
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergetic enhancement of organic solar cell thermal stability by wire bar coating and light processing†
Ching-Hong
Tan
ac,
Him Cheng
Wong
cde,
Zhe
Li
abc,
David G.
Bucknall
f,
James R.
Durrant
*abc and
João T.
Cabral
*cd
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK
bSPECIFIC, College of Engineering, Swansea University, Baglan Bay Innovation, Knowledge Centre, Central Avenue, Baglan SA12 7AX, UK
cCentre for Plastic Electronics, Imperial College London, SW7 2AZ, UK. E-mail: j.cabral@imperial.ac.uk; j.durrant@imperial.ac.uk
dDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
eEngineering Product Development, Singapore University of Technology and Design, 8 Somapah Road, 487372 Singapore, Singapore
fMaterials Science and Engineering, Georgia Institute of Technology, 801 Ferst Dr NW, Atlanta, GA 30332, USA
First published on 13th August 2015
Abstract
We demonstrate that organic solar cells can exhibit different morphological and performance stability under thermal stress depending upon the processing technique employed, without compromising initial device efficiency. In particular, we investigate benchmark PCDTBT:PC60BM solar cells fabricated by wire bar coating (a technique attractive for commercial manufacture) and the more widely employed, lab scale, technique of spin coating. For this system, wire bar deposition results in superior device stability, with lifetime improvements in excess of 20-fold compared to spun cast devices. Neutron reflectivity reveals that the enhanced PC60BM segregation to the top interface in the slower, wire bar, casting process is likely responsible for the hindered PC60BM nucleation at tens of nm length scale, characterized by atomic force microscopy (AFM), and thus enhanced morphological stability. Modest light exposure of the active layer (at approximately 10 mW cm−2), known to reversibly photo-oligomerize fullerenes and thus impart morphological stability, is found to further improve device stability by a factor of 10. The combined effects of wire bar coating and light processing are highly synergetic, resulting in solar cells which are overall 200 times more stable than devices prepared by spin casting without light processing.
Introduction
Organic solar cells (OSCs), generally comprising a nanostructured thin film of a conjugated polymer donor and a fullerene derivative acceptor, provide an increasingly attractive technology for energy harvesting and solar energy conversion. While single-junction solar cells are now approaching 10%, often regarded as the threshold for commercialization,1–4 significant challenges remain in attaining the long-term OSC stability required for commercial application5 and ensuring low cost, reliable manufacturing and scale-up. Our paper considers the role of representative photoactive layer deposition and processing approaches on the morphological and performance stability of OSC under 80 °C thermal stress, relevant to device operation conditions.The vast majority of highly efficient polymer/fullerene OSCs reported to date has been fabricated by spin coating (SC).6–8 This is intrinsically a batch technique, and unsuitable for large scale manufacturing.7 Further, SC typically wastes 90% of precursor solution and is incompatible with device patterning. A number of alternative deposition methods for solution processable OSCs have been proposed, including spray drying,9 flow and wire bar coating (WBC), which appear compatible with industrial roll-to-roll (R2R) manufacturing.10,11 WBC, focused on herein, employs a wire wound bar that translates over the substrate at a well-defined height, depositing the active layer solution; this processing technique allows control of velocity and temperature profiles with minimal waste and high reproducibility.11,12 The impact of solution deposition in OSC morphology and performance is known to be significant,10,13–16 related in part to the different flow fields and temporal drying profiles experienced during film formation. SC is generally faster, while WBC is comparatively slower and can lead to the development of travelling fronts, surface templating, structural rearrangement and component stratification.7,10,11,17 During the photoactive layer film formation process, the development of a nano-phase separated bicontinuous morphology (a ‘bulk heterojunction’) is required for device performance, and in particular for charge generation and carrier transport.2 Film deposition, in addition to subsequent thermal,18 solvent19 and light processing,20,21 plays a key role in engineering a functional nanostructured photoactive layer.
Solar cell operation inherently involves the cyclic temperature and illumination variations which occur in the environment. Prolonged thermal annealing has been found to cause degradation of OSC device efficiency, which has been related to the diffusion, aggregation and large-scale crystallization of the fullerene derivatives, coarsening of the nanophases and delamination of electrodes, amongst others.13,18,22 Hence, a thermally stable (either thermodynamically-stable or ‘frozen’ out-of-equilibrium) morphology is desirable for long-term OSC device performance.
Both micron- and nano-scale [6,6]-phenyl-C61-butyric-acid-methyl-ester (PC60BM) crystallization have been extensively documented both in neat films and when blended with a donor polymer,23–29 and found to be dependent on solution deposition method and solvent, substrate, polymer matrix and regioregularity, polymer composition profile, and subsequent annealing temperature. For instance, slow evaporation of neat PC60BM solutions was found to favour micron-size crystals, by contrast with nanocrystals formed by faster spin coating,23 highlighting the importance of evaporation and fullerene self-organization kinetics in film formation. The mechanism of PC60BM crystallization, including overall crystal size and crystal structure, has been shown to be mediated by interaction with the polymer matrix and templated by the substrate (mica, in one study).27 Micron- and nano-scale PC60BM crystal formation can happen simultaneously or exclusively through a competitive bimodal crystallization process.29 Abundant formation of large crystals generally corresponds to a scarcity of nano-crystals and vice versa.20,21,27,29
Blend films of donor polymers with PC60BM, the most widely studied acceptor for OSCs, often exhibit only limited thermal stability, depending on the donor polymer employed.30–32 For example, our recent thermal stability studies of OSC's employing the benchmark donor polymer poly[[9-(1-octylnonyl)-9H-carbazole-2,7-diyl]-2,5-thiophenediyl-2,1,3-benzothiadiazole-4,7-diyl-2,5-thiophenediyl] (PCDTBT) showed a retention of only 65% of the initial device performance after 130 h of thermal annealing at 80 °C.21 Strategies to enhance OSC thermal stability have focused on chemically modifying the polymer donor, additives or fullerenes (recently reviewed by Rumer and Mcculloch33). For instance, enhanced OSC thermal stability has been recently demonstrated by crosslinking the polymer donor (achieved 0.9 internal quantum yield),34 copolymer (power conversion efficiency (PCE) of 4.7% after 72 h of 150 °C thermal annealing),35 functionalization of donor polymer (PCPDTBT) side chain36 and by UV-crosslinking the fullerene phase37 as well as the addition of film additive to improve OSC thermal stability.38 We have recently reported20,21 that modest white light illumination (<10 mW cm−2) prior to thermal annealing, can also result in the OSC thermal stability increasing ∼10 fold due to a light-induced fullerene oligomerization process.39 This process bonds at least two PC60BM molecules and, beyond a certain threshold, effectively suppresses fullerene crystallization during thermal annealing, which in turn results in an enhanced device thermal stability. We21 and others40 have shown that this light induced morphological stabilization effect is observable across PC60BM mixtures with a range of benchmark donor polymers including P3HT, DPP-TT-T and PTB7, and can also be induced by the addition of chemically synthesized fullerene dumbbells.41 Unexpectedly, we found that only the nucleation – but not the growth – rate of PC60BM macroscopic crystals is affected by light exposure. Fullerene nucleation appears thus to be effectively frustrated by PC60BM oligomerization, with approximately 20% conversion fully suppressing crystallization. Further, light induced oligomerization was found to be reversible,21 as the fullerene–fullerene bond can be fully dissociated (dissociation energy approx. 1 eV) at elevated temperatures (150 °C for 60 minutes). We also noted a significant role of the substrate on the PC60BM crystallization in the film and its thermal stability: micron-sized crystallization is significant on SiOx but infrequent on poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) under identical annealing conditions. Instead, nano-sized crystal formation is observed on PEDOT:PSS. Prior illumination was found to be effective in suppressing both micro and nano-sized crystallization under thermal annealing.21
With these ideas in mind, we now directly compare the industrially relevant film processing technique of WBC to the benchmark SC process in terms of film morphology, OSC device performance and thermal stability. In addition, we implement the previously established light-induced thermal stability enhancement20,21 on WBC and SC devices to evaluate the combined effects of the two processing approaches, and demonstrate an overall (synergetic) lifetime enhancement of approximately 200× for PCDTBT:PC60BM OSCs.
Experimental
Device fabrication and thermal stability studies
PCDTBT (Mw ∼ 21.6 kg mol−1; PDI ∼ 5.5) and PC60BM were supplied by 1-Materials, Inc. and Nano-C, Inc. respectively. A solution of PCDTBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC60BM in 1:2 ratio was dissolved in CB solvent with a total solution concentration of 25 g L−1. This solution was stirred for 24 h in N2 environment followed by filtration through a 0.2 μm filter prior to applying.
:PC60BM in 1:2 ratio was dissolved in CB solvent with a total solution concentration of 25 g L−1. This solution was stirred for 24 h in N2 environment followed by filtration through a 0.2 μm filter prior to applying.
ITO glass substrates were cleaned with acetone, iso-propyl alcohol and deionized water sequentially and underwent an oxygen plasma treatment at 100 W for 7 min. PEDOT:PSS layers of 35 nm were spin coated on the glass substrates followed by annealing at 150 °C for 20 min in air. The filtered PCDTBT and PC60BM solution was deposited on the substrates by spin coating and wire bar coating to achieve a thickness of 75–80 nm, as determined with a Dektak 6M profilometer. Film drying kinetics was monitored in situ by a CCD camera and automated image analysis, and ‘film formation time’ was estimated based on cessation of colour change (Fig. S1, ESI†). The films were then placed into an evaporation vacuum chamber for 25 nm of calcium and 100 nm of aluminium deposition through a 6-pixel mask (each pixel area is 0.045 cm2) respectively. Selected samples were illuminated under a fluorescent lamp prior to electrode deposition, corresponding to ‘light’ conditions, while some ‘dark’ control films were kept in the dark conditions; both in a N2 glove box. The deposited devices were annealed at 80 °C while the device performance was measured. Annealing temperature was measured and calibrated using a Kane-May KM330 temperature sensor. The devices current density–voltage (J–V) characteristics were measured using a Xenon lamp at AM 1.5 solar illumination (Oriel Instruments) calibrated to a silicon reference cell with a Keithley 2400 source meter. A fluorescent lamp was used as light source, with light irradiance (≈10 mW cm−2) and wavelength distribution calibrated with photospectrometer (StellarNet EPP2000).
Morphology and neutron reflectivity studies
SiOx substrates were purchased from Compart Technology and cleaned with N2 before deposition of the same PCDTBT:PC60BM solution by SC and WBC techniques. For few selected substrates, 35 nm of PEDOT:PSS layers were spun coated on SiOx substrates before solution deposition. Film thicknesses were confirmed by profilometry. The blend films were illuminated and annealed at 80 °C and 140 °C in a N2 glove box. The same fluorescent lamp was applied as the light source. The morphology of the films was measured with a reflection optical microscopy (Olympus BX 41M), equipped with an XY stage and CCD camera (AVT Marlin). The surface topography of annealed films was measured with atomic force microscopy (Innova, Bruker AXS) in tapping-mode, using super sharp TESP-SS tips.Results and discussion
Fig. 1 illustrates our processing route, including the deposition of active layer (PCDTBT:PC60BM) with SC and WBC techniques along with a light treatment (and dark control) prior to an evaluation of thermal stability. The device employs a conventional architecture ITO/PEDOT:PSS/PCDTBT:PC60BM/Ca/Al together with an optimized solvent, polymer:fullerene ratio and solution concentration, as described in the Experimental section, and following previous work;42,43 these parameters are therefore kept constant throughout this study.
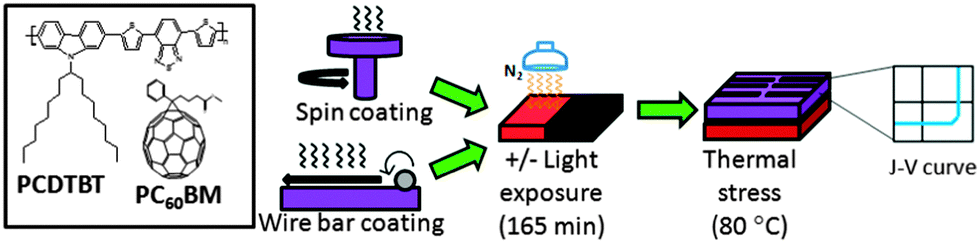 | ||
| Fig. 1 Schematic representation of processing PCDTBT:PC60BM blend with different coating techniques (SC and WBC) and light treatment. Chemical structures of PCDTBT and PC60BM are shown on the left box. Modest illumination (<10 mW cm−2) is irradiated on film for a constant duration of 165 minutes prior electrode (Ca/Al) vacuum deposition. The devices and films without and with illumination are designated as “dark” (black colour) and “light” (red colour) conditions respectively in this study. The performance (J–V curve) of the fabricated devices is measured under 80 °C thermal stress. | ||
Our first observation is that WBC films exhibit a higher average drying time (13–15 s), compared to SC films (around 5 s) to achieve a similar film thickness of 75–80 nm PCDTBT:PC60BM blend film.42 Drying time was estimated by film colour (Fig. S1, ESI†); this trend is expected due to the rapid solution removal during the initial stages of SC, followed by drying. The volume required for WBC is only 15 μL, compared to 35 μL for SC, due to the waste created during the spinning process, which is a major asset of the former processing route.
Device performance and stability
The performance of typical devices fabricated with SC and WBC techniques, in the absence of prior light processing, is shown in Fig. 2a (procedures detailed in the Experimental section). Although these coating techniques involve different film formation mechanisms, the initial performance of these typical devices achieved using the SC and WBC techniques are identical for this system within measurement uncertainty (average PCE of 5.20 ± 0.2% and 5.22 ± 0.2% for SC and WBC respectively). These similar performances can be understood as a result of a compromise between JSC and FF: 9.82 mA cm−2 and 0.59 for SC and 9.29 mA cm−2 and 0.61 for WBC, for the devices shown. The differences in JSC and FF did not correlate with variations in film thickness (±5 nm) and the VOC achieved in both devices is the same (0.93 V) and typical for optimized PCDTBT:PC60BM systems.42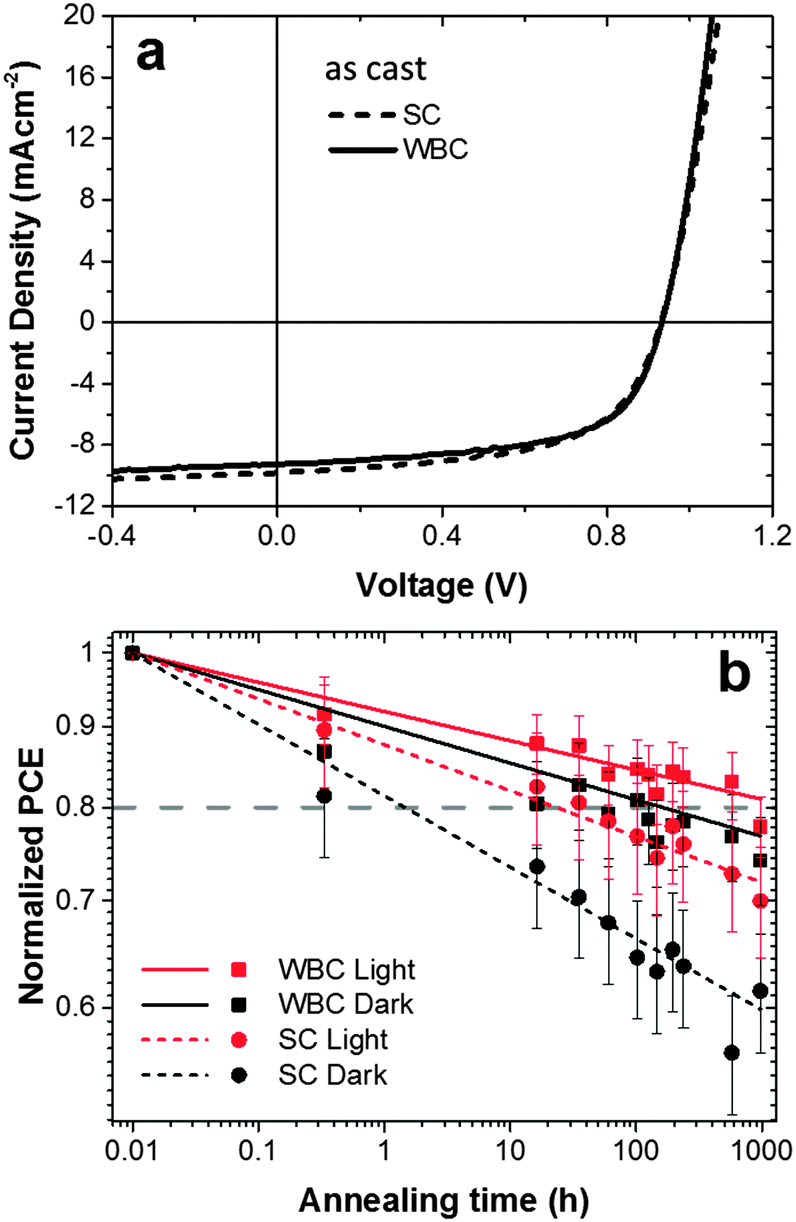 | ||
| Fig. 2 (a) Current density (J)–voltage (V) curves for typical PCDTBT:PC60BM blend devices processed with SC and WBC (prior to light processing) exhibiting PCE of 5.33% and 5.26% respectively (b) log:log plot of normalized PCE vs. annealing time during thermal stress at 80 °C for devices fabricated via SC and WBC under dark and light processing conditions. | ||
From the UV-visible absorption and polymer photo-luminescence (PL) quenching measurements (Fig. S2 and S3, ESI†), we find that films fabricated by SC and WBC have similar optical absorption, indicating similar PCDTBT:PC60BM compositions and similar PL quenching, showing similar polymer exciton quenching efficiencies, indicative of a similarly intermixed system. This verification is needed to ensure no composition bias is introduced during film deposition. The SC devices do however exhibit higher External Quantum Efficiencies (EQE) (Fig. S4, ESI†), consistent with their higher JSC (Fig. 2a). On the other hand, the higher FF attained for the WBC devices suggests a better percolation pathway for efficient charge transport in the morphology obtained by WBC.
These devices, along with their light-processed analogues, were thermally annealed at 80 °C for 1000 hours in the dark in a glove box, and their room temperature performance measured periodically under a simulated AM 1.5 irradiation. These thermal stress PCE results are summarized in Fig. 2b. ‘Light processed’ devices, were exposed to ≈10 mW cm−2 white light for 165 min, prior to thermal annealing, following a procedure reported previously20,21 and illustrated in Fig. 1. We have found this modest and brief light pre-treatment to yield no significant loss of the initial device performance,20,21 and that the thermal ageing profile remained unchanged before and after top electrode deposition,21 (by contrast with the top electrode interfacial degradation reported by McGehee and co-workers,44 for 60 h under 1 sun conditions). For the current devices, we recorded a modest (≈5%) drop in initial device performance, attributed to minor glove box contamination over the period of these experiments. The PCEs obtained are averaged over 3 pixels each from two different devices. We note that the PCE variation (obtained from 6 distinct measurements) of SC devices is somewhat larger than that of WBC devices (approximately by ≈10–20%); this might be caused by the faster kinetics of SC film formation, but further work would be required to establish statistically significant deviations.
The decays of the device PCEs in the four conditions shown in Fig. 2b are mainly due to changes in FF and JSC (data not shown). In all cases, the drop in device efficiency is, within measurement uncertainty, well described by a single power law decay, compatible with a single degradation process (and timescale).
We quantify relative device thermal stability by estimating the OSC ‘lifetimes’ corresponding to a 20% drop of the initial PCE under 80 °C annealing. The SC device without prior light exposure (‘dark’ control) degrades to 80% of its initial performance after only 3 h under thermal stress. Compared to this control, all the other processing conditions resulted in significant improvements in stability. SC devices exhibit an approximate 10 fold stability improvement after light processing, consistent with our previous findings of photo-induced PC60BM oligomerization.20,21 Interestingly, WBC devices, even in the absence of light processing, exhibit approximately 20 times thermal stability enhancement compared to the control SC devices. This improvement is further enhanced by light processing by a factor of around 10, bringing it to a total of approximately 200× lifetime enhancement for light processed WBC devices over dark processed SC devices in dark conditions, with a 20% reduction of PCE only being observed after 1000 hours of thermal stress at 80 °C. Similar lifetime enhancements are obtained with different degradation criteria, e.g. by defining the time associated with a 10% drop in efficiency. The devices manufactured by WBC and light processing have thus substantially improved stability compared to both SC and WBC dark counterparts.
By comparing the relative magnitudes of lifetime enhancement, we find that there is a compound effect of solution casting technique and photo-induced oligomerization, well above a simple additive rule. A possible explanation for this synergy is that fullerene oligomerization reactions are strongly concentration-dependent and not directly proportional to light intensity due to a partial of back-reaction from excited triplet states.45 An enhancement of WBC device stability after light treatment could indicate a higher PC60BM concentration available for oligomerization thereby restricting the degradation of performance. However, UV-vis data (Fig. S2, ESI†) confirm that the average PCDTBT and PC60BM composition remains similar regardless of solution casting technique. We therefore hypothesize that longer drying times involved in WBC may result in enhanced fullerene segregation, which locally raises fullerene concentration and, in turn, increase the propensity for photo-oligomerization.
Morphological stability
In order to investigate the stability enhancement of devices fabricated by WBC, the film nanostructure was investigated by optical and AFM as well as specular neutron reflectivity (NR). To allow a direct comparison with OSC performance data, the active layers (supported by PEDOT:PSS, but without electrode deposition) were annealed at 80 °C for 1 h, the AFM results are shown in Fig. 3. As expected, we observed the formation of nano-sized PC60BM crystals (∼100 nm)46 on the SC film on PEDOT:PSS (Fig. 3a). Evidently, below the Tg of both neat polymer and fullerene phases, the composite films enable sufficient PC60BM mobility (corroborated by interdiffusion41 and morphological studies21) to yield nano-sized aggregation. These nano-crystals do not form when the film is illuminated prior to thermal annealing (Fig. 3b), which we have rationalized in terms of the suppressed nucleation associated with photo-induced oligomerization.21 Our recent results on nano-sized PC60BM crystallization suppression by chemically synthesized PC60BM dimers within the active layer41 supports this interpretation. Unexpectedly, no nano-crystals are observed on blends produced by WBC on PEDOT:PSS substrates with or without light processing, as shown in Fig. 3c and d. The films have similar topography and roughness, with maximum amplitude of 10.3 nm and 11.9 nm in ‘dark’ and ‘light’ conditions respectively. Negligible micron-sized crystallization is observed for all samples on PEDOT:PSS (cf. optical microscopy data in Fig. S5 and S7, ESI†), interpreted as due to a bias towards nano-sized crystal formation on PEDOT:PSS in the context of PC60BM's competitive bimodal crystallization.27,29 The inhibition of nano-crystal formation with WBC coating and light processing correlates qualitatively with our device thermal stability results, whereby SC device performance in dark conditions is the least stable under thermal stress.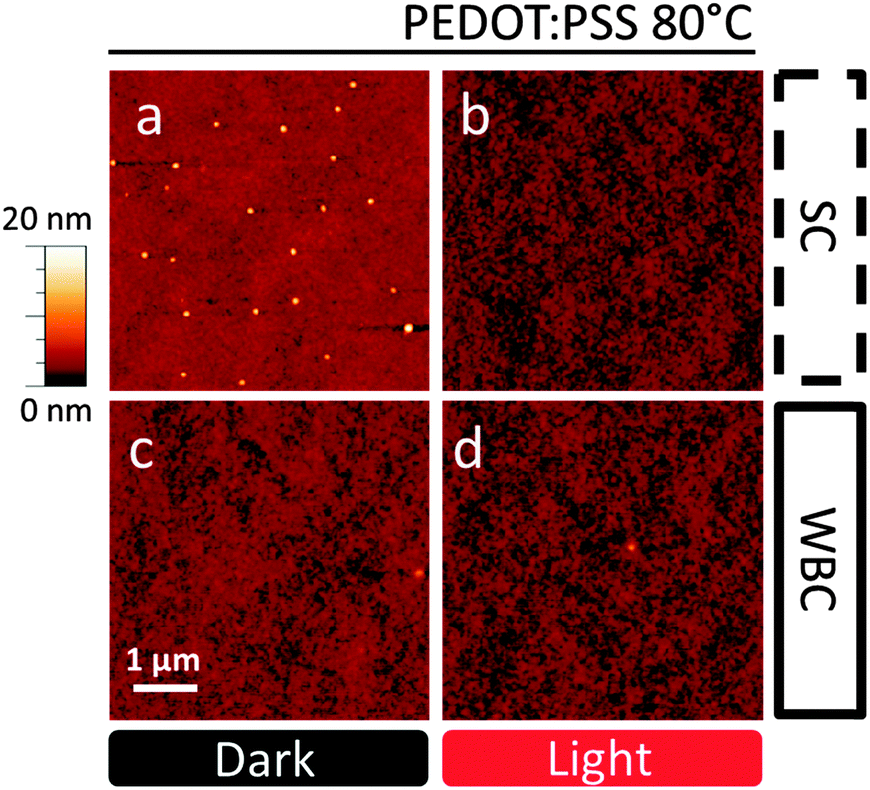 | ||
| Fig. 3 AFM images of PCDTBT:PC60BM blend on PEDOT:PSS substrate annealed at 80 °C for l h. Films cast by (a) and (b) SC and (c) and (d) WBC and processed without (a) and (c) and with (b) and (d) prior light exposure. | ||
Intriguingly, we find that morphological studies of thermally induced PC60BM crystallization of the same blends when cast onto SiOx substrates, as used in several previous studies,20,21,32,47–51 does not correlate with our device stability data. Representative optical micrographs of SC and WBC films annealed for 1 h at 140 °C, with or without prior light treatment, are shown in Fig. 4. As cast films on SiOx are uniform in all cases (Fig. S6, ESI†) in agreement with previous findings,21 and annealing at 80 °C does not suffice to induce crystallization, as seen by AFM and optical microscopy. Thermal annealing of thin blend films supported by SiOx substrates above glass transition temperature at 140 °C results exclusively in micron-sized fullerene crystals,20,21,32,47–51 in contrast to nano-sized on PEDOT:PSS substrates.21 The number density of micron-scale crystals in WBC films is now considerably higher (Fig. 4c) than in SC films (Fig. 4a) for ‘dark’ control blends. Partial suppression of micron-sized PC60BM crystallization is observed for both SC and WBC films upon prior light exposure, as shown in Fig. 4b and d, and the crystal number density remains higher in the latter. While morphology and OSC performance are often correlated, and specifically large scale crystallization is generally associated with poor performance, these results on SiOx show the opposite trend with device PCE stability between WBC and SC devices. We find this to be a significant observation as, owing to the good optical contrast and simplicity sample preparation, SiOx substrates are often employed in surrogate optical microscopy morphological stability studies for OSCs under thermal stress; annealing temperatures higher than typical operating temperatures are also often employed to facilitate phase identification.20,40 In this case, we show that morphological results for annealed SiOx supported films are inversely correlated with OSC stability. However, we do find a positive correlation between nano-sized crystallization and the loss of device performance under thermal stress, at the same annealing temperature (80 °C) and for films supported by the same substrate (PEDOT:PSS), as shown by Fig. 2 and 3.
 | ||
| Fig. 4 Optical microscope images of PCDTBT:PC60BM blend on PEDOT:PSS substrate annealed at 140 °C for l hour. Films deposited by (a) and (b) SC and (c) and (d) WBC and processed without (a) and (c) and with (b) and (d) prior light exposure. | ||
In order to rationalize these findings, we turn to NR to evaluate the film composition depth profiles for both SC and WBC techniques, light exposure and substrate types (SiOx and PEDOT:PSS). Our hypothesis is that PC60BM stratification during film formation and the effectiveness of light-induced oligomerization at high fullerene concentration may, jointly and individually, contribute to thermal stability. We recall that the topochemical criteria for photo-induced fullerene 2+2 cyclo-addition requires parallel carbon double bond alignment of adjacent fullerenes and fullerene proximity below 4.2 Å.52 In first approximation, photo-oligomerization is therefore more likely to be satisfied at a higher fullerene concentration, thus upon a stronger segregation, suggesting that local PC60BM concentration at both interface is likely to be critical to film and device thermal stability.
NR was employed to measure the scattering length density (and thus composition) profile of thin films along the direction orthogonal to the film surface (i.e. momentum transfer along qz, the vertical direction). We investigate the influence of SC and WBC on the film depth profile, the role of the substrate (namely, SiOx and PEDOT:PSS) and that of moderate light exposure (and ‘dark’ control). Fig. 5a compares the reflectivity and depth profile of as cast films deposited on PEDOT:PSS by WBC and SC. As cast depth profile of PCDTBT:PC60BM deposited on PEDOT:PSS using SC technique has been previously studied using NR53,54 where PC60BM enrichment at the top interface, with diminishing concentration towards the substrate interface, are observed. Our SC results (bottom row in Fig. 5a) generally agree with previous reports. Compared to SC, we find that WBC films exhibit higher PC60BM enrichment at the air interface as well as more PC60BM depleted at the substrate interface (top data in Fig. 5a), consistent with the longer time scale for film formation and thus component segregation. This finding is also consistent with the higher device FF, with the PC60BM enrichment at the active layer–cathode interface likely to improve charge collection as shown in Fig. 2a. This segregation is not detrimental to overall device performance as similar PCE values were obtained for devices fabricated with both techniques. Also, such segregation correlates well with expected higher efficiency for photo-oligomerization and thus the observed superior device thermal stability upon light exposure. Indeed, we observe the illuminated WBC condition offers the most stable device performance of all four conditions tested (Fig. 2b). In short, our results suggest that the longer drying time associated with WBC result in enhanced compositional segregation, and thus a film depth profile which is more thermally stable and more susceptible to photo-oligomerization compared to SC films.
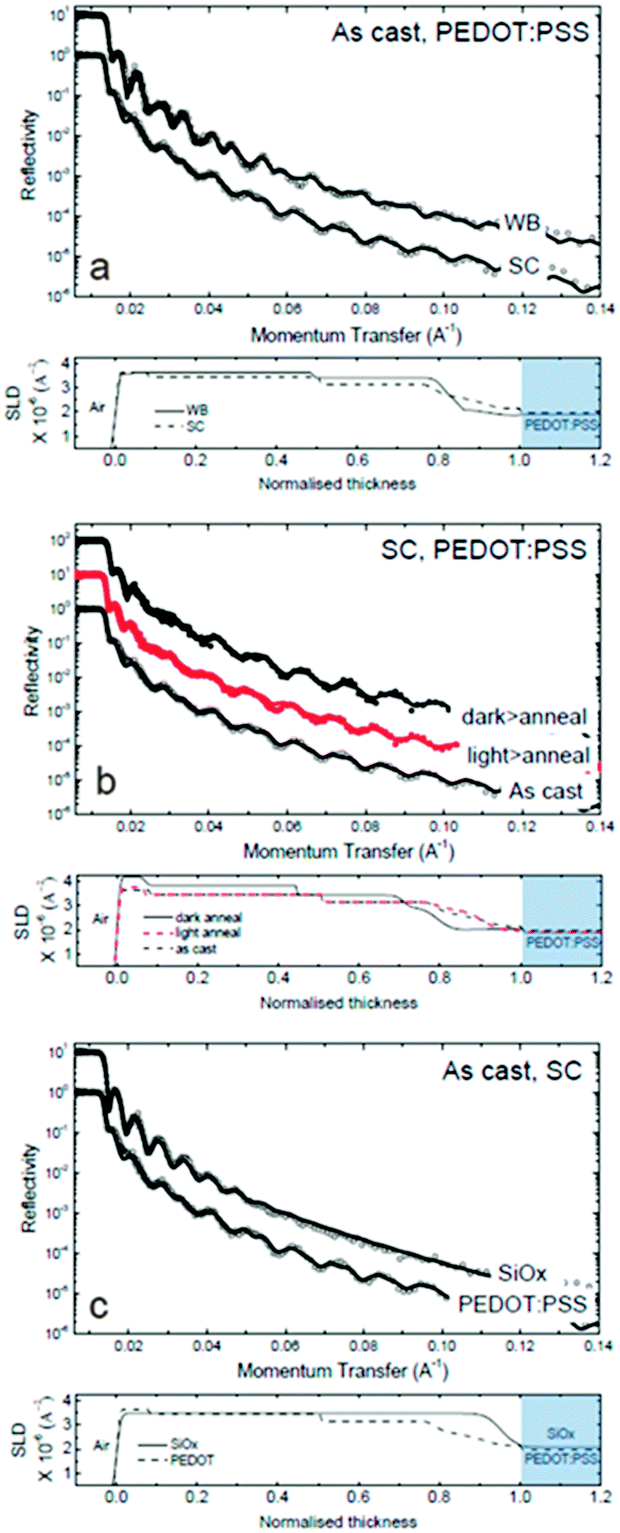 | ||
| Fig. 5 Neutron reflectivity data of PCDTBT:PC60BM system: (a) as cast SC and WBC films on PEDOT:PSS substrate; (b) SC films on PEDOT:PSS substrate in ‘dark’ and ‘light’ conditions after thermal annealing at 80 °C for 1 hour; (c) as cast SC films on PEDOT:PSS and SiOx substrates. | ||
In agreement with our previous findings on polystyrene(PS):C60,55 we find that light exposure does not alter the film reflectivity profiles, and thus the composition depth profile, of polymer:fullerene films. Here we show the scattering length density (SLD)/PC60BM composition depth profiles of light-processed spun cast films also remain largely unchanged upon 80 °C annealing (red filled data), compared to as cast films (black unfilled data) as shown in Fig. 5b. This robustness to annealing illustrates the role of PC60BM photo-oligomerization in OSC film stability and contrasts with the depth profile changes of films annealed without prior illumination (black filled data) (‘dark’ SC). Indeed, we find enhanced PC60BM surface segregation in ‘dark’ SC films upon annealing and, in AFM which probes the lateral structure, we observe the formation of PC60BM nano-sized crystals (Fig. 3a).
We finally compare the composition depth profile of films supported by PEDOT:PSS and SiOx substrates and a more uniform PC60BM profile in the latter, compared to the former (Fig. 5c). This stratification is mediated by the lower surface energy of PEDOT:PSS (≈47 mN m−1), compared to SiOx (≈73 mN m−1).56 The device thermal instability appears to correlate with the formation of nano-sized crystals in the active layer, whose inhibition (e.g. by light-induced oligomerization of PC60BM) yields OSCs with enhanced thermal stabilities. The presence of a trivial fraction of PC60BM oligomers (10–20%) appears to effectively suppress nucleation and thus film crystallization.20,21,41
Evidently, WB films form over longer timescales compared to SC films, which appears to yield enhanced film composition segregation as shown by NR, which we believe hinders PC60BM diffusion. Light exposure further improves both WBC and SC devices yielding solar cells with enhanced lifetimes. This synergistic effect is particularly significant for WBC cells, whose lifetime improvement appears to be approximately multiplicative (instead of additive, for example) with respect to SC ‘dark’ devices.
Conclusions
In summary, we examine the morphology and performance of PCDTBT:PC60BM organic solar cells fabricated by two significant solution casting techniques: wire bar coating (WBC), compatible with large scale manufacturing, and spin coating (SC), well established for lab-scale fabrication and employed for most OSC research. We find that both WBC and SC solar cells yield similar initial performance, despite slight variations of JSC and FF, as measured by PCE (≈5%). WBC utilizes a much lower solution volume than that required by SC of equivalent active layer films. Further, we find that WBC cells exhibit improved device thermal stabilities, measured over 1000 h annealing at 80 °C, which appears to stem from enhanced PC60BM segregation, towards the top interface during deposition, which is likely due to longer film drying timescales. To the best of our knowledge, this is the longest time frame for thermal stability investigated with this system. We show that both WBC and SC cells respond favorably to a light processing step prior to thermal annealing, which increases device lifetime due to a morphological stabilization caused by PC60BM photo-oligomerization. The overall PCE lifetimes for WBC light processed solar cells is found to be nearly 200 times longer compared to conventional SC devices (manufactured without light processing).We found that widely used correlations between morphological stability and device lifetime can be misleading if morphological studies are not performed at the same temperature and on the same device substrate, due to impact of the substrate in film vertical segregation and lateral morphology. For example, the morphological results of WBC and SC on SiOx actually suggest the opposite trend for OSC thermally stability, since WBC films are found to result in a much greater fullerene crystal number density at 140 °C on SiOx but yield OSC with much improved thermal stability on PEDOT:PSS. In this case, morphological studies of micron-sized crystallization on SiOx thus provide neither insight into OSC morphology on PEDOT:PSS nor prediction for its thermal stability and lifetime.
We do find that OSC lifetime under thermal stress correlates with nano-sized crystal formation on PEDOT:PSS at the relevant annealing temperature. Polymer–fullerene demixing is well-known to impact OSC performance57 and we find that WBC results in more stable devices compared to SC. The timescales for film formation can be tuned by employing a controlled atmosphere, mixed solvents and additives, or by vapor annealing, opening possibilities for further improvement. Similarly the light-induced stabilization depends on wavelength, exposure duration and temperature, of which only one condition was explored in this work, resulting in substantial device lifetime enhancement. There are therefore realistic prospects that optimization of these processing parameters will enable further improvements of thermal stability of OSCs and relieve a major bottleneck for their current commercial viability.
Acknowledgements
The authors thank the EPSRC (EP/I019278/1 and KTS, DPF funding for ZL and HCW) and the Welsh government (Sêr Cymru) as well as the EU project CHEETAH for financial support. The authors also thank Marie-Beatrice Madec (Solvay SA), and Pabitra Shakya and Shahid Ashraf (Imperial) for help in device fabrication. We are grateful to the Institut Laue Langevin (ILL), Grenoble for beamtime and Dr P. Gutfreund (ILL) and Dr A. Guilbert (Imperial) for experimental support.Notes and references
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, U297–U295 CrossRef PubMed.
- F. Liu, Y. Gu, J. W. Jung, W. H. Jo and T. P. Russell, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1018–1044 CrossRef CAS PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS PubMed.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- F. C. Krebs, in Organic photovoltaics 6, ed. Z. H. Kafafi and P. A. Lane, SPIE Proceedings Series, International Society for Optical Engineering, Bellingham, 2005, vol. 5938, pp. 234–244 Search PubMed.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS PubMed.
- K. Norrman, A. Ghanbari-Siahkali and N. B. Larsen, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2005, 101, 174–201 RSC.
- T. Wang, N. W. Scarratt, H. Yi, A. D. F. Dunbar, A. J. Pearson, D. C. Watters, T. S. Glen, A. C. Brook, J. Kingsley, A. R. Buckley, M. W. A. Skoda, A. M. Donald, R. A. L. Jones, A. Iraqi and D. G. Lidzey, Adv. Energy Mater., 2013, 3, 505–512 CrossRef CAS PubMed.
- J. Ouyang, T. F. Guo, Y. Yang, H. Higuchi, M. Yoshioka and T. Nagatsuka, Adv. Mater., 2002, 14, 915–918 CrossRef CAS.
- C. E. Murphy, L. Yang, S. Ray, L. Yu, S. Knox and N. Stingelin, J. Appl. Phys., 2011, 110, 093523 CrossRef PubMed.
- Y. Galagan, I. G. de Vries, A. P. Langen, R. Andriessen, W. J. H. Verhees, S. C. Veenstra and J. M. Kroon, Chem. Eng. Process., 2011, 50, 454–461 CrossRef CAS PubMed.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653–5667 CAS.
- X. Zhang, J. F. Douglas and R. L. Jones, Soft Matter, 2012, 8, 4980–4987 RSC.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- C. Koidis, S. Logothetidis, A. Ioakeimidis, A. Laskarakis and C. Kapnopoulos, Org. Electron., 2013, 14, 1744–1748 CrossRef CAS PubMed.
- B. Dan, G. C. Irvin and M. Pasquali, ACS Nano, 2009, 3, 835–843 CrossRef CAS PubMed.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS PubMed.
- G. Li, Y. Yao, H. Yang, V. Shrotriya, G. Yang and Y. Yang, Adv. Funct. Mater., 2007, 17, 1636–1644 CrossRef CAS PubMed.
- Z. Li, H. C. Wong, Z. Huang, H. Zhong, C. H. Tan, W. C. Tsoi, J. S. Kim, J. R. Durrant and J. T. Cabral, Nat. Commun., 2013, 4, 2227 Search PubMed.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297–1308 CrossRef CAS PubMed.
- B. A. Collins, J. R. Tumbleston and H. Ade, J. Phys. Chem. Lett., 2011, 2, 3135–3145 CrossRef CAS.
- X. Yang, J. K. J. van Duren, M. T. Rispens, J. C. Hummelen, R. A. J. Janssen, M. A. J. Michels and J. Loos, Adv. Mater., 2004, 16, 802–806 CrossRef CAS PubMed.
- A. Swinnen, I. Haeldermans, M. vande Ven, J. D'Haen, G. Vanhoyland, S. Aresu, M. D'Olieslaeger and J. Manca, Adv. Funct. Mater., 2006, 16, 760–765 CrossRef CAS PubMed.
- C. H. Woo, B. C. Thompson, B. J. Kim, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 16324–16329 CrossRef CAS PubMed.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS PubMed.
- L. Zheng, J. Liu and Y. Han, Phys. Chem. Chem. Phys., 2013, 15, 1208–1215 RSC.
- J. J. Richards, A. H. Rice, R. D. Nelson, F. S. Kim, S. A. Jenekhe, C. K. Luscombe and D. C. Pozzo, Adv. Funct. Mater., 2013, 23, 514–522 CrossRef CAS PubMed.
- D. Mon, A. M. Higgins, D. James, M. Hampton, J. E. Macdonald, M. B. Ward, P. Gutfreund, S. Lilliu and J. Rawle, Phys. Chem. Chem. Phys., 2015, 17, 2216–2227 RSC.
- E. Voroshazi, B. Verreet, T. Aernouts and P. Heremans, Sol. Energy Mater. Sol. Cells, 2011, 95, 1303–1307 CrossRef CAS PubMed.
- R. De Bettignies, J. Leroy, M. Firon and C. Sentein, Synth. Met., 2006, 156, 510–513 CrossRef CAS PubMed.
- B. J. Kim, Y. Miyamoto, B. Ma and J. M. J. Fréchet, Adv. Funct. Mater., 2009, 19, 2273–2281 CrossRef CAS PubMed.
- J. W. Rumer and I. McCulloch, Materials Today, 2015 DOI:10.1016/j.mattod.2015.04.001.
- B. Liu, R.-Q. Png, L.-H. Zhao, L.-L. Chua, R. H. Friend and P. K. H. Ho, Nat. Commun., 2012, 3, 1321 CrossRef PubMed.
- G. Griffini, J. D. Douglas, C. Piliego, T. W. Holcombe, S. Turri, J. M. J. Fréchet and J. L. Mynar, Adv. Mater., 2011, 23, 1660–1664 CrossRef CAS PubMed.
- J. Kesters, P. Verstappen, J. Raymakers, W. Vanormelingen, J. Drijkoningen, J. D’Haen, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, Chem. Mater., 2015, 27, 1332–1341 CrossRef CAS.
- L. Derue, O. Dautel, A. Tournebize, M. Drees, H. Pan, S. Berthumeyrie, B. Pavageau, E. Cloutet, S. Chambon, L. Hirsch, A. Rivaton, P. Hudhomme, A. Facchetti and G. Wantz, Adv. Mater., 2014, 26, 5831–5838 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- P. Zhou, Z.-H. Dong, A. M. Rao and P. C. Eklund, Chem. Phys. Lett., 1993, 211, 337–340 CrossRef CAS.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D'Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209–1214 CrossRef CAS PubMed.
- B. C. Schroeder, Z. Li, M. A. Brady, G. C. Faria, R. S. Ashraf, C. J. Takacs, J. S. Cowart, D. T. Duong, K. H. Chiu, C.-H. Tan, J. T. Cabral, A. Salleo, M. L. Chabinyc, J. R. Durrant and I. McCulloch, Angew. Chem., Int. Ed., 2014, 53, 12870–12875 CrossRef CAS PubMed.
- T.-Y. Chu, S. Alem, P. G. Verly, S. Wakim, J. Lu, Y. Tao, S. Beaupre, M. Leclerc, F. Belanger, D. Desilets, S. Rodman, D. Waller and R. Gaudiana, Appl. Phys. Lett., 2009, 95, 063304 CrossRef PubMed.
- S. Wakim, S. Beaupre, N. Blouin, B.-R. Aich, S. Rodman, R. Gaudiana, Y. Tao and M. Leclerc, J. Mater. Chem., 2009, 19, 5351–5358 RSC.
- T. Heumueller, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, J. A. Bartelt, T. M. Burke, T. Ameri, C. J. Brabec and M. D. McGehee, Energy Environ. Sci., 2014, 7, 2974–2980 CAS.
- J. Wang, J. Enevold and L. Edman, Adv. Funct. Mater., 2013, 23, 3220–3225 CrossRef CAS PubMed.
- We note the formation of nano-sized crystal was found to be dependent upon PCDTBT batch.
- J. Jo, S.-S. Kim, S.-I. Na, B.-K. Yu and D.-Y. Kim, Adv. Funct. Mater., 2009, 19, 866–874 CrossRef CAS PubMed.
- M. A. Ruderer, S. Guo, R. Meier, H.-Y. Chiang, V. Körstgens, J. Wiedersich, J. Perlich, S. V. Roth and P. Müller-Buschbaum, Adv. Funct. Mater., 2011, 21, 3382–3391 CrossRef CAS PubMed.
- O. Oklobia and T. S. Shafai, Sol. Energy Mater. Sol. Cells, 2013, 117, 1–8 CrossRef CAS PubMed.
- Z. Wu, T. Song, Y. Jin and B. Sun, Appl. Phys. Lett., 2011, 99, 143306 CrossRef PubMed.
- S. Khiev, L. Derue, G. Ayenew, H. Medlej, R. Brown, L. Rubatat, R. C. Hiorns, G. Wantz and C. Dagron-Lartigau, Polym. Chem., 2013, 4, 4145–4150 RSC.
- A. M. Rao, M. Menon, K.-A. Wang, P. C. Eklund, K. R. Subbaswamy, D. S. Cornett, M. A. Duncan and I. J. Amster, Chem. Phys. Lett., 1994, 224, 106–112 CrossRef CAS.
- J. W. Kingsley, P. P. Marchisio, H. Yi, A. Iraqi, C. J. Kinane, S. Langridge, R. L. Thompson, A. J. Cadby, A. J. Pearson, D. G. Lidzey, R. A. L. Jones and A. J. Parnell, Sci. Rep., 2014, 4, 5286 CAS.
- P. A. Staniec, A. J. Parnell, A. D. F. Dunbar, H. Yi, A. J. Pearson, T. Wang, P. E. Hopkinson, C. Kinane, R. M. Dalgliesh, A. M. Donald, A. J. Ryan, A. Iraqi, R. A. L. Jones and D. G. Lidzey, Adv. Energy Mater., 2011, 1, 499–504 CrossRef CAS PubMed.
- H. C. Wong, A. M. Higgins, A. R. Wildes, J. F. Douglas and J. T. Cabral, Adv. Mater., 2013, 25, 985–991 CrossRef CAS PubMed.
- C. He, D. S. Germack, R. Joseph Kline, D. M. Delongchamp, D. A. Fischer, C. R. Snyder, M. F. Toney, J. G. Kushmerick and L. J. Richter, Sol. Energy Mater. Sol. Cells, 2011, 95, 1375–1381 CrossRef CAS PubMed.
- B. A. Collins, E. Gann, L. Guignard, X. He, C. R. McNeill and H. Ade, J. Phys. Chem. Lett., 2010, 1, 3160–3166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SC and WBC drying kinetics measurements. UV-vis absorption spectra of SC and WBC PCDTBT:PC60BM blend layers. PL spectra of SC and WBC blended layers and their respective PCDTBT neat layers. EQE of blend devices deposited by SC and WBC techniques. Optical microscopy images of the blend layer deposited by SC and WBC on SiOx (as cast) and PEDOT:PSS (at 80 °C and 140 °C thermal annealing temperature). Data supporting this publication can be obtained on request from E-mail: polymer-microfluidics@imperial.ac.uk; . See DOI: 10.1039/c5tc01624j |
| This journal is © The Royal Society of Chemistry 2015 |