
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile approach to synthesize an oxo-functionalized graphene/polymer composite for low-voltage operating memory devices†
Zhenxing
Wang‡§
a,
Siegfried
Eigler‡
*b,
Yoshitaka
Ishii
*c,
Yichen
Hu
c,
Christian
Papp
d,
Ole
Lytken
d,
Hans-Peter
Steinrück
d and
Marcus
Halik
*a
aOrganic Materials & Devices (OMD), Institute of Polymer Materials, Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Martensstraße 7, 91058 Erlangen, Germany. E-mail: Marcus.Halik@fau.de
bDepartment of Chemistry and Pharmacy and Institute of Advanced Materials and Processes (ZMP), Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Dr.-Mack-Straße 81, 90762 Fürth, Germany. E-mail: Siegfried.Eigler@fau.de
cDepartment of Chemistry, University of Illinois at Chicago, 845 W Taylor St, Chicago, IL 60607, USA. E-mail: YIshii@uic.edu
dInstitute of Physical Chemistry II and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Egerlandstraße 3, 91058 Erlangen, Germany
First published on 16th July 2015
Abstract
Memory devices are a key technology of our era and one of the constant challenges is the reduction of their power consumption. Herein, we demonstrate that graphene oxide with very few defects, that is, about 1 nm thin oxo-functionalized graphene derivative, can be used in memory devices operating at 3 V. A memory device stores charges in the material of the active channel. Thereby, writing and erasing information can be performed at low voltage, facilitating low power consumption. To enable operation at low voltage, a novel synthetic approach is necessary. We find that the selective non-covalent electrostatic functionalization of mainly organosulfate ions is possible with dodecylammonium. This functionalization allows the non-covalent coating of flakes with a polystyrene-derivative as nm-thin dielectric medium. The resulting polymer-wrapped composite has a height of about 5 nm. We find that the thin coating of a few nm is mandatory to make the memory device work at low voltage. Furthermore, a self-assembled monolayer of an imidazolium derivative further enhances the function of the memory device. The prepared composite materials are characterized by state-of-the-art analysis including solid state nuclear magnetic resonance spectroscopy and thermogravimetric analysis coupled with gas chromatography, mass spectroscopy or infrared spectroscopy. Reference experiments prove the importance of the controlled synthesis to enable the function of the memory device.
Introduction
Graphene attracts much attention all over the world due to its excellent performance in electronic devices.1–6 The good conductivity along with the low thickness allows for its application as a transparent electrode.7 Field-effect transistors based on 2-dimensional materials have already been realized.1,2,5 Due to the mechanical flexibility of graphene, the device technology can be transferred to plastic substrates, e.g. for wearable electronics.8–11 Graphene is also used for electrodes and active transistor channel materials and it plays an important role in constructing nanoelectronic memory devices.3 Especially in the latter, graphene can be introduced as a floating gate, in which the carriers are stored during the operation of the memory device.12,13 Due to the high cost of graphene grown by chemical vapor deposition (CVD), reduced graphene oxide (rGO) is considered to be a cost-effective alternative, in particular, since its resulting performance as a floating gate for memory devices is as good as that of CVD-grown graphene.14–19 Even graphene oxide (GO) can be directly used as a floating gate as long as there are sufficient charge traps in the layer.20 Besides the floating gate configuration, GO can also be used as a resistive memory switch, which only needs two terminals for operation.21–28 An advantage of GO is its better processability, when using solvents such as water or alcohols.29 For a memory configuration with a floating gate, a tunneling dielectric is placed between the floating gate and the active channel in order to isolate them.14–20,30 However, the insertion of any extra dielectric layer usually increases the operation voltage to several tens of volts, which lead to an accordingly higher power consumption.14,20,31 A reduced thickness of the dielectric layer is challenging, but nevertheless mandatory for a memory device to work at low voltage. Although several functionalization concepts of graphene have been developed, presently no composite material is available that provides sufficient isolation for the operation of memory devices at a low voltage of 3 V.24In this work, we use a graphene oxide with an improved quality, that is, with an almost intact carbon framework, which in addition to hydroxyl and epoxy groups also bears organosulfate groups. We use oxo-G1 as abbreviation for this oxo-functionalized graphene, following a systematic formula recently introduced (see the ESI†).32,33 Herein, we present for the first time the synthesis of a novel composite of a polymer-wrapped oxo-functionalized graphene (oxo-G1) derivative, which we directly used as a floating gate in memory devices without including an extra tunneling dielectric layer (Fig. 1). The oxo-G1-functionalized composite is synthesized by using electrostatic interactions to control the chemical structure. It is soluble in tetrahydrofuran and enables the interaction of the dielectric layer, a polystyrene derivative, with the surface of the flakes. We found that a special design of the oxo-G1 composite material is necessary in order to facilitate the efficient wrapping of the polystyrene derivative as a dielectric around the oxo-G1 flakes, thereby sufficient isolation for the charges stored in the oxo-G1 flake is provided, yielding a good retention property for the memory device. Despite the fact that the thickness of the dielectric is only about 2–3 nm, we demonstrate that its sufficient isolation from the substrate can be achieved by incorporating a self-assembled monolayer containing an imidazolium moiety, denoted as IMI-SAM, between the Al2O3-layer and the polystyrene derivative-wrapped oxo-G1 (Fig. 1B). The composite is examined by state-of-the-art methods, including solid state NMR (ssNMR), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA) coupled with Fourier transform infrared spectroscopy (FTIR), and mass spectrometry (MS), also combined with gas chromatography (GC). The intention of this work is to overcome the current limits of graphene oxide-based electronics by controlled chemical synthesis of novel derivatives of oxo-G1.
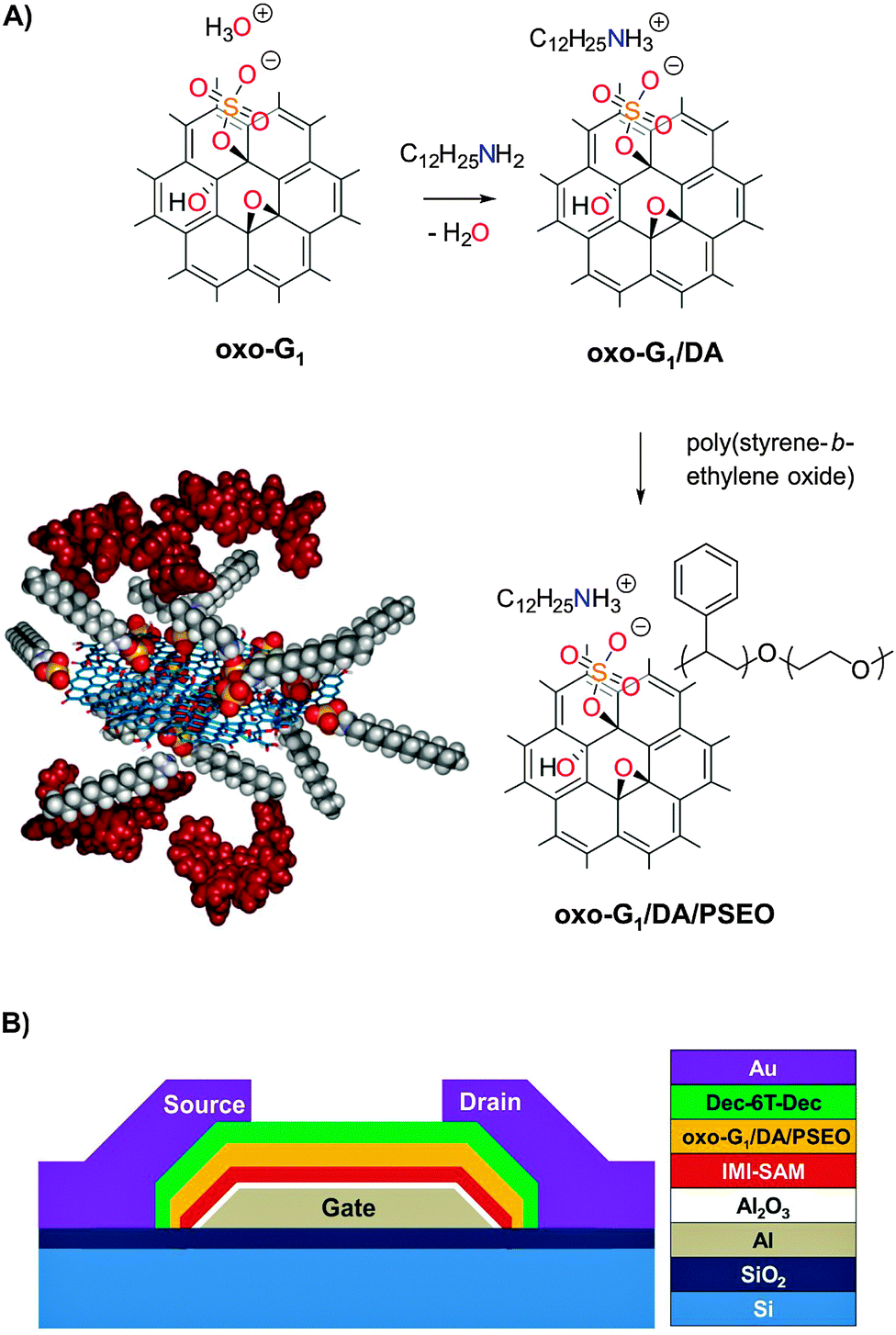 | ||
| Fig. 1 (A) Reaction scheme of the non-covalent functionalization of oxo-G1 followed by an additional non-covalent functionalization with poly(styrene-b-ethylene-oxide), (PSEO) and structure model of the composite oxo-G1/DA/PSEO. (B) Memory device architecture. | ||
Results and discussion
Synthesis of oxo-functionalized graphene (oxo-G1)
The quality of graphene oxide depends strongly on the preparation conditions,33 as not only the contribution of functional groups but also the integrity of the carbon framework differs from one synthesis condition to another. A variation in the density of defects up to several % is well known.33,34 In this work, we use oxo-G1, which exhibits an average density of defects of only 0.3% within the carbon skeleton (Fig. S1A, ESI†).33 Since the average density of defects is quite low, this material enables the functionalization of chemical groups at the basal plane, with functionalization at defect sites playing only a minor role; we therefore call this material oxo-functionalized graphene (oxo-G1). The functional groups in oxo-G1 have been analyzed previously:35–38 they mostly are hydroxyl, epoxy and organosulfate groups, located on both sides of the basal plane, as illustrated in Fig. 1A.38–42 Recently, we proved that the carbon framework in oxo-G1 is thermally stable up to 100 °C, and that the functionalization of oxo-G1 is possible without harming the carbon framework.36,43,44 Oxo-G1 is prepared from natural graphite by the controlled oxidation in sulfuric acid using potassium permanganate and sodium nitrate as oxidants at a temperature of 5–10 °C, followed by aqueous work-up at the same temperature (Experimental details, see the ESI†). This procedure largely prevents the formation of defects. Hydrogen peroxide is subsequently used to make manganese salts soluble. The multilayered oxo-functionalized material, oxo-Gn (graphite oxide), is purified by repeated centrifugation and redispersion in cold water, until the pH of the supernatant is about 6. This oxo-Gn was already partially delaminated in water and successive sonication of the dispersion yielded oxo-G1 flakes by exfoliation. According to elemental analysis (EA), the carbon and sulfur contents are about 46% and 6.4% in weight, respectively. We have demonstrated that the sulfur content is a structure giving motif of oxo-G1 in the form of organosulfate.38 While TGA gives no evidence for significant amounts of inorganic sulfate, we estimate one organosulfate group per ∼19 C-atoms. Recently, we proposed that hydronium ions act as counter ions of organosulfate and therefore, the exchange with other cations is feasible.38Since the organosulfate groups are located at the carbon skeleton, they can be used to covalently modify oxo-G1 at the basal plane, e.g. by azide ions.36 Here, we demonstrate that they allow for electrostatic functionalization of oxo-G1 with dodecylamine forming a composite that enables the wrapping of a polystyrene derivative as a dielectric, while maintaining individual flakes.
Considerations in the synthesis of oxo-G1/dodecylamine and related composites
Here, we target a controlled synthesis of a composite of functionalized oxo-G1 and a block-copolymer poly(styrene-b-ethylene oxide). While the direct formation of a composite has failed, it turns out that an electrostatic modification of oxo-G1 with dodecylamine (DA) as a linker enables the subsequent introduction of poly(styrene-b-ethylene oxide). Since the chemical structure of defined amounts of DA bound to purified oxo-G1 is unknown, we need to investigate the interaction of oxo-G1 and DA in detail. There are several reports in the literature addressing the interaction of graphene oxide and DA, and the ability to form dispersions of single layers of graphene oxide in organic solvents. The synthetic procedures in many of the previous studies used excess DA or apply DA at low pH. Thereby, composites with undefined chemical compositions were produced. While the focus of the previous reports was on the physical properties in applications, we will outline below that a controlled chemical functionalization is mandatory to attain the desired function.Another issue to be considered is the choice of solvent in the synthesis. While oxo-G1 is well dispersible in polar solvents, it forms aggregates in less polar organic solvents, such as tetrahydrofuran (THF). To overcome this challenge, it was demonstrated for defect-rich graphene oxide that alkyl ammonium species enable dispersibility in organic solvents.45 Electrostatic bonding between organosulfate and dodecyl ammonium molecules was proposed,46 and is plausible at a pH of about 0. The binding of alkyamines, e.g. octylamine to graphene oxide, was used to process graphene oxide in THF and to incorporate it into polymer matrices, although the exact binding was not known.38,47
Synthesis of an oxo-G1/dodecylamine composite
For a controlled synthesis of the composite, we first determined the pKa value of oxo-G1 to enable a controlled reaction of dodecylamine (DA) and acidic groups of oxo-G1. The intention of adding DA to oxo-G1 is the generation of a hydrophobic surface. Previous studies by us showed that the carbon framework of oxo-G1 is not stable against bases, such as sodium hydroxide, at 40 °C; however, at 10 °C, where chemical reactions such as organosulfate cleavage, still occur, the carbon framework stays intact.44 The instability of the carbon framework of graphene oxide with defects on the % scale towards water and base was also described.48 Thus, it is of utmost importance to control the reaction conditions, in order not to harm the carbon framework.44,49First, we performed a titration experiment of oxo-G1 in a 1/1 mixture of i-propanol and water with sodium hydroxide to determine the pKa, which was found to be about 5.5 (Fig. S1B, ESI†). The titration of a stirred dispersion starts at a pH of 3.6 and a pH of 6 is reached after adding about 1 hydroxide molecule on 20 carbon atoms. Note that this ratio is comparable to the molar ratio of S and C (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19), as determined by EA. Although the stoichiometry fits, the expected pKa value of organosulfate is much lower. However, in an earlier study on sulphur species in graphene oxide we found similar pKa values, and reported evidence for hydronium ions as counterions of organosulfate.38 Therefore, we suggest that the experimentally determined pKa value may be a result of the strongly negatively charged and partially π-conjugated macromolecule. Similar effects have been found in multiply charged FeIII–porphyrin systems that bear up to 16 negative charges that prevent the deprotonation of coordinated water, yielding a pKa of 9.9.50 However, we do not exclude the possibility that other functional groups of oxo-G1 may partially contribute to the acidity. The pH value of 6.0 remains stable with time (data not provided); however, if the dispersion is titrated up to a pH of 8.3, the decline of the pH to 7.4 occurs within one hour. This observation is in line with the observation and explanation of Dimiev et al., who found that the pH change was accompanied by the decomposition of the carbon framework.49
:19), as determined by EA. Although the stoichiometry fits, the expected pKa value of organosulfate is much lower. However, in an earlier study on sulphur species in graphene oxide we found similar pKa values, and reported evidence for hydronium ions as counterions of organosulfate.38 Therefore, we suggest that the experimentally determined pKa value may be a result of the strongly negatively charged and partially π-conjugated macromolecule. Similar effects have been found in multiply charged FeIII–porphyrin systems that bear up to 16 negative charges that prevent the deprotonation of coordinated water, yielding a pKa of 9.9.50 However, we do not exclude the possibility that other functional groups of oxo-G1 may partially contribute to the acidity. The pH value of 6.0 remains stable with time (data not provided); however, if the dispersion is titrated up to a pH of 8.3, the decline of the pH to 7.4 occurs within one hour. This observation is in line with the observation and explanation of Dimiev et al., who found that the pH change was accompanied by the decomposition of the carbon framework.49
Organosulfate groups (and possibly other organic acid functional groups) in oxo-G1 are located on both sides of the basal plane and should enable homogeneous functionalization. Therefore, we titrated oxo-G1 with DA in a 1/1 mixture of i-propanol and water. After the addition of about one DA molecule per 18 C-atoms, a pH of about 6 was reached (Fig. 2A and B). The determined pKa value is about 5.5, comparable to the titration experiment with sodium hydroxide (Fig. S1B, ESI†). Although an organic amine is a weak base, the determined ratio of amine molecules to carbon atoms is similar to that found in the titration experiment with hydroxide. This experiment suggests that (a) hydronium ions, the counterions of organosulfate, react with DA in an acid/base reaction and are therefore exchanged with dodecylammonium (Fig. 2A), and (b) the DA remains adsorbed on the surface of oxo-G1. Thus, the formation of a defined complex between organosulfate and dodecylammonium is plausible. We added one DA per 18 C-atoms of oxo-G1 (at an oxo-G1 concentration of 100 μg ml−1) to form a defined oxo-G1/DA composite. Although the dispersion of oxo-G1 is quite diluted, the composite precipitates in the solvent mixture of i-propanol and water. The composite is subsequently isolated by centrifugation. It was previously described that the surface of graphene oxide may not be accessible at higher concentrations than 35 μg ml−1.51 However, according to our titration experiment of oxo-G1 and DA, we found that 100 μg ml−1 of oxo-G1 was sufficient to introduce DA groups while maintaining single layers. Furthermore, our recent study on the correlation of the lateral dimension of oxo-G1 and the sedimentation coefficient proves that single layers of oxo-G1 were exclusively present at a concentration of 100 μg ml−1.52 The oxo-G1/DA composite was purified by repeated washing and centrifugation with a 1/1 mixture of i-propanol/water. Finally, the composite was suspended in water and freeze-dried for analysis.
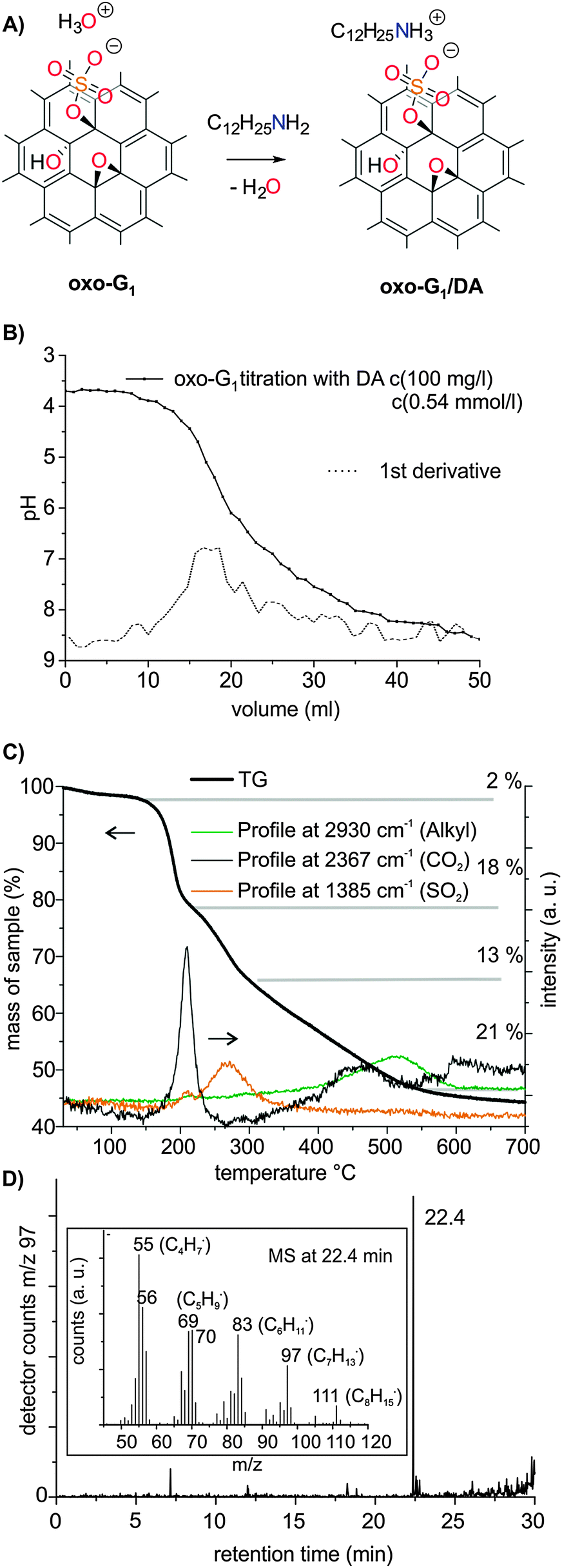 | ||
| Fig. 2 (A) Reaction scheme of the formation of oxo-G1/DA. (B) pH-titration of oxo-G1 with DA. (C) TGA of oxo-G1/DA between 30 °C and 700 °C and the temperature-profiles of cleaved alkyl, CO2 and SO2 formation, identified by FTIR. (D) GC chromatogram of m/z 97 of oxo-G1 decomposing at 430 °C, and the inset shows the MS of the decomposing product at 22.4 min retention time that can be related to dodecene. | ||
Our elemental analysis (EA) of the sample suggested dodecylammonium in small excess bound to organosulfate in oxo-G1/DA (Experimental, ESI†). Although the interaction between organosulfate and dodecylammonium should be mainly of electrostatic nature, some sp2 carbons of oxo-G1 may also interact with the aliphatic group of dodecylammonium, as deduced from solid-state nuclear magnetic resonance (ssNMR) data (see below). The oxo-G1/DA composite was further analyzed by thermogravimetric analysis (TGA) coupled with Fourier transform infrared spectroscopy (FTIR) and mass spectrometry (MS). In addition, gases evolving from the decomposition of the material were separated by gas chromatography (GC) and subsequently analyzed by MS, a method that was only recently introduced.53,54 The TGA plot reveals three decomposition stages (Fig. 2C). Up to about 200 °C, oxo-functional groups decompose, in agreement with recent results.38,55 Furthermore, organosulfate decomposes up to 350 °C, followed by the continuous decomposition that is related to dodecylammonium. The organic residue, assumedly dodecene, can be identified by monitoring the alkyl signal in FTIR, with a maximum of the decomposition rate at about 500 °C (Fig. 2C and Fig. S3, ESI†). A mixture of several other products is formed in the course of the decomposition of the carbon framework proceeding in this temperature regime. However, the gases can be separated by GC and the decomposition pattern of dodecene is well resolved by MS (Fig. 2D).
We further elucidated the binding of DA, using 15N-labelled DA, in the oxo-G1/15N–DA composite by ssNMR; this method provides information about the regiochemistry, which is not accessible by other techniques. The electrostatic interaction of 15N–DA and conventionally prepared graphene oxide (with defects within the carbon framework on the % scale) was recently analyzed by ssNMR.46 However, the synthetic protocol differed from our approach, because DA was added to the reaction mixture after the synthesis of graphite oxide (after H2O2 addition) in an acidic solution at a pH of 0. The authors suggested that the interaction of dodecylammonium with the functional groups is electrostatic, because they find no evidence for a reaction of epoxide groups with DA. In our experiment, we find a single 15N ssNMR signal at 33.5 ppm, referenced to NH3, which is very similar to the reported 15N signal of graphene oxide/DA at 33 ppm (or −347 ppm referenced to nitromethane).46 This 15N NMR signal is assigned to dodecylammonium, which is consistent with the electrostatic interaction between DA and organic acid groups of oxo-G1 indicated in our titration experiments. The 15N shift is also consistent with the ab initio calculated shift for dodecylammonium (33.4 ppm), rather than with that for the neutral amine form of DA (18.9 ppm, Fig. S4, ESI†). These data reveal that dodecylammonium is indeed formed and that no additional amine is present in the composite (Fig. 3). The presence of the ammonium ion was also confirmed by XPS: the main peak at 401.3 eV in the N 1s region is assigned to the ammonium moiety (Fig. 3F). The presence of a smaller peak at 399 eV is attributed to amine.56 As XPS is a surface sensitive method, we suggest that some amines may be present at the surface of oxo-G1 as a consequence of deprotonation of DA, assumedly induced by adsorbed water or induced by the X-ray illumination. We also conducted a 13C–{15N} REDOR ssNMR experiment; the data give no evidence for the formation of a bond between 15N of dodecylammonium and any carbon species in oxo-G1. Simulations of the REDOR signal dephasing curves for a simplified model of oxo-G1 and an 15NH3+ ion indicate that 15N of the ammonium ion should be located at a distance of 3.5 Å to the carbon basal plane (Fig. 3E, Fig. S5, ESI†). This suggests that the majority of the ammonium ions do not directly interact with the carbon species of the basal plane. In addition, our 2D 15N–1H correlation experiment (Fig. 3C) shows a single peak at (ωH, ωN) = (6.8 ppm, 33.3 ppm); the peak is attributed to a cross peak between 15N of the ammonium ion and ammonium 1H, because of the relatively short cross-polarization (CP) contact time (0.4 ms) employed in this experiment. Nevertheless, in a 2D 13C–1H correlation ssNMR spectrum, a cross-peak signifying the proximity between 1H of aliphatic chains of dodecylammonium groups and 13C of sp2 carbons of oxo-G1 is observed. This observation suggests the possibility of an additional stabilizing hydrophobic interaction between DA and oxo-G1, besides electrostatic interaction in the composite.
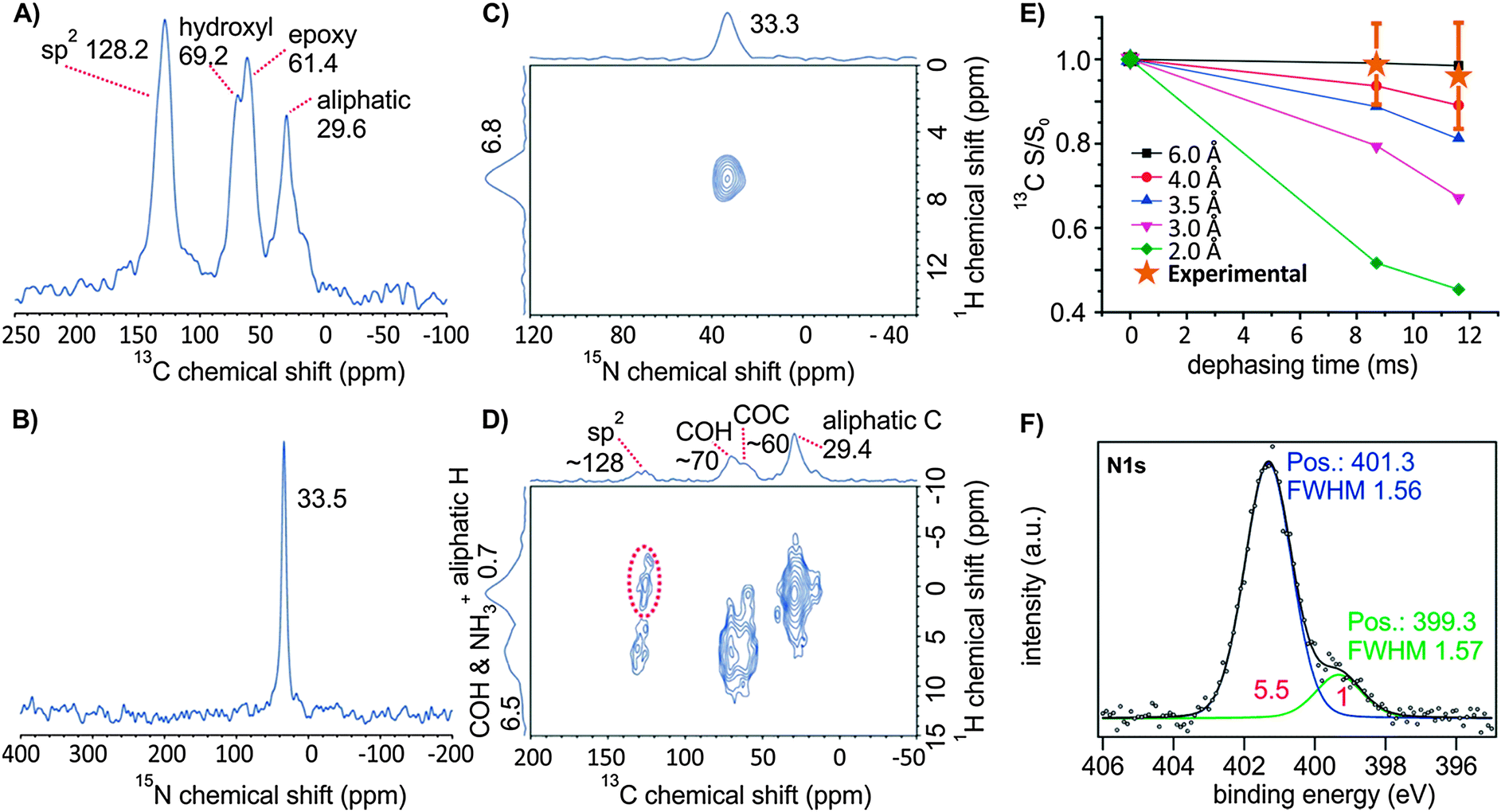 | ||
| Fig. 3 (A) 13C ssNMR and (B) 15N ssNMR MAS spectra of the oxo-G1/DA, where DA was 15N-labeled. (C) 2D 15N–1H ssNMR and (D) 2D 13C–1H ssNMR spectra of oxo-G1/DA. The peaks in the dotted red circle indicate contacts between aliphatic 1H in DA and 13C of sp2. (E) A 13C–{15N} REDOR dephasing curve for oxo-G1/15N–DA with simulation curves for varied distance R. The data were collected at a spinning speed of (A and B) 20 kHz and (C–E) 22 kHz. (F) N 1s signal in XPS of oxo-G1/DA. | ||
The oxo-G1/DA composite can be easily dispersed in THF and individual flakes of the composite can be placed on 300 nm SiO2/Si substrates for further analysis by simple drop-casting. Atomic force microscope (AFM) pictures revealed that individual flakes with lateral dimensions of about 2–10 μm were formed with an average height of about 2 nm (Fig. S6, ESI†). As the length of DA is ∼1.6 nm, a height of ∼4 nm is expected, assuming that DA is oriented perpendicular to the basal plane via electrostatic interactions with organosulfate from both sides of oxo-G1. Thus, we conclude that DA should be tilted toward the basal plane, in order to explain the thickness of ∼2 nm determined by AFM in Fig. S6 (ESI†). This finding is further confirmed by structural optimization, where a DA group (with –NH3+ facing an organosulfate group) that was initially perpendicular to an oxo-G1-flake was found to turn towards a parallel arrangement, which seems to be a more stable structure (Fig. S5C, ESI†). This model agrees well with the AFM thickness profile, if we consider DA groups to be located on both sides of the oxo-G1 flake. In addition, this finding also well explains the proximity of the DA aliphatic chains with sp2 carbons of oxo-G1, as mentioned before. Furthermore, in this model, the 15N to graphene basal plane distance is ∼4.5 Å, which is consistent with our finding from REDOR simulations (>3.5 Å) in Fig. 3E. Thus, the chemical structure and geometry of oxo-G1/DA presented here are well supported by the ssNMR and AFM data.
Synthesis of an oxo-G1/dodecylamine/poly(styrene-b-ethylene oxide) composite
The formation of oxo-G1/DA proceeded in a defined way and was used as a precursor for wrapping with a dielectric layer. We avoided excess of DA that might induce other binding motifs or chemical reduction. As a dielectric material we chose the block-copolymer poly(styrene-b-ethylene oxide) (PSEO) with a molecular weight contribution ratio of 105000/3000. PSEO is soluble in THF and can be mixed with oxo-G1/DA homogeneously. We used a simple mixing procedure and found that PSEO adsorbs on the surface of oxo-G1/DA non-covalently. After mixing, the new composite (oxo-G1/DA/PSEO) is purified by repeated centrifugation and redispersion in THF to remove excess of PSEO. The yielded composite is well dispersible in THF and contains both dodecylammonium and PSEO. The purification procedure is necessary to remove free PSEO. In Fig. S8 (ESI†), free PSEO is visible between the flakes of the composite.
The final oxo-G1/DA/PSEO composite is analyzed by FTIR (Fig. S9A, ESI†), EA and TGA (Fig. 4). The analytical results reveal that DA and PSEO are part of the composite and the molar ratio of N/S is about 0.9 indicating that the complex between organosulfate and DA in the composite is still present. The S-content in the composite is 0.58% (S-mass content oxo-G1: 6.47% and in oxo-G1/DA: 2.40%). It is difficult to relate the S-content to the portion of oxo-G1. Nevertheless, the data suggest that some DA and sulfate were partially cleaved during the mixing of oxo-G1/DA with PSEO in THF. We propose that dodecylammonium sulfate or dodecylammonium hydrogensulfate is formed and some organosulfate was cleaved. Anyway the polymer is wrapped around the flakes of oxo-G1/DA and can be identified by TGA-GC-MS. Between 400 and 500 °C, a mass loss of 39% is found (Fig. 4A). The gas produced at 450 °C is analyzed by GC coupled to MS, and fragments of polystyrene are clearly identified by MS due to the signals at m/z 78. Furthermore, aromatic C–H vibrations are detected by coupling TGA and FTIR, further corroborating the decomposition temperature of the polymer at about 470 °C in the composite, as illustrated in Fig. 4B and C.
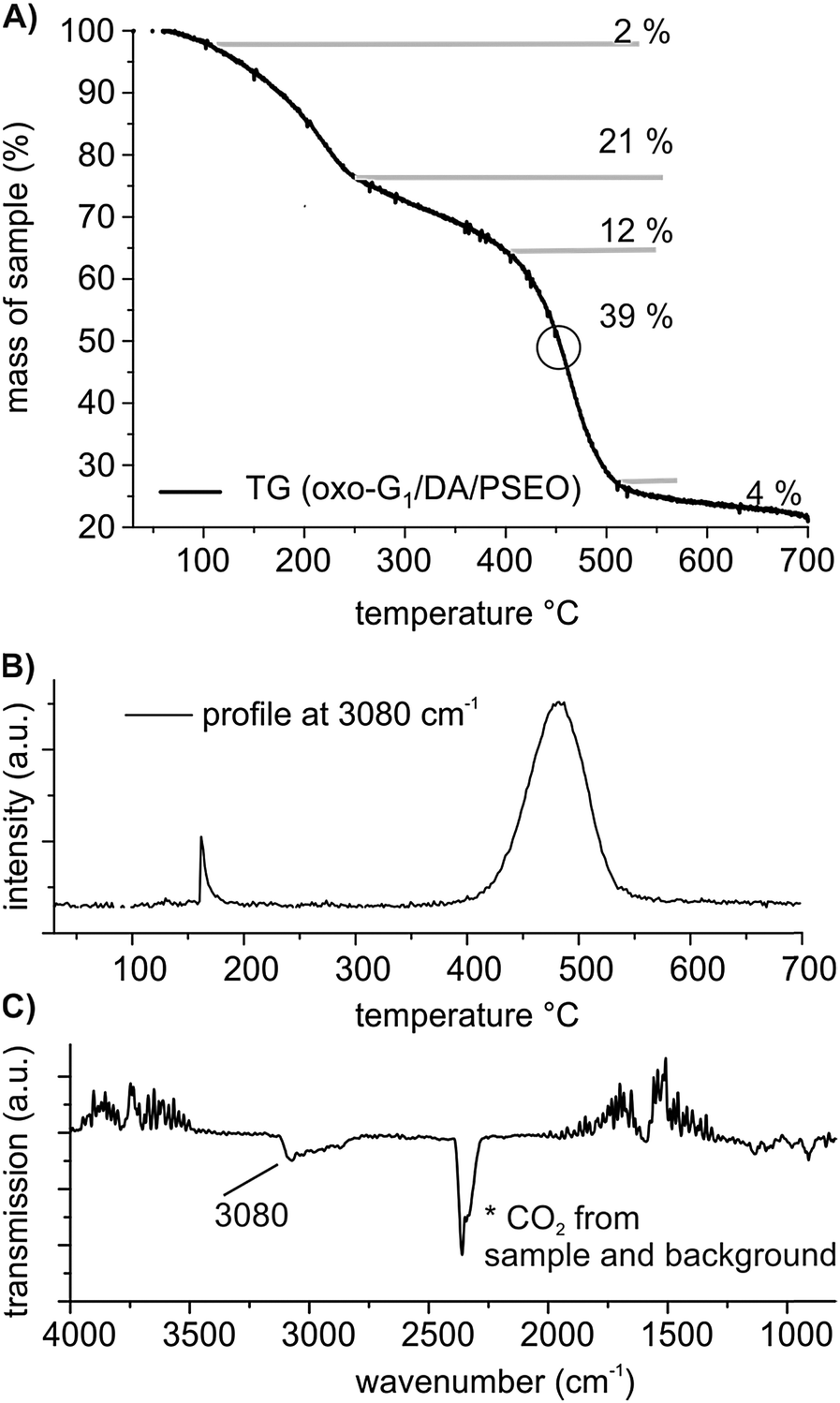 | ||
| Fig. 4 (A) TGA of oxo-G1/DA/PSEO between 30 °C and 700 °C and (B) the temperature-profiles of cleaved aromatic compounds identified by FTIR. (C) FTIR spectrum of gases produced at 470 °C. | ||
The oxo-G1/DA/PSEO composite is well dispersible in THF and individual flakes of the composite can be deposited on 300 nm SiO2/Si substrates by drop-casting from a 0.01 mg ml−1 dispersion. AFM images reveal a final flake thickness below 6–10 nm, as depicted in Fig. 5. PSEO is adsorbed on the surface of flakes of oxo-G1/DA. Thus, the procedure described here indeed leads to a composite with a wrapped dielectric material and the layer thickness is roughly 3 nm on each side, according to AFM. This thin coating facilitates operation of memory devices at low-voltage, as we will describe below.
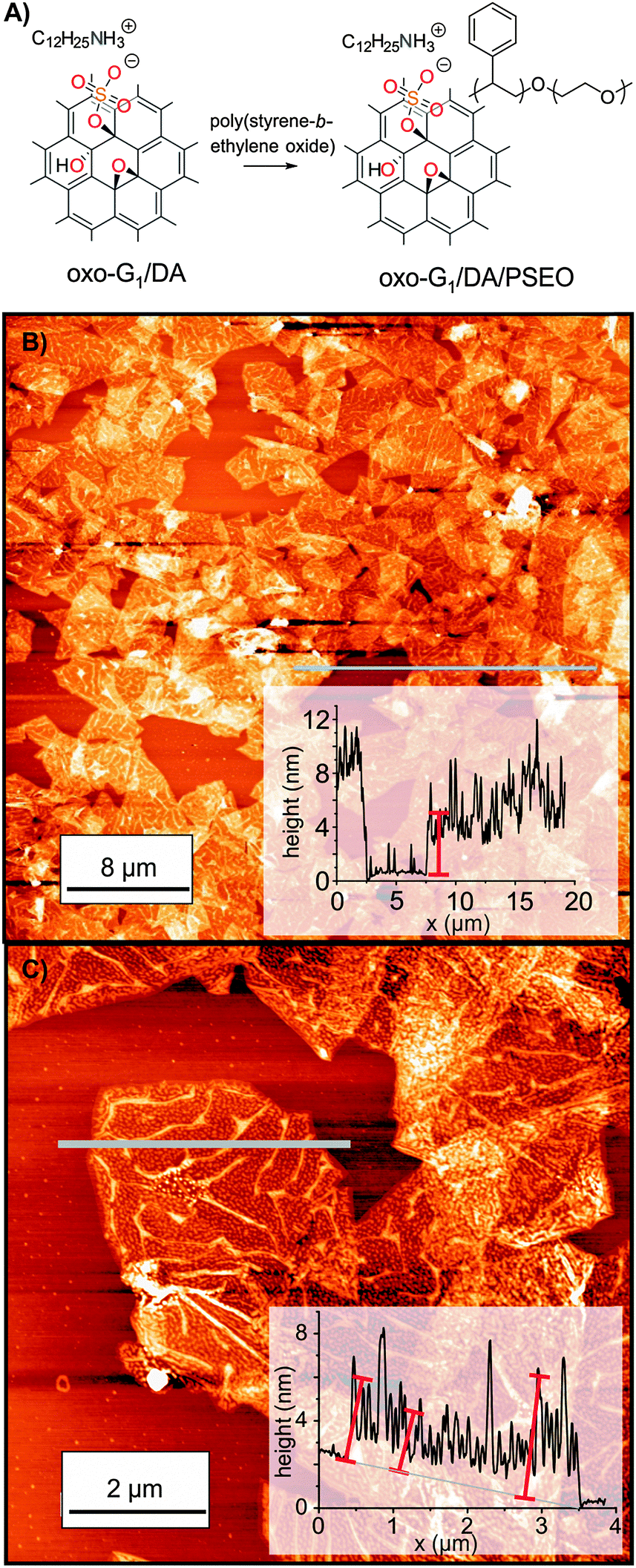 | ||
| Fig. 5 (A) Formation of the oxo-G1/DA and poly(styrene-block-ethylene oxide) (PSEO) composite in THF. (B) and (C) AFM images of oxo-G1/DA/PSEO on SiO2/Si with the height profile along the grey line. | ||
Synthesis of reference composites
Reference experiments reveal that the composite architecture of oxo-G1/DA/PSEO is mandatory and skipping oxo-G1/DA formation does not yield flat single layers of composites. The direct formation of an oxo-G1/PSEO composite failed, although a solvent-exchange method was used to exchange water against THF.57 Thereby the obtained final oxo-G1/PSEO composite is only barely dispersible in THF, and AFM images reveal a crumbled structure of the flakes (Fig. S7B, ESI†). Thus, processing and applying oxo-G1/PSEO are not favored.Furthermore, the interaction of DA and organosulfate in oxo-G1 is essential to form a functional device. Cleaving organosulfate, accompanied by the introduction of structural defects, leads to non-functional devices. As a proof, a reference composite of graphene oxide with a moderate amount of defects and only about one organosulfate per 200 C-atoms (def-GO) was prepared. First, def-GO was prepared from oxo-G1 by sodium hydroxide treatment. The base cleaves organosulfate and also causes C–C bond cleavage, as described in the literature and confirmed by statistical Raman microscopy (SRM, Fig. S1, ESI†). The density of defects was investigated by SRM on films of flakes of chemically reduced def-GO. The reduction was performed with vapor of hydroiodic acid and trifluoroacetic acid, a method that is described elsewhere.44,58,59 The final defect density was determined by analyzing the full-width at half-maximum (Γ) of the 2D peak in the Raman spectra that can be related to the defect density.60–62
The average defect density in def-GO increased from 0.3% to about 0.8% in average.60,61 A def-GO/DA/PSEO composite was prepared using the same protocol as described above for the oxo-G1/DA/PSEO composite. It is expected that the interaction of dodecylammonium with def-GO is due to acidic groups also present predominantly at defect sites or due to dispersive interaction of DA with the π-system. In contrast to oxo-G1/DA and oxo-G1/DA/PSEO, no stable dispersions formed in THF for def-GO/DA and def-GO/DA/PSEO and thus, aggregates and crumbled flakes formed in THF, as determined by optical microcopy and AFM (Fig. S10, ESI†). Analytical results by means of TGA-MS or TGA-FTIR and FTIR of bulk material are depicted in Fig. S9B and S11 (ESI†). These analyses in combination with EA reveal that DA is either arbitrary adsorbed on def-GO or assumedly bound by interacting with functional groups at defect sites, and no stoichiometric derivative is formed.
Memory device fabrication and characterization
Taking advantage of such a defined composite (oxo-G1/DA/PSEO), we are able to fabricate transistors with a floating gate, which can be utilized as a memory device. Oxo-G1 flakes can be considered as charge traps while charge carriers are transported in the semiconductor channel. However, oxo-G1 must also be isolated from the channel, which is achieved by polymer wrapping of the composite as a tunneling barrier. Now, the trapping layer can be charged or discharged depending on the potential applied on the gate electrode. In this way, a memory operation can be realized and this concept has been widely used in memory devices.12–20 The device fabrication is based on a shadow mask approach; a detailed step-by-step illustration can be found in Fig. S12 (ESI†). The gate, source, and drain electrodes, as well as the organic semiconductor layer are prepared in a thermal evaporator. The 30 nm thick aluminum gate electrodes are first patterned, followed by an oxidation process in an oxygen plasma chamber (3 min, pressure 0.2 mbar, power 200 W). Afterwards, a self-assembled monolayer (SAM) of 3-methyl-1-(12-phosphonododecyl)imidazolium bromide (IMI-PA) is formed on top of aluminum oxide by immersing the sample in the SAM isopropanol solution (0.025 mM) for 4 hours. This kind of special SAM has shown a good performance in such FET configurations.11,63 The composite dispersion of oxo-G1/DA/PSEO is then spin-coated on the sample at a speed of 500 rpm. The sexithiophene derivative, α,α′-didecylsexithiophene (Dec-6T-Dec), is used as the active channel material, with a thickness of 30 nm. The last step is the evaporation of the source and drain gold electrodes of 30 nm thickness. The final layout of the device is shown in Fig. 6A. The device is electrically characterized under ambient conditions on a DC probe station (Micromanipulator), which is connected to a semiconductor analyzer (Agilent Model B1500A). | ||
| Fig. 6 The memory device based on GO with different wrapping. (A) The device layout with source (S), drain (D) and gate (G) electrodes marked. The channel length is 40 μm and the channel width is 600 μm. (B) Transfer and (C) retention performance of the memory device based on oxo-G1/DA/PSEO. Vds = −1 V. (D) Transfer performance of the device based on oxo-G1 flakes, Vds = −1 V. (E) Transfer performance of the device based on oxo-G1/DA, Vds = −1 V. (F) Transfer performance of the device based on def-GO/DA/PSEO, Vds = −1 V. | ||
The transfer performance of the memory device is shown in Fig. 6B. The applied drain voltage is −1 V. Based on the model of thin-film organic transistors, the mobility of holes can be calculated to be 1.42 × 10−2 cm2 V−1 s−1.64 The hysteresis of the threshold voltage ΔVth is about 0.8 V. The retention property of the memory device is shown in Fig. 6C. After applying an erase signal (Vgs = −3 V) for 20 seconds, the drain current is sampled at Vgs = −0.5 V every 6 seconds for 5 minutes. After applying a write signal (Vgs = 2 V), the drain current is also sampled similarly. The current ratio after writing and erasing varies from 2.9 to 1.9. To figure out the function of the outer shell of a polymer, we made a comparison to a device, in which the oxo-G1/DA/PSEO composite layer is replaced by an oxo-G1 layer without any wrapping, which is assembled by a self-assembled process.11,53 Alternatively, we replaced the composite layer with an oxo-G1/DA layer. The transfer performances of the two devices are shown in Fig. 6D and E respectively. Without the isolation of the DA or PSEO wrapping, the device cannot be used as a memory device, since there is no hysteresis present. The charges may get into the GO layer quite easily but cannot be stored due to the lack of the dielectric layer. This comparison strongly underlines the significance of the wrapping shells, both by DA and PSEO.
Moreover, we have to point out that the GO quality plays an important role. Although the dispersion of def-GO/DA/PSEO is less stable in THF, a freshly prepared dispersion can be applied to produce devices. If we use def-GO (def-GO/DA/PSEO) instead of oxo-G1, we do not observe any pronounced hysteresis in the transfer curve as shown in Fig. 6F. Also the device with def-GO/DA does not deliver observable hysteresis in the transfer properties (Fig. S13A, ESI†). Due to the loss of carbon of def-GO, defects such as in-plane OH or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are most likely formed. Thus, we suggest that the plane is locally charged, which makes a discharge from the def-GO layer quite difficult due to a high energy barrier. Consequently, we barely observe any hysteresis in the transfer properties. However, we do not want to exclude morphological reasons based on aggregation processes occurring due to the lower stability of the def-GO/DA/PSEO composite dispersion in THF.
O bonds are most likely formed. Thus, we suggest that the plane is locally charged, which makes a discharge from the def-GO layer quite difficult due to a high energy barrier. Consequently, we barely observe any hysteresis in the transfer properties. However, we do not want to exclude morphological reasons based on aggregation processes occurring due to the lower stability of the def-GO/DA/PSEO composite dispersion in THF.
We also have to note that the thickness of the dielectric layer is rather critical for memory applications. In this study, both the wrapping outer shell of oxo-G1 and the SAM layer serve as the dielectric. To make the point clearer, we spin-coated oxo-G1/DA/PSEO twice to increase the layer thickness, and we indeed observe a decrease in the channel capacitance from 0.93 μF cm−2 (one time spin-coating) to 0.67 μF cm−2 (two times spin-coating). The corresponding transfer curve in Fig. S13B (ESI†) shows that the hysteresis of the curve decreases significantly. As expected, a thicker floating gate composite layer decreases the field-effect efficiency of the gate voltage and hence the charge transfer between the channel and the floating gate. Therefore, the design of the composite is very important to achieve sufficient hysteresis for the memory application. On the other hand, we also tried to remove the SAM of IMI-PA, which also yielded almost hysteresis-free transfer properties as shown in Fig. S13C (ESI†). In this case, we simply used the bare aluminum/aluminum oxide as the gate dielectric without any SAMs. The SAM plays an important role in the memory setup, since this layer also prevents the flow of the charges in the floating gate tunnel through the gate dielectric back to the gate electrode. As we can see, only our special design, with the combination of the imidazolium SAM and the polymer wrapped oxo-G1, enables the memory operation at a low voltage of about 3 V.
Conclusions
We have demonstrated that it is possible to design composite materials based on functionalized graphene to facilitate low voltage operation and, therefore, low power consumption of memory devices. In the course of our studies it turned out that a controlled chemical approach is essential to enable the function of the final devices at about 3 V. The storage of charges in oxo-G1 without undesired discharging requires an efficient isolation, which is realized by the combination of two concepts. The first is the surface functionalization of the device structure by an imidazolium SAM; the second is the isolation of flakes of oxo-G1 with a polymer. Thereby, the thickness of the isolation layer is of crucial importance for low voltage operation. Furthermore, we demonstrated the synthesis of flakes of the oxo-G1/DA/PSEO composite with a thickness of a few nm by the rational design of the precursor composite of oxo-G1/DA. The synthesis utilizes the stoichiometric electrostatic interaction of mainly organosulfate groups and dodecylammonium due to an out-of-plane electrostatic interaction. 2D NMR spectroscopy in solids using labelled 15N dodecylamine enabled the identification of the regiochemistry and the electrostatic binding of the dodecylammonium also includes interactions with the π-system of oxo-G1. State-of-the-art thermogravimetric analysis coupled with FTIR, GC and MS, in combination with EA and XPS, was used to thoroughly analyze the materials. Furthermore, we presented a series of reference experiments, which all lead to non-satisfying performance, in order to underline the importance of the controlled synthesis of the composite. This example of a memory device based on a composite of oxo-G1, which operates at 3 V, is only possible by controlling the chemistry of precursors. Thus, we believe that further developing the controlled synthesis of derivatives of oxo-G1 will overcome actual performance limits of graphene oxide-based applications.Acknowledgements
We gratefully acknowledge the German Research Council (DFG) with the Collaborative Research Center (SFB-953), Grant no. EI 938/3-1, and the Erlangen Graduate School of Molecular Science (GSMS) for funding and Prof. Dr Andreas Hirsch for his support at FAU Erlangen-Nürnberg. This work was also supported by the Cluster of Excellence ‘Engineering of Advanced Materials’ (EAM) funded by the DFG. The ssNMR structural analysis of oxo-G1/DA was supported by the U.S. National Science Foundation (NSF CHE-1310363) for YI.References
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS PubMed.
- Z. Wang, Z. Zhang and L. Peng, Chin. Sci. Bull., 2012, 57, 2956–2970 CrossRef CAS.
- Y. Chen, B. Zhang, G. Liu, X. Zhuang and E. T. Kang, Chem. Soc. Rev., 2012, 41, 4688–4707 RSC.
- A. N. Grigorenko, M. Polini and K. S. Novoselov, Nat. Photonics, 2012, 6, 749–758 CrossRef CAS.
- N. O. Weiss, H. Zhou, L. Liao, Y. Liu, S. Jiang, Y. Huang and X. Duan, Adv. Mater., 2012, 24, 5782–5825 CrossRef CAS PubMed.
- H. Xu, Z. Zhang, R. Shi, H. Liu, Z. Wang, S. Wang and L. M. Peng, Sci. Rep., 2013, 3, 1207 Search PubMed.
- T.-H. Han, Y. Lee, M.-R. Choi, S.-H. Woo, S.-H. Bae, B. H. Hong, J.-H. Ahn and T.-W. Lee, Nat. Photonics, 2012, 6, 105–110 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- S. Lee, K. Lee, C. H. Liu, G. S. Kulkarni and Z. Zhong, Nat. Commun., 2012, 3, 1018 CrossRef PubMed.
- Z. Wang, S. Eigler and M. Halik, Appl. Phys. Lett., 2014, 104, 243502 CrossRef.
- A. J. Hong, E. B. Song, H. S. Yu, M. J. Allen, J. Kim, J. D. Fowler, J. K. Wassei, Y. Park, Y. Wang, J. Zou, R. B. Kaner, B. H. Weiller and K. L. Wang, ACS Nano, 2011, 5, 7812–7817 CrossRef CAS PubMed.
- S. M. Kim, E. B. Song, S. Lee, J. Zhu, D. H. Seo, M. Mecklenburg, S. Seo and K. L. Wang, ACS Nano, 2012, 6, 7879–7884 CrossRef CAS PubMed.
- A. Rani, J.-M. Song, M. Jung Lee and J.-S. Lee, Appl. Phys. Lett., 2012, 101, 233308 CrossRef.
- S. T. Han, Y. Zhou, C. Wang, L. He, W. Zhang and V. A. Roy, Adv. Mater., 2013, 25, 872–877 CrossRef CAS PubMed.
- A. Mishra, A. Janardanan, M. Khare, H. Kalita and A. Kottantharayil, IEEE Electron Device Lett., 2013, 34, 1136–1138 CrossRef CAS.
- S. T. Han, Y. Zhou, Q. D. Yang, L. Zhou, L. B. Huang, Y. Yan, C. S. Lee and V. A. Roy, ACS Nano, 2014, 8, 1923–1931 CrossRef CAS PubMed.
- D. S. Chung, S. M. Lee, J. Y. Back, S. K. Kwon, Y. H. Kim and S. T. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 9524–9529 CAS.
- C. Kim, J. M. Song, J. S. Lee and M. J. Lee, Nanotechnology, 2014, 25, 014016 CrossRef PubMed.
- T.-W. Kim, Y. Gao, O. Acton, H.-L. Yip, H. Ma, H. Chen and A. K. Y. Jen, Appl. Phys. Lett., 2010, 97, 023310 CrossRef.
- G. L. Li, G. Liu, M. Li, D. Wan, K. G. Neoh and E. T. Kang, J. Phys. Chem. C, 2010, 114, 12742–12748 CAS.
- G. Liu, Y. Chen, R.-W. Li, B. Zhang, E.-T. Kang, C. Wang and X. Zhuang, ChemElectroChem, 2014, 1, 514–519 CrossRef.
- Z. S. Wang, F. Zeng, J. Yang, C. Chen, Y. C. Yang and F. Pan, Appl. Phys. Lett., 2010, 97, 253301 CrossRef.
- B. Zhang, Y. Chen, Y. Ren, L. Q. Xu, G. Liu, E. T. Kang, C. Wang, C. X. Zhu and K. G. Neoh, Chemistry, 2013, 19, 6265–6273 CrossRef CAS PubMed.
- B. Zhang, G. Liu, Y. Chen, L. J. Zeng, C. X. Zhu, K. G. Neoh, C. Wang and E. T. Kang, Chemistry, 2011, 17, 13646–13652 CrossRef CAS PubMed.
- B. Zhang, Y. L. Liu, Y. Chen, K. G. Neoh, Y. X. Li, C. X. Zhu, E. S. Tok and E. T. Kang, Chemistry, 2011, 17, 10304–10311 CrossRef CAS PubMed.
- X. Zhuang, Y. Chen, L. Wang, K.-G. Neoh, E.-T. Kang and C. Wang, Polym. Chem., 2014, 5, 2010–2017 RSC.
- X.-D. Zhuang, Y. Chen, G. Liu, P.-P. Li, C.-X. Zhu, E.-T. Kang, K.-G. Noeh, B. Zhang, J.-H. Zhu and Y.-X. Li, Adv. Mater., 2010, 22, 1731–1735 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. T. Han, Y. Zhou and V. A. Roy, Adv. Mater., 2013, 25, 5425–5449 CrossRef CAS PubMed.
- T. Mosciatti, S. Haar, F. Liscio, A. Ciesielski, E. Orgiu and P. Samori, ACS Nano, 2015, 9, 2357–2367 CrossRef CAS PubMed.
- F. M. Koehler and W. J. Stark, Acc. Chem. Res., 2013, 46, 2297–2306 CrossRef CAS PubMed.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 ( Angew. Chem. , 2014 , 126 , 7852–7872 ) CrossRef CAS PubMed.
- S. Eigler, Chem. Commun., 2015, 51, 3162–3165 RSC.
- S. Eigler, Y. Hu, Y. Ishii and A. Hirsch, Nanoscale, 2013, 5, 12136–12139 RSC.
- S. Eigler, S. Grimm, F. Hof and A. Hirsch, J. Mater. Chem. A, 2013, 1, 11559–11562 CAS.
- S. Eigler, M. Enzelberger-Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Rockert, J. Xiao, C. Papp, O. Lytken, H. P. Steinrück, P. Müller and A. Hirsch, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS PubMed.
- S. Eigler, C. Dotzer, F. Hof, W. Bauer and A. Hirsch, Chem. – Eur. J., 2013, 19, 9490–9496 CrossRef CAS PubMed.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Lerf, H. Heb, T. Riedl, M. Forster and J. Klinowskib, Solid State Ionics, 1997, 101–103, 857–862 CrossRef CAS.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- S. Eigler, S. Grimm and A. Hirsch, Chem. – Eur. J., 2014, 20, 984–989 CrossRef CAS PubMed.
- S. Eigler, S. Grimm, M. Enzelberger-Heim, P. Müller and A. Hirsch, Chem. Commun., 2013, 49, 7391–7393 RSC.
- J. W. Kim, D. Kang, T. H. Kim, S. G. Lee, N. Byun, D. W. Lee, B. H. Seo, R. S. Ruoff and H. S. Shin, ACS Nano, 2013, 7, 8082–8088 CrossRef CAS PubMed.
- P. Feicht, D. A. Kunz, A. Lerf and J. Breu, Carbon, 2014, 80, 229–234 CrossRef CAS.
- F. Beckert, A. M. Rostas, R. Thomann, S. Weber, E. Schleicher, C. Friedrich and R. Mülhaupt, Macromolecules, 2013, 46, 5488–5496 CrossRef CAS.
- A. M. Dimiev and T. A. Polson, Carbon, 2015, 93, 544–554 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- J. E. Jee, S. Eigler, N. Jux, A. Zahl and R. van Eldik, Inorg. Chem., 2007, 46, 3336–3352 CrossRef CAS PubMed.
- P. Montes-Navajas, N. G. Asenjo, R. Santamaría, R. Menéndez, A. Corma and H. García, Langmuir, 2013, 29, 13443–13448 CrossRef CAS PubMed.
- J. Walter, T. J. Nacken, C. Damm, T. Thajudeen, S. Eigler and W. Peukert, Small, 2015, 11, 814–825 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. – Eur. J., 2014, 20, 1871–1877 CrossRef CAS PubMed.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S.-E. Wu, S.-F. Chen, C.-P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS PubMed.
- S. Eigler, C. Dotzer, A. Hirsch, M. Enzelberger and P. Müller, Chem. Mater., 2012, 24, 1276–1282 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- X. Zhang, A. C. Coleman, N. Katsonis, W. R. Browne, B. J. v. Wees and B. L. Feringa, Chem. Commun., 2010, 46, 7539–7541 RSC.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- S. Eigler, Phys. Chem. Chem. Phys., 2014, 16, 19832–19835 RSC.
- M. M. Lucchese, F. Stavale, E. H. M. Ferreira, C. Vilani, M. V. O. Moutinho, R. B. Capaz, C. A. Achete and A. Jorio, Carbon, 2010, 48, 1592–1597 CrossRef CAS.
- L. G. Cancado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef CAS PubMed.
- S. Eigler, F. Hof, M. Enzelberger-Heim, S. Grimm, P. Müller and A. Hirsch, J. Phys. Chem. C, 2014, 118, 7698–7704 CAS.
- Z. Wang, S. Mohammadzadeh, T. Schmaltz, J. Kirschner, A. Khassanov, S. Eigler, U. Mundloch, C. Backes, H. G. Steinruck, A. Magerl, F. Hauke, A. Hirsch and M. Halik, ACS Nano, 2013, 7, 11427–11434 CrossRef CAS PubMed.
- M. C. Hamilton, S. Martin and J. Kanicki, Chem. Mater., 2004, 16, 4699–4704 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details on synthesis, Raman analysis, titration experiments, TGA, FTIR, GC, MS, ssNMR and simulations, AFM, optical microscope images, device preparation and characterization. See DOI: 10.1039/c5tc01861g |
| ‡ Both authors contributed equally to this work. |
| § Current address: Advanced Microelectronic Center Aachen (AMICA), AMO GmbH, Otto-Blumenthal-Straße 25, 52074 Aachen, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |