
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-area wet-chemical deposition of nanoporous tungstic silica coatings
K. H.
Nielsen
a,
K.
Wondraczek
b,
U. S.
Schubert
cd and
L.
Wondraczek
ad
aLaboratory of Glass Science, Otto Schott Institute of Materials Research, University of Jena, Fraunhoferstrasse 6, 07743 Jena, Germany. E-mail: lothar.wondraczek@uni-jena.de
bLeibniz Institute of Photonic Technology e.V., Albert Einstein Strasse 9, 07745 Jena, Germany
cLaboratory of Organic and Macromolecular Chemistry, University of Jena, Humboldtstrasse 10, 07743 Jena, Germany
dCenter of Energy and Environmental Chemistry (CEEC), University of Jena, Max-Wien-Platz 1, 07743 Jena, Germany
First published on 28th August 2015
Abstract
We report on a facile procedure for synthesis of nanoporous coatings of tungstic silica through wet-chemical deposition and post-treatment of tungsten-doped potassium silicate solutions. The process relies on an aqueous washing and ion exchange step where dispersed potassium salt deposits are removed from a 150 nm silicate gel layer. Through an adjustment of the pH value of the washing agent within the solubility regime of a tungstic salt precursor, the tungsten content of the remaining nanostructured coating can be controlled. We propose this route as a universal approach for the deposition of large-area coatings of nanoporous silica with the potential for incorporating a broad variety of other dopant species. As for the present case, we observe, on the one hand, antireflective properties which enable the reduction of reflection losses from float glass by up to 3.7 percent points. On the other hand, the incorporation of nanoscale tungstic precipitates provides a lever for tailoring the coating hydrophilicity and, eventually, also surface acidity. This may provide a future route for combining optical performance with anti-fouling functionality.
1. Introduction
Nanoporous SiO2-based coatings assume a key role in functional glass substrates, for example, as anti-reflective (AR) thin films for cover glasses in solar energy conversion systems,1–3 or in biomedical slides for the immobilization of biomolecules.4 For example, in photovoltaic systems, about 25% of the employed cover glasses comprise AR coatings, with an anticipated increase in market penetration to more than 80% by 2020. The functionality of such cover glasses is usually judged in terms of the the glass efficiency factor, ηGL, which derives from a convolution of light transmission weighted over cell sensitivity, angle-dependence of light transmission, and sensitivity to photolysis and solarization. A value of ηGL = 1 represents the reference case where no light is lost at any angle of incidence due to reflection, scattering, internal absorption or gradual degradation of the glass cover.5 The best-in-class single-side coated low-Fe glasses today reach an ηGL of ∼0.98, which requires the AR coating to provide a transmission increase of >3 percent-points in the wavelength regime of ∼500–700 nm. Today, although AR coatings are commercially available in a variety of types (usually differentiated through the process of deposition), they still constitute a topic of intense research interest. Beyond further improving the optical performance, the interest is to mostly target alternative strategies of synthesis and post-processing which enable, e.g., tailored porosity and pore-size distribution, enhanced mechanical, thermal or chemical stability, and secondary doping and addition of further functionality. Besides physical vapor deposition, the most prominent deposition processes involve sol–gel methods, where a coating of colloidal silica is generated from solutions or suspensions of alkali silicates,3 silicon alkoxides,6 or silica nanoparticles.7 To ensure the reliability of the coatings, in particular, during outdoor exposure over an expected lifetime of up to 40 years8 (such as in the case of solar modules), mechanical1 and chemical stability9,10 are very important parameters. Accumulation of dirt and dust significantly reduces the performance of photovoltaic modules, and self-cleaning has become an interesting feature for further improvement of long-term performance. Hence, coatings have been modified by tailoring their morphology,7 hydrophobicity,2 or photocatalytic properties.6 Also, biofilms and immobilized microorganisms – while specifically targeted in some applications – are very likely to cause corrosion and irreversible degradation, for example, impeding the light transmission of solar glasses,11,12 or providing a humid surface which is susceptible to increased dust adhesion.7 Therefore, anti-bacterial functionality has become yet another attractive property of cover and substrate glasses. Such anti-bacterial effects can be achieved through tailoring interaction of the surface glass with microbial deposits, e.g., by adding bactericidal metals.13 In addition, a photocatalytic functionality may be considered for controlling the adhesion and passive removal of organic deposits. For the latter, addition reactions of TiO2 are usually considered.6,14 As an additional advantage, the incorporation of transition metal oxides such as TiO2 and ZrO2 may also provide the coating with enhanced chemical stability.9Compounds with high surface acidity, such as molybdenum and tungsten oxide, have been proven to be a durable and efficient way of reducing microbial growth.15,16 Their functioning relies on an acidic surface reaction with ambient humidity, locally creating a low pH value,16
| WO3 + H2O ↔ H2WO4 | (1) |
| H2WO4 + 2H2O ↔ 2H3O+ + WO42− | (2) |
Tungsten trioxide is well-known for its high surface acidity. It has therefore been proposed for use as an undoped material17 or as a dopant in, e.g., porous SiO2 matrices in catalysis.18 Making use of the electronic properties of the polyvalent tungsten ion, thin films of pure WO3 can further be used as electron donors or acceptors, for example, in electro-chromic devices.19,20
Here, we focus on a versatile procedure for creating nanoporous films of tungstic silica, which, as noted above, may act as a component in a variety of applications. Momentarily, however, we focus on the potential for creating high surface acidity, such as is of key importance in an antibacterial and anti-fouling functionality. We use the fact that tungstic acid is highly soluble in alkaline solutions, yielding the tungstate anion, WO42−, which readily condensates upon lowering the pH value19 to result in various kinds of {WO3} species, such as colloids,21 hydrates, or gels.22 Furthermore, alkaline dissolved tungstate can be precipitated by divalent cations such as calcium.23 This behavior is comparable to that of aqueous silicates24 and suggests that it could be combined for the preparation of novel types of thin films. We start with a previously developed procedure for generating large-area nanoporous and antireflective silica coatings from alkali silicate solutions.3
Whereas such silicate coatings are normally washed with demineralized water to remove the alkali species,3 here, we conduct an ion exchange process in which the combined coatings undergo an acid wash to precipitate and embed tungsten oxide in the thin film. A similar effect may be brought about by an ion exchange process of the thin gel film25 with the aforementioned divalent cations. We demonstrate an efficient route and mechanism of synthesis, suitable for generating anti-reflective and auto-acidic thin films containing SiO2 and WO3 at low cost.
The present analysis aims to characterize the optical properties, the morphology and interaction with atmospheric water of such coatings so as to provide a perspective of the versatility of the general synthesis procedure.
2. Materials and methods
2.1 Coating synthesis
Precursor solutions were prepared from 29 wt% aqueous potassium silicate solutions with a SiO2/K2O molar ratio of 4 (pH = 11) (BASF, Düsseldorf, Germany), and analytical-grade sodium tungstate dihydrate, Na2WO4·2H2O (Reachim Ltd, Soju Chimexport, Russia) through dilution with demineralized water and mixing in polyethylene bottles with subsequent ageing under magnetic stirring for up to six days. Silicate precursor solutions containing 6 wt% K2Si4O9 were used for the experiments. The dry mass constituents of the doped precursor solutions are given in the chart in Fig. 1(A). An undoped solution is long-term stabile and remains free of precipitates, whereas the doped solution was optically stable over weeks, but eventually formed a white precipitate.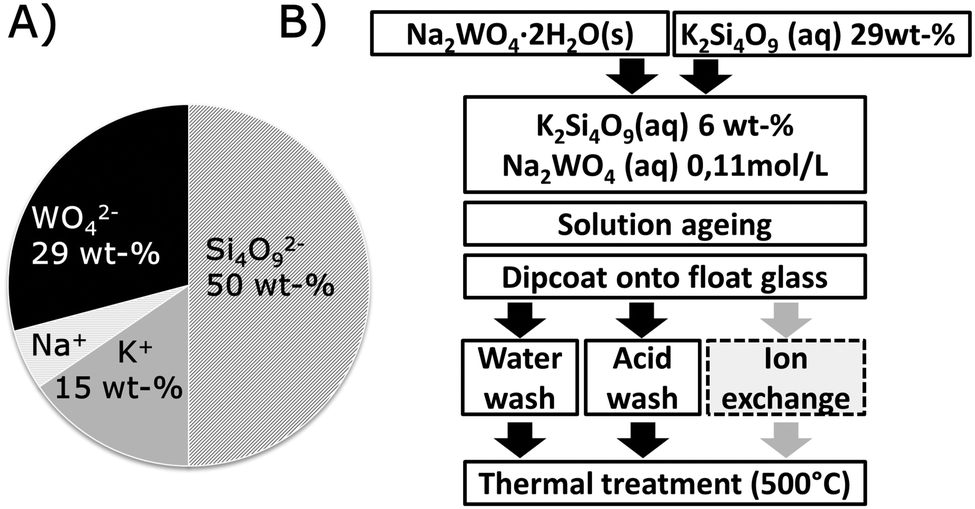 | ||
| Fig. 1 (A) Nominal composition by weight for the dry constituents of the precursor solution. (B) Preparation scheme for sol–gel derived tungstic acid doped SiO2 rich coatings. | ||
Sol–gel coatings were subsequently applied from such solutions by dip coating onto commercial low-iron float glass (2 mm), and allowed to dry for 5 minutes under ambient conditions. The thus-formed gel was then washed for one minute to remove alkali ions3 in either demineralized water, or in a 0.1 M HCl solution prepared from analytical grade 37 wt% HCl (AnalaR NORMALAPUR, VWR International, Dresden, Germany). Reference samples were prepared in parallel from precursor solutions without tungstate addition. After the washing step, the samples were allowed to dry vertically for at least an hour under ambient conditions, and eventually thermally treated for 20 min at 500 °C in a muffle furnace (LM312, Linn high Therm, Eschenfelden, Germany). High temperature treatment serves to sinter and dehydrate the silicate coating3 to eventually form tungstic oxide phases,19 but it is limited by the substrate glass (Tg = 566 °C). The full procedure is schematically summarized in Fig. 1(B). As the first step of characterization, the thickness of each coating was determined by atomic force microscopy (AFM, Ultrascan, Surface Imaging Systems GmbH, Herzogenrath, Germany), typically lying within 95 to 150 nm for water- and acid-washed coatings, respectively, which corresponds to the thickness observed for comparable coatings of pure potassium silicate.3
2.2 Coating analysis
The tungsten content of the coated samples was investigated using laser ablation (LSX-213G2+, Cetac, Omaha, USA) inductively coupled plasma mass spectroscopy (X-Series II, ThermoFisher Scientific, Bremen, Germany) (LA-ICP-MS). Measurements are repeated five times for each sample over an ablation length of 2.6 mm, creating a crater with a depth of several micrometers. For estimating the tungsten content within the washed coating, the observed tungsten content of the unwashed coating is compared with that of the washed coatings. Similar experimental settings were used for all analyses procedures to ensure comparability of all collected data. Measurements were conducted in parallel reference glasses, coated glasses, and coated glasses which were stored for 72 h in 50 °C demineralized water to evaluate the chemical stability of the incorporated tungsten species and, in particular, to judge eventual losses through reactions with ambient humidity.Transmission spectra of the coated glass were obtained on a Cary 5000 spectrophotometer equipped with a 150 mm integration sphere (Agilent, Santa Clara, United States), considering both direct and integral transmission.
Scanning electron micrographs (SEM) were obtained using a JSM7001F (Jeol Ltd, Tokyo, Japan) equipped with a TTL detector. Prior to measurements, the sample was coated with a thin carbon layer (Auto 306, Edwards, Crawley, United Kingdom). In addition and as already noted, AFM surface scans were obtained using an Easyscan 2 (Nanosurf, Langen, Germany) equipped with PPP-NCLR probes (Nanosensors, Neuchâtel, Switzerland) in dynamic force mode (non-contact measurement).
The effect of the washing process on the gel phase which was formed after drying was investigated on a dissipative quartz crystal micro-balance (Q-Sense E1, LOT-QuantumDesign, Darmstadt, Germany). For this, the precursor solution was applied by spin coating to gold coated sensors treated with amino propyl triethoxy silane for increasing the hydrophilicity. The coated sensors were measured before and after washing in a flow cell with either demineralized water or 0.1 M HCl. The resonance frequency shift of the treated QCM sensor compared to the untreated sensor is directly related to the mass change, as described in the simplified Sauerbrey equation:26
| Δm = −C/nΔf, | (3) |
Coating interaction with atmospheric humidity is necessary for the acidic surface reaction to take place and may give information on porosity of the coating. Investigations commenced by recording in situ measurements of variations of the optical pathlength as a function of the surrounding humidity on an interference microscope (Jenaval Interphako u.map, Carl Zeiss, Jena, Germany) equipped with a THMS600 heating stage (Linkam Scientific Instruments Ltd, Surrey, UK) in a controlled atmosphere. Humid air and dry argon were flushed over the samples in cycles of 90 min in the case of dry argon, and 60 or 90 min in the case of humid air. During such cycling, the coating interacts with ambient water through physical or chemical sorption, which alters its optical properties. Experiments were conducted at 30 °C on microscope slides coated on one side with a tungsten doped coating and a reference coating, both having undergone an acid wash and subsequent thermal treatment prior to measurement. This experimental set-up was previously developed27 for the study of capillary condensation in similar but undoped nanoporous films.
2.3 Coating solution analysis
Investigations on the precursor solutions were conducted using DLS (λ = 633 nm) using a Zetasizer Nano Zs (Malvern Instruments, Herrenberg, Germany) at 25 °C. The measurement was repeated three times for 15 seconds with counts collected at 173°. The given particle size values are extracted from the number particle size distribution. Samples were prepared in glass vials and kept under magnetic stirring for up to seven days before measurements. At a first view, the obtained data confirmed the predominant presence of finely dispersed particles with diameters below 100 nm. After prolonged storage for seven days, the solutions became more complex and, in particular, showed evidence of phase separation.2.4 Powder synthesis
For investigations on the mechanism of tungsten incorporation in the coating, powder samples were also prepared from tungstate-doped and undoped solutions of potassium silicates by drying the precursor material in a furnace (Memmert, Schwabach, Germany) at 80 °C. The powder was subsequently ground, sieved to obtain the 15 to 320 μm fraction and washed in a Büchner funnel equipped with filter paper with either demineralized water or 0.1 M HCl to simulate washing of the coating gel. Before measurements or thermal treatment, the powders were dried again. Some of the powdered samples were further heat treated at 550 °C in a fine-controlled furnace (DIL 402ES, Netzsch Gerätebau, Selb, Germany). The thermal treatment serves to mimic that of the coated glass. The slightly higher temperature for the powder samples derives from a transition observed in the thermal analysis at 450 °C.2.5 Powder analysis
Powder samples were characterized by X-ray diffraction (D 5000, Siemens, Erlangen, Germany), and also by thermal analysis in combination with simultaneous mass spectrometry (MS) (STA 449F1, Netzsch, Selb, Germany with QMS 403, Netzsch, Selb, Germany) for the detection of H2O (n = 18) and CO2 (n = 44) release up to 600 °C at a heating rate of 10 K min−1. Finally, a Raman microscope (inVia, Renishaw, Pliezhausen, Germany) was used to collect the spectra from the powdered samples excited with a 532 nm laser.3. Results and discussion
3.1 Effect of washing on the tungsten content in the coating
The results of the elemental analysis by LA-ICP-MS are given in Table 1. The data demonstrate that after acid-wash, tungsten remains in the coatings, whereas in the case of synthesis via washing in water, practically no tungsten remains. It has to be reminded that the analysis method not only analyzes the coating alone but, through deep ablation, comprises contributions from the substrate as well. However, since the substrate contains neglectable amounts of tungsten, the measured totality practically represents the amount of tungsten trapped in the coating alone.| Sample | W content (ppm) |
|---|---|
| Reference glass | 0.3 |
| Coated glass, unwashed | 1030 ± 70 |
| Sample | Solution age (days) | After 500 °C treatment | After leaching |
|---|---|---|---|
| Coated glass, acid wash | 0 | 430 ± 12 | 420 ± 39 |
| 1 | 470 ± 12 | 556 ± 6 | |
| 6 | 420 ± 30 | 188 ± 4 | |
| Coated glass, water wash | 0 | 3.3 ± 0.3 | |
| 1 | 9 ± 1 | ||
| 6 | 10.1 ± 0.7 | ||
The values summarized in Table 1 indicate that the acid washed coatings contain ∼40 wt% of the original amount of the W dopant, corresponding to an equivalent WO3 content of ∼18 wt%, assuming no loss of SiO2 during washing. Furthermore, the tungsten content appears to be independent of solution ageing time, indicating the absence of a reaction between tungsten and silica compounds upon solution ageing. Table 1 also shows data for samples that were leached in demineralized water over 72 h at 50 °C. Leaching was done to obtain a rapid estimate of the coating's sensitivity to weathering under humid conditions. The acid-washed coatings largely retain the tungsten component after leaching, although there is some variation between the samples. Thus the acid wash enables the incorporation of tungsten in a form that after thermal treatment is stable towards hydrolysis and leaching. The variation observed after leaching may derive from lateral heterogeneities in the coating.
3.2 Effect of washing on optical properties
The transmission spectra of the coated samples are presented in Fig. 2(A). The morphological effect of the different types of washing which impact these transmission properties is sketched-out in Fig. 2(B). | ||
| Fig. 2 (A) Optical transmission spectra of washed and thermally treated coating at solution age of 0 days. Blue: water wash. Red: acid wash. Black: reference (uncoated substrate). The inset shows the transmission increase maximum as a function of solution ageing time. (B) Models illustrating the observed effect of the water and acid wash on coating properties, with the water wash causing a loss of W-species and gel compaction. | ||
Coatings prepared using either acid or water wash increase the maximum direct optical transmission of float glass by 2.6 ± 0.2 and 3.7 ± 0.2 percent points, with a transmission maximum at ∼570 nm and ∼520 nm, respectively. For comparison, coatings derived from an undoped silicate solution increased transmission by 3.0 ± 0.2 and 3.2 ± 0.2 percent points when prepared with water and with acid wash, respectively. Within the observed range of solution ageing, the transmission increase appears to be independent of ageing time, see the inset in Fig. 2(A). This is in good agreement with the observations of the effect of washing on the overall W-content as discussed in the previous paragraph. In general, the W-doped coatings exposed to washing with demineralized water have a slightly higher transmission than those exposed to acid wash. This indicates a somewhat higher porosity for these coatings, apparently due to the absence of tungsten precipitates.
The antireflective properties of thin films derive from destructive interference3 and a low reflective index of the coating. While the index is largely affected by porosity, the wavelength position of maximum transmission λ depends on the coating thickness d via d = λ/4. The blue shift of maximum transmission observed on acid and water washed coating, that is, from 570 to 520 nm, clearly reflects a lower coating thickness observed for the water-washed coating, as also observed by AFM. The lower thickness most probably reflects more pronounced gel compaction during coating consolidation. As will be discussed later, this is not in contrast to the observation of lower refractive index and higher coating porosity.
Amorphous silica and tungsten oxide have refractive indices of 1.46 and ∼2.1 (at 750 nm),28 respectively. Considering the refractive index of the glass substrate which is ∼1.52, this does not by intuition lead to significantly increased transmission because the selection rule for optimal compensation of Fresnel reflection is not fulfilled, i.e., the coating indices are notably larger than the square root of the substrate index. However, the measured higher transmission values do indicate an overall lower refractive index of the washed coatings which is ascribed to an increased porosity of the coatings. We have to note here that in contrast to what is often done, we may not readily assume that all pores are filled with air. Instead, capillary condensation of ambient water has to be taken into account.29
Fig. 2(B) illustrates the possible effects of the two types of washing on the morphology of the resulting coating structure; subsequent heat treatment further densifies the films. The higher transmission value after water-wash indicates an overall lower refractive index which is ascribed to the more or less complete removal of the tungsten species resulting in larger interstitial volumes within the SiO2-network as compared to the acid-wash (which leaves about 40 wt% of the W-species trapped in the coating). The evidence of the antireflective properties of the acid-washed coating is attributed to the still high porosity, despite the remaining WO3 in the coating. Interestingly, at this point, while washing in water removes tungsten from the consolidated coating, its resulting refractive index is lower as compared to the reference coating prepared from pure potassium silicate solution. This indicates that the addition of tungstic salts acts on colloid formation in a way that results in a subsequently more open coating structure.
3.3 Morphology of doped and undoped films
Fig. 3(A) exemplarily shows micrographs on two different length scales of the coating to reveal microscopic film homogeneity and the assembly of constituting colloids on the sub-micrometric scale. On a scale of about 10 μm × 10 μm (and above), there are no indications of particle agglomeration, phase separation, delamination or other film defects. However, the AFM surface scans on the 1 μm × 1 μm scale (Fig. 3(B)) clearly indicate the existence of particles or agglomerates for both the acid and water washed coatings. This is consistent with earlier measurements on undoped coatings;3 the particles are expected to be pure SiO2. Roughness, as determined from AFM scans, was comparable for both types of coatings, rrms = 2.8 nm. Results of a particle size analysis applying a watershed algorithm is given in Fig. 3(C). The particles were found to be approximately 30 nm, with the particles of the acid washed coating being very slightly larger compared to the water-washed films. For the acid washed samples, no secondary particle distribution which could derive from the incorporation of particulate WO3 species was observed.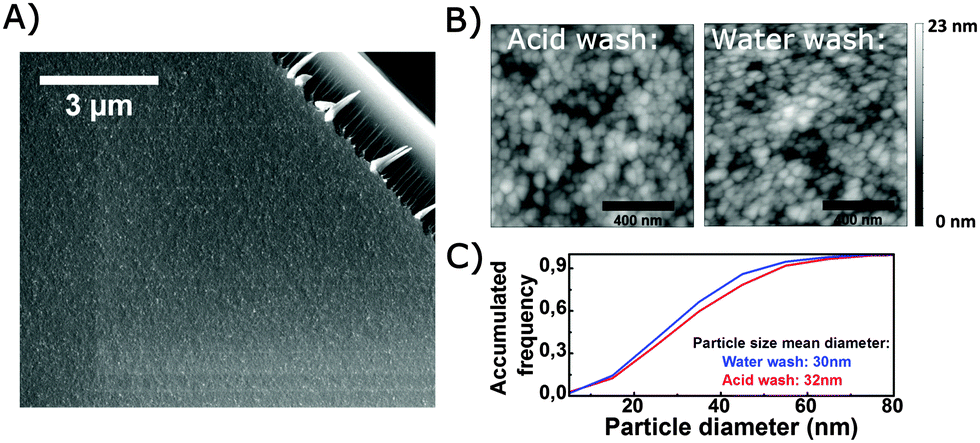 | ||
| Fig. 3 (A) SEM micrograph of an acid washed coating. The upper right corner is a fractured glass surface. (B) AFM surface scans of an acid and water washed sample (C) accumulated frequency for particles on AFM scans. Particle size is estimated by a watershed algorithm. | ||
3.4 Effect of humidity on coating performance
In the following, the behavior of the doped coating upon exposure to humidity is investigated to evaluate the interaction with water.The so-called optical breathing effect of the coating when exposed to humidity is shown in Fig. 4(A). The cycling reflects nicely the previously reported steps of isothermal water desorption from a porous thin silica film, comprising (i) initially water-filled capillaries under humid conditions, (ii) the onset of capillary desorption, and finally (iii) convergence to a new equilibrium condition at low humidity.30 At first sight, the tungsten-containing film has a similar response to what was earlier reported for an undoped coating.27 This again confirms that the doped coating is somewhat hydrophilic and porous. The response amplitude, however, is smaller than that of an undoped coating, as shown by the red data points in Fig. 4(A). At first hand, this apparently lower degree of water adsorption can be taken to be a result of lower porosity, or of the different pore structure of the tungsten containing coating. Water uptake occurs in a time scale of a few minutes. However, when the water loading time is changed from 60 min to 90 min, the water release time is also increased. The water release in terms of the optical pathlength as a function of time has been fitted using the Avrami equation,27 and the estimated half-life times of the fit curves are plotted in Fig. 4(B). While the optical response to adsorption of water is very fast, the kinetics of desorption strongly depend on the preceding water loading time. This indicates that water desorption follows a multi-step route, involving a faster process (e.g. physical desorption/evaporation) and a subsequent, slower process (e.g. capillary decondensation or chemical desorption). This could indicate that chemisorbed water is more strongly bound to the more acidic surface. However, a similar but less pronounced effect was observed for the undoped coating (not shown) and corresponds to earlier observations by infrared spectroscopy on anti-reflective coatings made from other SiO2 precursors.31
 | ||
| Fig. 4 (A) Optical pathlength for the tungstic acid doped coating subjected to three cycles of water loading (60 min each) and release. For comparison, the first cycle response for an undoped coating is plotted in red. (B) Half lifetime of water release for the tungstic acid doped coating for two different water loading durations: blue: 90 min, red: 60 min. | ||
3.5 Mechanism of gel washing
As concluded from the previous observations, the washing step plays an essential role in the overall route of film consolidation.To monitor online the different effects of washing with H2O or HCl, a set of QCM experiments was performed for which the coating was applied on gold layers supported on a quartz substrate.
The initial thickness of the spin-cast, doped water glass layer on the QCM sensor was determined from the frequency shift between coated and uncoated quartz, which was typically df/n = −1300 Hz. Using eqn (3) (and assuming a film density of 1 g cm−3) an equivalent Sauerbrey thickness of 230 nm was obtained. The as-cast film appeared rigid as no change of dissipation was monitored.
An example of QCM analysis and scan is shown in Fig. 5. The frequency scans carried out on the dry sensor before coating, after coating and after washing with water are shown in Fig. 5(A). A negative shift of the resonance curve towards lower frequencies indicates a loading of the quartz with mass, i.e. after spin coating of the water glass. The observed positive shift of frequency between the coated and the washed film indicates that washing removes the load from the quartz. This is an indication that the water glass film on the glass loses mass during the washing process and part of the film constituents are washed out. Since the bandwidths of the three resonance curves remain the same, the film can be considered as rigid before and after washing.
 | ||
| Fig. 5 (A) Resonance frequency scans for a coated quartz sensor. Black curve: resonance curve of the bare quartz; red curve: as-deposited gel-coating, blue curve: water washed gel-coating. (B) Normalized frequency shift df/n as a function of washing time as measured during a washing experiment carried out on a doped coating. | ||
In situ changes upon washing can be monitored via QCM from the change of frequency shift which is exemplarily shown in Fig. 5(B). The kinetics of water-washing are notably faster than those of the acid-wash. Also, the overall increase of frequency shift is larger in the case of using water as compared to acid, indicating again that more load is removed with water than with acid washing. Analysis of the films after equilibrated washing conditions in the dry state via frequency scans yields an overall mass loss of about 48% in the case of the water wash, and of about 33% in case of the acid wash. The as-deposited film had a nominal content of 49 wt% of alkali and tungstic acid ions, which in the case of the water wash are hence completely removed. For the acid wash, only 33% is removed, which reflects the remaining fraction of WO42−.
3.6 Precursor analyses
DLS analysis of the precursor solutions shows that one single type of colloidal particles is present in both the doped and undoped solutions. This is most likely pure silica. Fig. 6 shows the mean particle diameter as a function of solution ageing time as determined from DLS. The colloidal particles are initially in the range of 2 nm but increase to 6 nm after four days of ageing. Measurements obtained after seven days of ageing showed a very complex picture of the different particle size fractions and agglomerates (not shown). Further solution stabilization lies outside the present scope, but may be a subject of subsequent work. A large fraction of silica at around 2 nm in concentrated silicate solutions has been observed before.32 Such colloidal silica particles are known to grow upon dilution due to a decreased concentration of hydroxyl ions.24 A secondary peak, found only in the doped solution, indicates another class of colloidal particles that also display an increase in their particle size from 5 to 23 nm over time. This may be explained by agglomeration which is caused by the higher ionic strength generated through the addition of sodium tungstate. According to earlier observations, agglomeration at high pH value with monovalent cations should be reversible.24 That the extra peak in the doped solution is not caused by formation of, e.g., silicotungstic acid, can be assumed from the fact that this peak intensity is also increasing with time. The particles observed by DLS in solution are larger than the ones observed by AFM on the coating (but in a similar range). This divergence may originate from the different types of measurements (solution–thin film), as well as certainly from the drying and thermal treatment of the coating which leads to further growth and agglomeration. | ||
| Fig. 6 Particle sizes in solution (<100 nm) as a function of solution ageing time as determined by dynamic light scattering. The larger particles in the doped solution are interpreted as agglomeration as illustrated. Blue: W-doped solution. Black: undoped solutions. Lines are guides for the eye. | ||
The optical transmission measurements indicated that tungsten doping increases the porosity of the water-washed coating when compared to coatings derived from undoped solutions. Such increased porosity is taken to be a result of the observed agglomeration of colloidal silica in the precursor solutions.
3.7 Powder samples and bulk analyses
The powder obtained from drying and washing the coating solution was analyzed by XRD for its structure, and further by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), simultaneous MS and ex situ Raman spectroscopy. The results of the XRD analysis are shown in Fig. 7. The as dried powder (gel) contains at least one predominant tungstate phase which we identify as a (K, Na) tungsten hydrate. After washing, however, and regardless of whether in acid or water, none of the samples includes a detectable crystalline tungstate species anymore. Instead, an amorphous hump dominates the diffractogram, accompanied by traces of a crystalline potassium hydrogen silicate, KHSi2O5 (a phase which has also earlier been observed in aqueous potassium silicate upon drying).33 Upon thermal treatment, the KHSi2O5 phase also disappears, and some minor amounts of tridymite remain as the only crystalline phase. Detection of the potassium silicate indicates that not all of the K+ is removed during the washing of the powder, which corresponds to earlier observations on the thin films.3 The signals related to the crystalline potassium silicate are less strong in the acid-washed sample, further indicating that the acid-wash is either more efficient in the removal of K+, or that it prevents crystallization. The presence of silicate crystallites in thermally treated thin film coated glass samples was not observed at any time. | ||
| Fig. 7 Diffraction patterns for powder samples based on dried tungstic acid doped potassium silicate. Diffraction patterns are obtained for dry material, water washed material, acid washed material, and acid washed thermally treated material respectively. | ||
DSC and TGA curves are given in Fig. 8. The extracted temperatures and events as derived from MS and TGA are summarized in Table 2. All samples exhibit mass and water loss at around 110 °C, related to physically bound water or water from thermal degradation of bicarbonates, as will be explained later. The water-washed sample further loses water at 239 °C, which may be related to loss of crystal water from the observed KHSi2O5 phase. For crystalline Na2Si2O5, this reaction occurs at 220 °C24 and similar temperatures were reported for other crystalline silicates.34 For the water-washed sample, another endothermic peak is present ∼450 °C which is also associated with the release of H2O. This higher temperature correlates with an observed condensation of Si–OH groups from crystalline silicates34 and could correspond to a transformation of the potassium silicate to amorphous SiO2 or tridymite as indicated by the XRD analysis, or to another amorphous or crystalline phase.34 The suggested transformations are also illustrated in Fig. 8. The absence of these thermal events in the acid-washed powder agrees with the previous observations, in particular, with XRD analysis, where the acid-washed samples contain less crystallites. Carbonation is expected to occur during drying of the gel3 and may explain the low temperature detection (106 and 119 °C) of CO2 as a degradation of bicarbonates,35 for example,
| 2NaHCO3 → Na2CO3 + CO2 ↑ + H2O ↑ | (4) |
 | ||
| Fig. 8 DSC and TGA curves for powdered precursor material washed with water and acid. The detected release temperatures of gases for the different samples are given of Table 2. The figures illustrate the interpretation of the scans in terms of the release of adsorbed water, loss of crystal water and recrystallization of the silica species. Release of adsorbed CO2 is not illustrated. | ||
| Temperature (°C) | ||
|---|---|---|
| Acid wash | Water wash | |
| Mass loss | 96 | 106 |
| 239 | ||
| 451 | ||
| H2O release | 106 | 119 |
| 236 | ||
| 449 | ||
| CO2 release | 107 | 121 |
| 221 | 289 | |
| 355 | ||
Raman spectra for the powdered samples are presented in Fig. 9. Peak assignments are done based on sol–gel derived silicate glasses36 and the KHSi2O5 phase37 as observed by XRD; they are summarized in Table 3. Before thermal treatment, all samples except for the acid-washed doped sample are dominated by signals from crystalline species, corresponding with the observations by DSC and XRD. After thermal treatment, the spectra of three of the samples much resemble those of amorphous silica, with pronounced D1 and D2 vibrations and a broad band centered at 443 cm−1, except for the undoped water washed sample. It is suggested that the crystallization of the precursor powder is prevented by the agglomeration of particles in the doped samples, as observed by DLS, and the acid wash, as observed by XRD. The single sharp peak at 518 cm−1 for the undoped water-washed sample corresponds well to a SiO2 crystalline phase.38 A shoulder observed at 943 cm−1 only for the acid-washed doped samples disappeared after thermal treatment and could not be assigned to a silicate species. It can rather be assigned to a W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, which gives a strong signal for H2WO4 but is weak for WO3.20 The strongest signal from WO3 should occur at 809 cm−1, overlapping with the Si–O–Si bending vibration from amorphous silica. It is not directly observable here.
O stretching vibration, which gives a strong signal for H2WO4 but is weak for WO3.20 The strongest signal from WO3 should occur at 809 cm−1, overlapping with the Si–O–Si bending vibration from amorphous silica. It is not directly observable here.
 | ||
| Fig. 9 Raman spectra of powdered samples with peak positions given (cm−1). Peak assignments are found in Table 3. | ||
| Peak position (cm−1) | Assignment | Ref. |
|---|---|---|
| 1095 | O–Si (s), crystalline | 37 |
| 1060 | SiO2 Q3 (s), amorphous | 36 |
| 1045, 1010 | O–Si (s), crystalline | 37 |
| 960 | SiO2 Q2 (s), amorphous | 36 |
| 943 | WO (s), H2WO4 |
20 |
| 902 | SiO2 Q1 (s), amorphous | 36 |
| 865 | SiO2 Q0 (s), amorphous | 36 |
| 809 | W–O–W (s), WO3 | 20 |
| 805 | Si–O–Si (b), amorphous | 36 |
| 785 | Si–O–Si (b), crystalline | 37 |
| 596 | D2 SiO2 rings (o), amorphous | 36 |
| 532 | O–H (s), crystalline | 37 |
| 518 | Si–O–Si (s), crystalline | 38 and 39 |
| 486 | D1 SiO2 rings (o), amorphous | 36 |
| 476 | Si–O–Si (b), crystalline | 37 |
| 443, broad | Si–O–Si (v), amorphous | 36 |
| 386 | Si–O–Si (s), crystalline | 39 |
| 311 | O–Si–O (b), crystalline | 37 |
| 280 | O–Si–O (b), crystalline | 37 |
| 211 | SiO2 (o), quartz | 36 |
4. Conclusions
In summary, wet chemical deposition of aqueous alkali silicate solutions presents an interesting route for the creation of large-area nanostructured coatings on glass at low cost. Through pH control and salt-doping of the precursor solution, combined with a dedicated washing and ion exchange process of the as-deposited gel layer, tungstic precipitates can be generated in the final coating which enable adjustment of the coating's porosity, hydrophilicity and surface acidity towards a combination of optical (antireflective) properties with a possible anti-fouling functionality. We propose that this route can be used in a universal, facile way for generating any type of salt-doped nanoporous silica film.Acknowledgements
We wish to thank our colleagues G. Hoffmann and F. Schmiedl at the Institute of Solid State Physics, F. Zimmermann and S. Nolte at the Institute of Applied Physics and T. Kittel at the Otto Schott Institute for Materials Research, all from the University of Jena, for assistance in the conduction of experiments. We further gratefully acknowledge financial support from the European Social Funds and the Thuringian State Ministry of Science and Education through the project NANOSOR.Notes and references
- S. C. Pop, V. Abbaraju, B. Brophy, Y. S. Yang, S. Maghsoodi and P. Gonsalves, in Photovoltaic Specialist Conference (PVSC), 2014 IEEE 40th, 2014, pp. 2715–2719.
- H. N. Noh and S. Y. Myong, Sol. Energy Mater. Sol. Cells, 2014, 121, 108–113 CrossRef CAS PubMed.
- K. H. Nielsen, D. K. Orzol, S. Koynov, S. Carney, E. Hultstein and L. Wondraczek, Sol. Energy Mater. Sol. Cells, 2014, 128, 283–288 CrossRef CAS PubMed.
- R. Gupta and N. K. Chaudhury, Biosens. Bioelectron., 2007, 22, 2387–2399 CrossRef CAS PubMed.
- L. Omlin, F. Ruesch and S. Brunold, in 28th EU PVSEC, 2013.
- G. Helsch and J. Deubener, Sol. Energy, 2012, 86, 831–836 CrossRef CAS PubMed.
- W. Glaubitt and P. Löbmann, J. Sol–Gel Sci. Technol., 2011, 59, 239–244 CrossRef CAS.
- J. Deubener, G. Helsch, A. Moiseev and H. Bornhöft, J. Eur. Ceram. Soc., 2009, 29, 1203–1210 CrossRef CAS PubMed.
- J. Li, Y. Lu, P. Lan, X. Zhang, W. Xu, R. Tan, W. Song and K.-L. Choy, Sol. Energy, 2013, 89, 134–142 CrossRef CAS PubMed.
- G. San Vicente, R. Bayón, N. Germán and A. Morales, Thin Solid Films, 2009, 517, 3157–3160 CrossRef CAS PubMed.
- S. Noack-Schönmann, O. Spagin, K. P. Gründer, M. Breithaupt, A. Günter, B. Muschik and A. A. Gorbushina, Int. Biodeterior. Biodegrad., 2014, 86(part C), 286–293 CrossRef PubMed.
- M. A. Shirakawa, R. Zilles, A. Mocelin, C. C. Gaylarde, A. Gorbushina, G. Heidrich, M. C. Giudice, G. M. B. Del Negro and V. M. John, J. Environ. Manage., 2015, 157, 160–167 CrossRef PubMed.
- P. Rivero, A. Urrutia, J. Goicoechea, C. Zamarreño, F. Arregui and I. Matías, Nanoscale Res. Lett., 2011, 6, 1–7 CrossRef PubMed.
- Ö. Kesmez, H. Erdem Çamurlu, E. Burunkaya and E. Arpaç, Sol. Energy Mater. Sol. Cells, 2009, 93, 1833–1839 CrossRef PubMed.
- S. Shafaei, M. Lackner, R. Voloshchuk, I. Voloshchuk, J. P. Guggenbichler and C. Zollfrank, Recent Pat. Mater. Sci., 2014, 7, 26–36 CrossRef.
- C. Zollfrank, K. Gutbrod, P. Wechsler and J. P. Guggenbichler, Mater. Sci. Eng., C, 2012, 32, 47–54 CrossRef CAS PubMed.
- A. Watcharenwong, W. Chanmanee, N. R. de Tacconi, C. R. Chenthamarakshan, P. Kajitvichyanukul and K. Rajeshwar, J. Electroanal. Chem., 2008, 612, 112–120 CrossRef CAS PubMed.
- P. Bhaumik, T. Kane and P. L. Dhepe, Catal. Sci. Technol., 2014, 4, 2904–2907 CAS.
- J. Livage and G. Guzman, Solid State Ionics, 1996, 84, 205–211 CrossRef CAS.
- B. Ingham, S. V. Chong and J. L. Tallon, J. Phys. Chem. B, 2005, 109, 4936–4940 CrossRef CAS PubMed.
- S. N. Chatterjee, J. Colloid Sci., 1958, 13, 61–66 CrossRef CAS.
- M. L. Freedman, J. Am. Chem. Soc., 1959, 81, 3834–3839 CrossRef CAS.
- J. I. Martins, J. L. F. C. Lima, A. Moreira and S. C. Costa, Hydrometallurgy, 2007, 85, 110–115 CrossRef CAS PubMed.
- R. K. Iler, The Chemistry of Silica, John Wiley and Sons, New York, 1979 Search PubMed.
- J. He, I. Ichinose, T. Kunitake and A. Nakao, Langmuir, 2002, 18, 10005–10010 CrossRef CAS.
- A. G. Hemmersam, K. Rechendorff, F. Besenbacher, B. Kasemo and D. S. Sutherland, J. Phys. Chem. C, 2008, 112, 4180–4186 CAS.
- K. H. Nielsen, T. Kittel, K. Wondraczek and L. Wondraczek, Sci. Rep., 2014, 4, 6595 CrossRef CAS PubMed.
- T. Nishide and F. Mizukami, J. Sol–Gel Sci. Technol., 1996, 6, 263–267 CrossRef CAS.
- L. Wondraczek, A. Dittmar, C. Oelgardt, F. Célarié, M. Ciccotti and C. Marlière, J. Am. Ceram. Soc., 2006, 89, 746–749 CrossRef CAS PubMed.
- K. Wondraczek, D. Johannsmann, L. Wondraczek and J. Deubener, Glass Technol.: Eur. J. Glass Sci. Technol., Part A, 2005, 46, 179–182 CAS.
- M. S. W. Vong and P. A. Sermon, Thin Solid Films, 1997, 293, 185–195 CrossRef CAS.
- D. Böschel, M. Janich and H. Roggendorf, J. Colloid Interface Sci., 2003, 267, 360–368 CrossRef PubMed.
- K. B. Langille, D. Nguyen, J. O. Bernt, D. E. Veinot and M. K. Murthy, J. Mater. Sci., 1991, 26, 695–703 CrossRef CAS.
- G. Lagaly, Adv. Colloid Interface Sci., 1979, 11, 105–148 CrossRef CAS.
- E. M. Barall Ii and L. B. Rogers, J. Inorg. Nucl. Chem., 1966, 28, 41–51 CrossRef.
- A. Kalampounias, Bull. Mater. Sci., 2011, 34, 299–303 CrossRef CAS.
- D. Schmidmair, V. Kahlenberg, L. Perfler and D. M. Többens, Mineral. Mag., 2014, 78, 609–622 CrossRef CAS PubMed.
- K. J. Kingma and R. J. Hemley, Am. Mineral., 1994, 79, 269–273 CAS.
- D. W. Matson, S. K. Sharma and J. A. Philüpotts, Am. Mineral., 1986, 71, 694–704 CAS.
| This journal is © The Royal Society of Chemistry 2015 |