PNIPAM | 30 ± 4 | 532 | 532 | 325 | −17.3 |
| Au10@Au20–PNIPAM | 41 ± 5 | 532 | 531 | 315 | −2.9 |
| Au20@Au20–PNIPAM | 60 ± 7 | 540 | 541 | 316 | −4.2 |
| Au36@Au20–PNIPAM | 92 ± 12 | 561 | 568 | 316 | −1.1 |
| Ag4@Au20–PNIPAM | 28 ± 3 | — | 380 | 336 | −11.5 |
| Ag13@Au20–PNIPAM | 46 ± 3 | 433 | 437 | 317 | −6.8 |
| Ag26@Au20–PNIPAM | 73 ± 5 | 458 | 464 | 329 | −9.3 |
| Ag40@Au20–PNIPAM | 100 ± 6 | 496 | 503 | 336 | −9.6 |
Fig. 2 shows TEM images of the whole set of synthesised particles, including the precursor particles (a). The spherical gold nanoparticles of the precursor particles are clearly visible as circles of much higher contrast (black) as compared to the PNIPAM-shell (grey). Note that each precursor particle contains only one gold core. The TEM micrographs in Fig. 2b–e show the particles after overgrowth with gold. The diameter of the spherical cores is clearly increasing from (b) to (e). This way, gold shell thicknesses between 5 and 36 nm are accessible, corresponding to total core sizes of 30 to 92 nm. This range of sizes is significantly larger as compared to the ones realized in a previous work where we performed overgrowth of already adsorbed particles (monolayer by spin-coating).37 Furthermore, much better control on the particle morphology upon overgrowth was achieved in the present work. Non-spherical side products and secondary nucleation were not observed. Similar overgrowth results were obtained with silver, as can be seen by the TEM micrographs j–m in Fig. 2. Spherical particles with silver shell thicknesses of 4 to 40 nm closely match the results of the gold overgrowth. The core dimensions from TEM analysis are listed in Table 1.
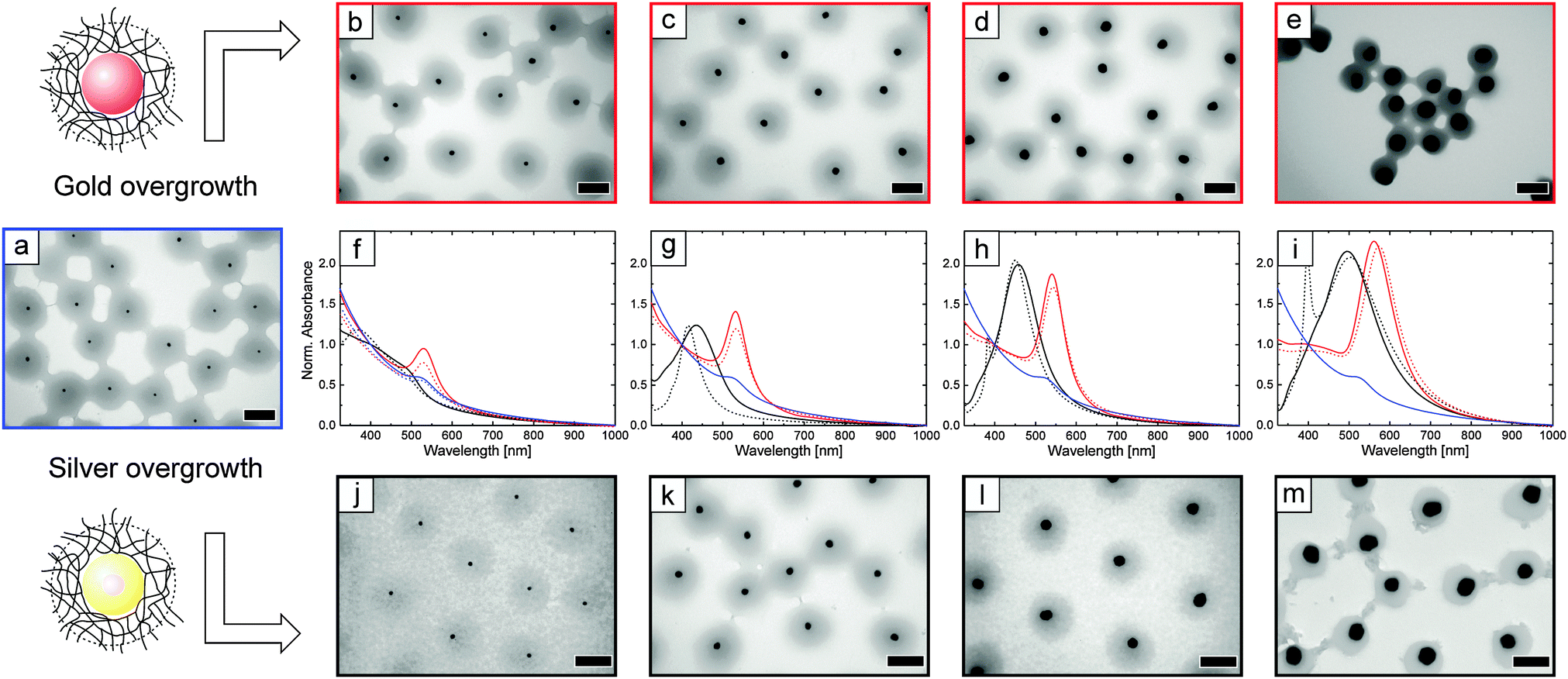 | ||
| Fig. 2 Microscopic and spectroscopic characterization of the plasmonic particles after different steps of core growth. (a) TEM image of the initial precursor particles, Au–PNIPAM. (b)–(e) TEM micrographs after overgrowth with gold. (j)–(m) TEM micrographs after overgrowth with silver. All scale bars in the TEM images correspond to 100 nm. The average core diameters of the Au@Au–PNIPAM particles are 30 nm (b), 41 nm (c), 60 nm (d) and 92 nm (e) and 28 nm (j), 46 nm (k), 73 nm (l), and 100 nm (m) for Ag@Au-PNIPAM. (f)–(i) UV-Vis absorbance spectra for the precursor particles (blue solid lines), the Au@Au–PNIPAM particles (red solid lines) and the Ag@Au–PNIPAM particles (black solid lines). Spectra for similar overall core sizes after overgrowth with gold and silver are plotted in one diagram for comparison. The dashed lines are theoretically calculated extinction spectra obtained by Mie theory. | ||
The ensemble absorbance spectra of the Au@Au–PNIPAM and Ag@Au–PNIPAM particles in aqueous dispersion are plotted in Fig. 2f–i. For comparison, spectra of Au@Au–PNIPAM and Ag@Au–PNIPAM colloids of similar core sizes are plotted in the same graphs along with theoretical extinction spectra. Additionally, the absorbance of the Au–PNIPAM precursor particles is included in each graph (blue solid lines). The spectra are normalised to the absorbance at 400 nm for an easier comparison. Almost all samples show a pronounced, single extinction peak that is attributed to the dipolar LSPR of the metal cores, with the exception of the precursor and the smallest overgrown silver particles. The small half width of the resonances manifests the low polydispersity of our particles despite several steps of sequential overgrowth. The absence of any other plasmonic modes further highlights that the core overgrowth did not produce any non-spherical particle morphologies as side products.
Having a closer look at the LSPR peak positions, a change from 523 to 561 nm is observed for gold as growth material. The resonance red shifts and the absorbance at the resonance increases with increasing core size. For silver as growth material a slightly different behaviour is observed for the first step of core growth. Overgrowth with a 4 nm-thick silver shell broadens and blue shifts the local absorbance maximum as compared to the LSPR of the initial Au–PNIPAM colloids. A clear LSPR peak cannot be clearly determined. This can be attributed to a synergistic effect between the two metals, where the gold core induces significant damping due to the mismatch of the dielectric function of the bimetallic core.53 Increasing the silver shell further to 13 nm leads to a significant change of the optical properties and a clear LSPR peak is now detectable. This LSPR is significantly blue shifted (by 90 nm) as compared to the Au–PNIPAM particles. For these particles, the bimetallic cores behave optically very similar to pure silver spheres due to the low volume fraction of the gold core (8%) and shallow skin depth of the surface plasmon. With a further increase in the silver shell thickness, the LSPR shifts to longer wavelength and becomes more intense. The LSPR positions of all particles with Ag@Au cores lie in a range of 433–496 nm. The LSPR wavelengths, λLSPR, of all samples are summarised in Table 1. The observed peak wavelengths of the resonances agree well with results from literature.32
Theoretical extinction spectra of spherical particles with core dimensions from TEM analysis are included in Fig. 2 as dotted lines. A very good match between the experimental and the calculated spectra is found for all core–shell particles with gold cores, including the Au–PNIPAM precursor particles. Table 1 shows that both sets of spectra provide nearly identical positions of the LSPR maximum, except for Au36@Au20–PNIPAM. In the latter case, small deviations of the spherical shape of these particles could explain the difference between the two LSPR positions. Simulations of Ag26@Au20–PNIPAM and Ag40@Au20–PNIPAM particles also match closely to the ensemble spectra. However, the simulated quadrupolar mode at 382 nm (Ag26@Au20–PNIPAM) and at 398 nm (Ag40@Au20-PNIPAM) is not observable in the experimental spectra. We attribute this to an irregular morphology of the silver shell in the overgrown particles. In Fig. 2m, the faceted character of the polycrystalline particles explains the deviation of the simulated spectra, which assume an ideal spherical geometry of the nanoparticles. In the case of Ag13@Au20–PNIPAM, simulation and experimental spectra differ significantly. An anisotropic growth of the silver shell or slight variation of core sizes could explain the observed deviations, which are even more pronounced for the Ag4@Au20–PNIPAM particles. We attribute this to the synergistic effect of the gold and silver as discussed above.
Plasmonic monolayer via colloidal assembly
In the next step, we used our library of core–shell colloids for the assembly into ordered monolayers. Our particles are mostly stabilised by steric interactions. The surface charge, stemming from the anionic radical initiator used in the shell growth, is small and slightly negative. Zeta potentials measured in aqueous dispersion at 25 °C reveal only small changes of the surface charge when the core size is changed (see Table 1) which might be a result of incomplete removal of CTAB or CTAC used in the core growth. Thus, we expect very similar particle–particle interactions for the whole series of particles independent of the core dimensions. For the assembly, particle dispersions in a 1:1 mixture (by volume) of H2O and ethanol were floated at the air/water interface, as described in the Experimental section.43 However, we want to stress here that the freely floating monolayer did not cover the whole accessible surface area of the air/water interface, which is rather different as compared to typical interface assemblies as realized in, e.g., a Langmuir trough. After immersing the particles at the interface, the spontaneously formed monolayers were transferred onto glass substrates by gentle lifting. Upon transfer onto glass slides, the monolayers were investigated by AFM and UV-Vis spectroscopy. AFM images were always recorded with different scan sizes and at completely independent, random sample positions. We did not find any significant deviation on the degree of order nor on the inter-particle distances at different sample positions. Representative results from these measurements are presented in Fig. 3. In addition, SEM images of the monolayer samples shown in Fig. 3a–d and i–l can be found in the ESI,† Fig. S1–S8. Images a–d show selected 10 × 10 μm2 AFM height profiles of Au@Au–PNIPAM arrays on glass, and similarly i–l the profiles of Ag@Au–PNIPAM arrays. Additionally, digital photographs of the particle monolayers on glass slides of approximately 1 cm2 are included as insets in Fig. 3a–d and i–l. The photographs show homogeneous coverage over the whole substrates as evident from the strong iridescence colours. As can be seen from the AFM images, all particles assembled into highly ordered, hexagonally packed monolayers. Large ordered domains are observable. Furthermore, a clear separation between the particles is found. This is related to the shrinkage of the previously highly swollen hydrogel shell upon drying of the monolayer. The particle density and nearest neighbour distance dc–c of the monolayers are summarised in Table 2.
 | ||
| Fig. 3 Characterization of Au@Au–PNIPAM and Ag@Au–PNIPAM monolayers. (a)–(d) Tapping mode AFM height profiles of Au@Au–PNIPAM particles. (e)–(h) UV-Vis spectra of the monolayers on glass. Spectra of Au@Au–PNIPAM and Ag@Au–PNIPAM arrays with comparable core sizes are combined in one graph (a&i, b&j, c&k and d&l). (i)–(l) AFM height profiles of Ag@Au–PNIPAM arrays. The insets show digital photographs of the monolayers on glass slides of approximately 1 cm2. The scale bars correspond to 5 mm. | ||
| Sample | d c–c [nm] | ρ [particles per μm2] | λ LSPR [nm] | Abs(λLSPR) |
|---|---|---|---|---|
| Au5@Au20–PNIPAM | 473 ± 15 | 4.49 | — | — |
| Au10@Au20–PNIPAM | 465 ± 14 | 4.76 | 536 | 0.02 |
| Au20@Au20–PNIPAM | 445 ± 15 | 4.94 | 547 | 0.053 |
| Au36@Au20–PNIPAM | 463 ± 19 | 4.45 | 573 | 0.133 |
| Ag4@Au20–PNIPAM | 461 ± 23 | 4.71 | — | — |
| Ag13@Au20–PNIPAM | 455 ± 22 | 4.77 | 484 | 0.022 |
| Ag26@Au20–PNIPAM | 453 ± 23 | 4.48 | 499 | 0.06 |
| Ag40@Au20–PNIPAM | 459 ± 20 | 4.67 | 527 | 0.166 |
| Binary sample | 455 ± 17 | 4.67 | 564 | 0.146 |
All monolayers have comparable particle densities with an average of 4.65 particles per μm2 and values of dc–c in the order of 460 nm. The inter-particle distance is large compared to the average hydrodynamic diameter of 323 nm of the particles measured with DLS (Table 2). This points to an anisotropic deformation of the hydrogel shell at the interface as has been observed previously for purely organic hydrogel particles adsorbed at a liquid/liquid interface.54 Since all monolayers show very similar values of dc–c and very regular, hexagonal packing of the particles independent on the size and composition of the plasmonic metal core, it is obvious that only the hydrogel shell drives the particle assembly. In other words, interface assembly of our metal–hydrogel core–shell particles allows the fabrication of highly ordered monolayers with nearly the same surface coverage from Au@Au and Ag@Au nanoparticles covering a diameter range from 20 to 100 nm. Because the monolayer structure and density is nearly the same for all samples, we can now investigate the dependence of the optical properties on the core size and material using far-field extinction spectroscopy.
Fig. 3e–h show UV-Vis extinction spectra of the monolayers on glass. For comparison, the absorbance of Ag@Au–PNIPAM and Au@Au–PNIPAM monolayers with comparable sized cores are plotted in one graph. The spectra show very similar results as compared to those recorded in solution (Fig. 2). It is worth noting that the illuminated area on the substrates is the same for all samples and that the spectra shown in Fig. 3 were not normalised nor shifted. Therefore, we conclude that the increase in absorbance is solely related to the increasing core volume, since roughly the same number of particles is probed for each sample.
The determined LSPR peak positions and values of absorbance at the resonance are listed in Table 2. For the smallest core sizes (Fig. 3e) the weak LSPR signal is related to the small absorption and scattering cross-sections of these rather small metal nanoparticle cores. With increasing core size, the plasmonic properties of the samples become more pronounced.
Fig. 4a shows the dependence of the LSPR position (λLSPR) as a function of the metal shell thickness obtained after each step of core growth, both of the particles in solution and of the monolayers on glass. Because of the high sensitivity of the plasmon resonance to the local refractive index, the peak positions of the particle monolayers on glass are shifted to longer wavelengths for both types of particles.26 This effect is stronger for the cores overgrown with silver. Furthermore, Fig. 4b shows that the absorbance of the different monolayers increases with the plasmonic shell thickness of the overgrown cores, independent of the growth material, i.e., silver and gold. For both sets of samples, the maximum shell thicknesses exhibit an absorbance increase by almost a factor of 10. This nicely underlines that our colloidal library not only allows the fabrication of plasmonic coatings with tailored resonance positions but also the tuning of the absorbance and consequently transmission of the monolayer.
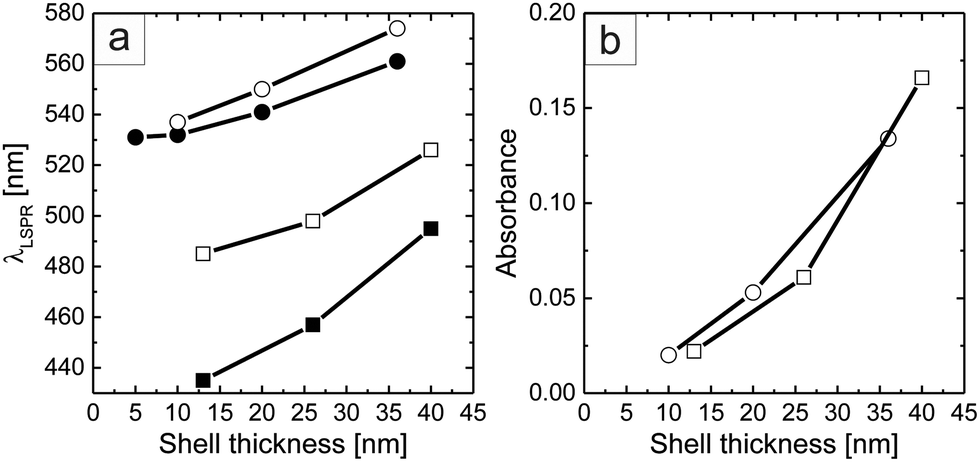 | ||
| Fig. 4 Results from analysis of the UV-Vis absorbance spectra. (a) LSPR peak wavelength, λLSPR, and (b) absorbance at the LSPR maximum as a function of the shell thickness obtained after sequential overgrowth of the gold cores of the precursor particles. Results are shown for spectra measured from aqueous dispersions (filled symbols) and monolayers on glass (open symbols) and for Au@Au–PNIPAM (circles), and Ag@Au–PNIPAM (squares). | ||
Mixed monolayer
To further demonstrate the tunability of the optical properties of our self-assembled particle arrays, we prepared a 1:1 mixed monolayer (by weight) of Ag40@Au20–PNIPAM and Au36@Au20–PNIPAM particles. Fig. 5a and b show an AFM height profile and a FE-SEM image of the binary monolayer, respectively. The images indicate homogeneous surface coverage and an ordered packing of the particles, very similar to the monolayers of single core species shown in Fig. 3. The average particle center-to-center distance of 452 ± 19 nm is similar to the previously discussed monolayers. Due to their same hydrodynamic dimensions (see Table 1) and hydrogel shell morphology, particles assemble into ordered structures with inter-particle distances and order parameters independent of the plasmonic core size and composition.
 | ||
| Fig. 5 Mixed particle monolayer on glass prepared from a 1:1 mixture (by weight) of Ag40@Au20–PNIPAM and Au36@Au20–PNIPAM particles. (a) AFM height profile, (b) FE-SEM image of the mixed monolayer, (c) UV-Vis absorbance spectra measured at five different positions of the sample (solid lines). Note, that the spectra look almost identical. For comparison, the absorbance of Ag40@Au20–PNIPAM (black dashed line) and Au36@Au20–PNIPAM (red dashed line) monolayers are included. | ||
Fig. 5c shows absorbance spectra of the mixed monolayer recorded at five different positions of the sample. The spectra match very nicely which manifests the random but homogeneous distribution of the Ag@Au–PNIPAM and Au@Au–PNIPAM particles throughout the whole monolayer.
Additionally, the absorbance spectra of monolayers prepared from the single systems, Ag40@Au20–PNIPAM (black dotted line) and Au36@Au20–PNIPAM (red dotted line), are included in the diagram for comparison. The peak wavelength and absorbance of the mixed monolayer are in between the properties of the individual colloidal building blocks. Thus, blending of Ag@Au–PNIPAM and Au@Au–PNIPAM particles is a quick and easy technique to further tune the absorption and scattering properties of the particle arrays.
Conclusions
With this work, we provide an experimental example of easily fabricated, macroscopic, plasmonic monolayers with controlled surface coverage, where the plasmonic properties are systematically varied through the chemical pre-treatment (core overgrowth) of one type of colloidal core–shell building blocks. Seeded growth was applied as a versatile protocol to tune the dimensions, composition, and hence, the optical properties of the plasmonic cores with almost nanometre precision. Our synthetic protocol based on Au–PNIPAM core–shell colloids produces neither secondary nucleation nor non-spherical side products, allowing the synthesis of a library of monodisperse, spherical metal particles with core sizes ranging from 20 to 100 nm in diameter. Furthermore, self-assembly at the air/water interface was applied as a sophisticated technique that allows for surface patterning with highly ordered particle monolayers. This approach works for all the particles in our library. The obtained monolayers have uniform surface coverage and inter-particle distances. Due to the difference in core size and composition, we can adjust the LSPR position of the monolayers between 484 and 573 nm by the choice of the building blocks. We demonstrated that blending of core-shell particles with different cores is easily possible, providing another handle on optically fine tuning the plasmonic properties of colloidal coatings. In the future, reducing the hydrogel shell dimensions of the core–shell particles may allow for the realization of smaller inter-particle separations in the monolayer. In this case, plasmon resonance coupling might be usable as another parameter to tune the plasmonic properties of the monolayer.Acknowledgements
MK and TH acknowledge financial support via the Deutsche Forschungsgemeinschaft (DFG) through the SFB 840. MK, JPSF, and KV are grateful for financial support via the Deutsche Forschungsgemeinschaft (DFG) through the Emmy Noether programme. The authors acknowledge Markus Retsch from the University of Bayreuth (Physical Chemistry I) for fruitful discussion and assistance with the interface assembly and Martina Heider (BIMF, University of Bayreuth) for her assistance with the FE-SEM investigations.References
- C. Hamon, S. Novikov, L. Scarabelli, L. Basabe-Desmonts and L. M. Liz-Marzan, ACS Nano, 2014, 8, 10694–10703 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- A. J. Haes, C. L. Haynes, A. D. McFarland, G. C. Schatz, R. R. Van Duyne and S. L. Zou, MRS Bull., 2005, 30, 368–375 CrossRef CAS.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS PubMed.
- N. Vogel, M. Jung, N. L. Bocchio, M. Retsch, M. Kreiter and I. Köper, Small, 2010, 6, 104–109 CrossRef CAS PubMed.
- T. Thai, Y. Zheng, S. H. Ng, S. Mudie, M. Altissimo and U. Bach, Angew. Chem., Int. Ed., 2012, 51, 8732–8735 CrossRef CAS PubMed.
- M. Mueller, M. Tebbe, D. V. Andreeva, M. Karg, R. A. Alvarez Puebla, N. Pazos Perez and A. Fery, Langmuir, 2012, 28, 9168–9173 CrossRef CAS PubMed.
- M. Karg, T. A. F. König, M. Retsch, C. Stelling, P. M. Reichstein, T. Honold, M. Thelakkat and A. Fery, Mater. Today, 2015, 18, 185–205 CrossRef CAS.
- H. Choi, J. P. Lee, S. J. Ko, J. W. Jung, H. Park, S. Yoo, O. Park, J. R. Jeong, S. Park and J. Y. Kim, Nano Lett., 2013, 13, 2204–2208 CrossRef CAS PubMed.
- H. L. Gao, X. W. Zhang, Z. G. Yin, H. R. Tan, S. G. Zhang, J. H. Meng and X. Liu, Appl. Phys. Lett., 2012, 101, 133903 CrossRef.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- P. Reineck, G. P. Lee, D. Brick, M. Karg, P. Mulvaney and U. Bach, Adv. Mater., 2012, 24, 4750–4755 CrossRef CAS PubMed.
- D. Chanda, K. Shigeta, S. Gupta, T. Cain, A. Carlson, A. Mihi, A. J. Baca, G. R. Bogart, P. Braun and J. A. Rogers, Nat. Nanotechnol., 2011, 6, 402–407 CrossRef CAS PubMed.
- O. Hess, J. B. Pendry, S. A. Maier, R. F. Oulton, J. M. Hamm and K. L. Tsakmakidis, Nat. Mater., 2012, 11, 573–584 CrossRef CAS PubMed.
- W. Lewandowski, M. Fruhnert, J. Mieczkowski, C. Rockstuhl and E. Gorecka, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- C. M. Soukoulis and M. Wegener, Nat. Photonics, 2011, 5, 523–530 CAS.
- Z. H. Jiang, S. Yun, F. Toor, D. H. Werner and T. S. Mayer, ACS Nano, 2011, 5, 4641–4647 CrossRef CAS PubMed.
- Q. Q. Gan, F. J. Bartoli and Z. H. Kafafi, Adv. Mater., 2013, 25, 2385–2396 CrossRef CAS PubMed.
- M. Arnold, V. C. Hirschfeld-Warneken, T. Lohmüller, P. Heil, J. Blümmel, E. A. Cavalcanti-Adam, M. López-García, P. Walther, H. Kessler, B. Geiger and J. P. Spatz, Nano Lett., 2008, 8, 2063–2069 CrossRef CAS PubMed.
- L. Jiang, C. Zou, Z. Zhang, Y. Sun, Y. Jiang, W. Leow, B. Liedberg, S. Li and X. Chen, Small, 2014, 10, 609–616 CrossRef CAS PubMed.
- M.-H. Lin, H.-Y. Chen and S. Gwo, J. Am. Chem. Soc., 2010, 132, 11259–11263 CrossRef CAS PubMed.
- A. Tao, P. Sinsermsuksakul and P. Yang, Nat. Nanotechnol., 2007, 2, 435–440 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- L. M. Liz-Marzan, Langmuir, 2006, 22, 32–41 CrossRef CAS PubMed.
- C. Novo, A. M. Funston, I. Pastoriza-Santos, L. M. Liz-Marzan and P. Mulvaney, J. Phys. Chem. C, 2008, 112, 3–7 CrossRef CAS.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781–1804 CrossRef CAS PubMed.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- S. J. Tan, M. J. Campolongo, D. Luo and W. Cheng, Nat. Nanotechnol., 2011, 6, 268–276 CrossRef CAS PubMed.
- J. Rodríguez-Fernández, J. Pérez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, Langmuir, 2006, 22, 7007–7010 CrossRef PubMed.
- N. Pazos-Perez, F. J. Garcia de Abajo, A. Fery and R. A. Alvarez-Puebla, Langmuir, 2012, 28, 8909–8914 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- M. Karg, S. Jaber, T. Hellweg and P. Mulvaney, Langmuir, 2011, 27, 820–827 CrossRef CAS PubMed.
- S. Jaber, M. Karg, A. Morfa and P. Mulvaney, Phys. Chem. Chem. Phys., 2011, 13, 5576–5578 RSC.
- M. B. Muller, C. Kuttner, T. A. F. Konig, V. V. Tsukruk, S. Forster, M. Karg and A. Fery, ACS Nano, 2014, 8, 9410–9421 CrossRef PubMed.
- R. Contreras-Caceres, J. Pacifico, I. Pastoriza-Santos, J. Perez-Juste, A. Fernandez-Barbero and L. M. Liz-Marzan, Adv. Funct. Mater., 2009, 19, 3070–3076 CrossRef CAS.
- J. Clara-Rahola, R. Contreras-Caceres, B. Sierra-Martin, A. Maldonado-Valdivia, M. Hund, A. Fery, T. Hellweg and A. Fernandez-Barbero, Colloids Surf., A, 2014, 463, 18–27 CrossRef CAS.
- N. Vogel, S. Goerres, K. Landfester and C. K. Weiss, Macromol. Chem. Phys., 2011, 212, 1719–1734 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- R. Contreras-Caceres, I. Pastoriza-Santos, R. A. Alvarez-Puebla, J. Perez-Juste, A. Fernandez-Barbero and L. M. Liz-Marzan, Chem. – Eur. J., 2010, 16, 9462–9467 CrossRef CAS PubMed.
- N. Vogel, C. Fernández-Lopez, J. Párez-Juste, L. M. Liz-Marzán, K. Landfester and C. K. Weiss, Langmuir, 2012, 28, 8985–8993 CrossRef CAS PubMed.
- M. Retsch, Z. Zhou, S. Rivera, M. Kappl, X. S. Zhao, U. Jonas and Q. Li, Macromol. Chem. Phys., 2009, 210, 230–241 CrossRef CAS.
- A. Moroz, Ann. Phys., 2005, 315, 352–418 CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Wiley-VCH Verlag GmbH, 2007 Search PubMed.
- G. M. Hale and M. R. Querry, Appl. Opt., 1973, 12, 555–563 CrossRef CAS PubMed.
- S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzán and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- J. Rodriguez-Fernandez, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, J. Phys. Chem. B, 2005, 109, 14257–14261 CrossRef CAS PubMed.
- B. Rodriguez-Gonzalez, A. Burrows, M. Watanabe, C. J. Kiely and L. M. Liz-Marzan, J. Mater. Chem., 2005, 15, 1755–1759 RSC.
- M. Tebbe, C. Kuttner, M. Mayer, M. Maennel, N. Pazos-Perez, T. A. F. König and A. Fery, J. Phys. Chem. C, 2015, 119, 9513–9523 CrossRef CAS PubMed.
- K. Geisel, L. Isa and W. Richtering, Langmuir, 2012, 28, 15770–15776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc02115d |
| This journal is © The Royal Society of Chemistry 2015 |