
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of (non)linear optical properties in tripodal molecules by variation of the peripheral cyano acceptor moieties and the π-spacer†
D.
Cvejn
a,
E.
Michail
b,
I.
Polyzos
b,
N.
Almonasy
a,
O.
Pytela
a,
M.
Klikar
a,
T.
Mikysek
c,
V.
Giannetas
b,
M.
Fakis
b and
F.
Bureš
*a
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz; Web: http://bures.upce.cz Fax: +420 46 603 7068; Tel: +420 46 603 7099
bDepartment of Physics, University of Patras, GR-26504, Patras, Greece
cDepartment of Analytical Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic
First published on 16th June 2015
Abstract
A series of twelve tripodal push-pull molecules with a central triphenylamine donor and peripheral cyano substituted acceptors has been prepared. These molecules possess systematically altered π-linkers as well as cyano acceptors. Based on the experimental properties measured by differential scanning calorimetry, electrochemistry, one and two photon absorption/emission spectroscopy, supported by the DFT calculations, thorough structure–property relationships were elucidated.
Introduction
Multiphoton absorbing (MPA)1 molecules are currently attracting considerable attention of organic and material chemists as well as physicists. The simultaneous absorption of two photons by a molecule has been theoretically predicted as early as 1931 by M. Göppert-Mayer and experimentally verified after the invention of the laser (1960). The fact that the excited state of a molecule can be populated by absorbing two/three photons of half/third the energy makes the two/three-photon absorption (2/3PA) a very attractive phenomenon.2 The use of a low-energy excitation source (NIR laser) along with the intensity-dependent MPA processes can be considered as two principal advantages that opened immense interest and prospective applications of MPA-active molecules across optoelectronics and photonics.3 Thus, materials exhibiting a large 2PA cross-section have been utilized in the fields ranging from materials science, biology and medicine to 2PA fluorescent microscopy while cellular imaging,4 photodynamic therapy,5 frequency up-converted lasing,6 3D optical data storage,7 3D microfabrication and lithography8 can be considered as the currently most successful applications.A joint feature of the most successful organic molecules is their push-pull and mostly tripodal (branched, star-shaped or octupolar) arrangement. In a push-pull D–π–A molecule (D = electron donor, π = conjugated system and A = electron acceptor), an intramolecular charge transfer (ICT) from the donor via the π-system to the acceptor occurs and the molecule becomes polarized and constitutes a dipole. The D–A interaction results in the formation of a new molecular orbital in which facile electron excitation by visible light takes place. Hence, push-pull molecules are also called chromophores.9 Apart from N,N-dialkylamino groups used as ordinary donors in linear D–π–A systems, triphenylamine (TPA) proved to be a very useful and readily available precursor for the synthesis of tripodal molecules. The central amino donor in TPA can easily be connected to various peripheral acceptors to gain D(–π–A)3 push-pull systems. In fact, D(–π–A)3 systems are much thoroughly investigated than inverse A(–π–D)3 molecules10 which is most likely due to their tedious and limited synthesis compared to possible modifications of the parent TPA (Fig. 1).
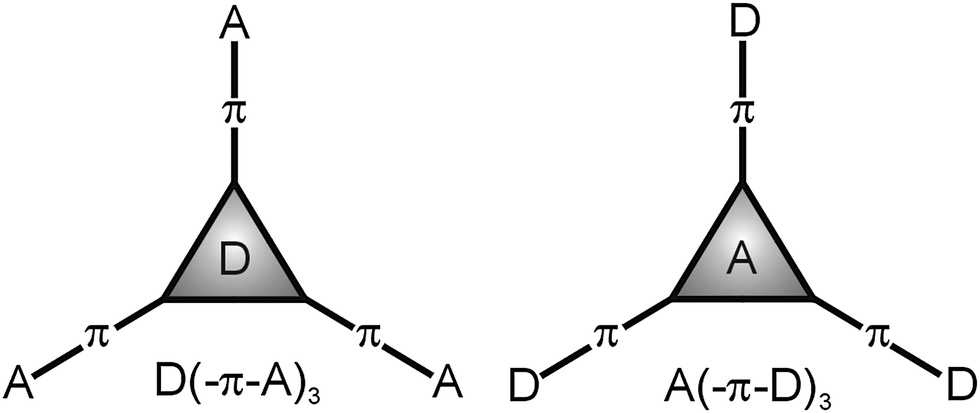 | ||
| Fig. 1 Two principal arrangements of tripodal push-pull molecules. | ||
The vast majority of tripodal molecules have been built on TPA and, since late 90s, an admirable number of articles reporting their preparation and 2PA activity have been published.11 These D(–π–A)3 molecules possess peripheral electron withdrawing groups such as nitro, ester, carbonyl, trifluoromethylsulfonyl etc. as well as (hetero)aromatic moieties such as fluorene, perylene diimide, dimesitylboryl, pyridine/pyridinium, triazole, oxadiazole, benzothiazole, benzothiadiazole and porphyrin. Some dendritic molecules have recently appeared as well.12 Further applications of such TPA-derived push-pull molecules range from 3PA,13 organic solar cells,14 molecular glasses,15 intercalates and metal–organic frameworks (MOF)16 to sensors used for the detection of ions,17 amines18 and explosives.19 In contrast to linear D–π–A systems,9,20 less tripodal compounds bearing peripheral cyano-substituted acceptor moieties were prepared and investigated as 2PA-active substances to date.21 Besides their exceptional 2PA activity, a few cyano-substituted TPA molecules were also utilized in solar cells.22
As only a few comprehensive structure–property relationship studies on tripodal TPA molecules were reported to date23 and as an extension of our recent efforts in the field of property tuning in push-pull molecules,9a we report herein on the design, synthesis and further studies of twelve model D(–π–A)3 molecules 1–12 (Fig. 2). These chromophores possess either one or two cyano groups connected to each particular TPA branch and vary in the arrangement, extension and composition of the π-linker. The impacts of such structural changes on the properties of 1–12 were studied by means of differential scanning calorimetry, electrochemistry, one and two photon absorption spectroscopy and were also supported by DFT calculations.
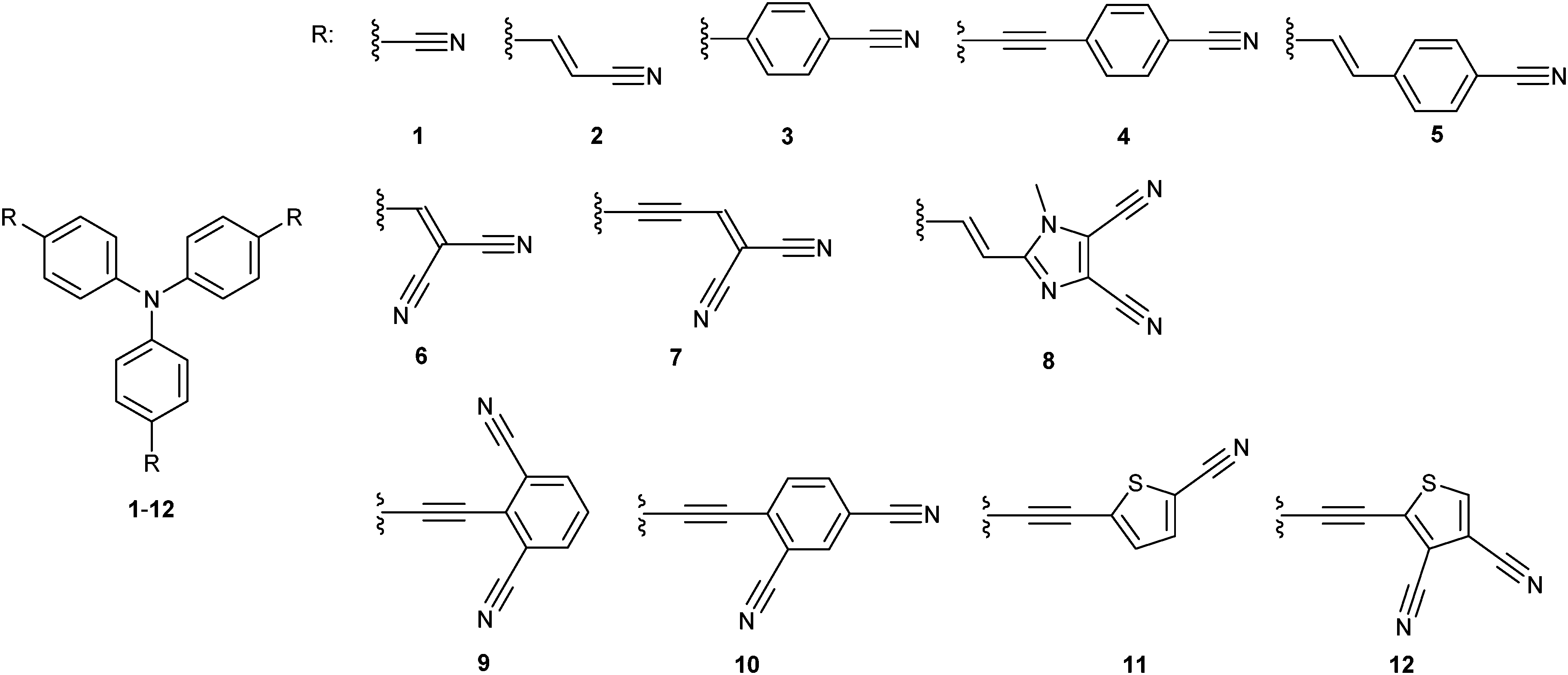 | ||
| Fig. 2 Molecular structure of all target chromophores 1–12. | ||
Results and discussion
Synthesis
In general, the synthesis of chromophores 1–12 involves three reaction steps: synthesis of the properly substituted TPA derivative, preparation of the acceptor moieties and their final combination via a cross-coupling reaction. Schemes 1 and 2 show the preparation of the particular D- and A-parts 13–16 and 23–33, respectively. Threefold Sonogashira cross-coupling of commercially available tris(4-iodophenyl)amine 13 with an excess of trimethylsilylacetylene (TMSA) and subsequent TMS group removal by tetrabutylammonium fluoride (TBAF) afforded smoothly tris(4-ethynylphenyl)amine 14 in 76% overall yield.23c A similar threefold Sonogashira reaction using propargyl alcohol afforded triole 15 which upon oxidation with the Dess–Martin reagent provided trialdehyde 16. The synthesis of 16 can also be accomplished via a Sonogashira reaction of 13 with 3,3-diethoxypropyn affording triacetal 34 (90%). Subsequent removal of the acetal protecting group afforded 16 in 61% yield. However, the overall yield is generally lower than that attained via the reaction path shown in Scheme 1 (see the ESI† for more details). The synthesis of di- and tricyanosubstituted benzenes 23–25 (Scheme 2) has been accomplished in a modular manner starting from di- and trimethylbromobenzenes which were oxidized to corresponding acids 17–19 using KMnO4 in tBuOH and subsequently treated with thionyl chloride and saturated with gaseous NH3 to afford amides 20–22. This conversion for 19 was accompanied by a halogen replacement and, therefore, chlorotriamide 22 was isolated instead of the expected bromotriamide. Final dehydration to nitriles 23–25 was carried out by trifluoroacetic anhydride (TFAA) in dioxane/pyridine. Other precursors of the electron withdrawing moieties in 1–12 are also shown in Scheme 2. They are either commercially available (26–28, 30 and 32) or can easily be synthesized via procedures reported in the literature (29 and 31).24,25 2-Iodothiophene-3,4-dicarbonitrile 33 was synthesized via a selective monoiodination of thiophene-3,4-dicarbonitrile which was achieved by lithiation at −78 °C and a subsequent reaction with molecular iodine.26 | ||
| Scheme 1 Modification of the TPA core leading to precursors 13–16. | ||
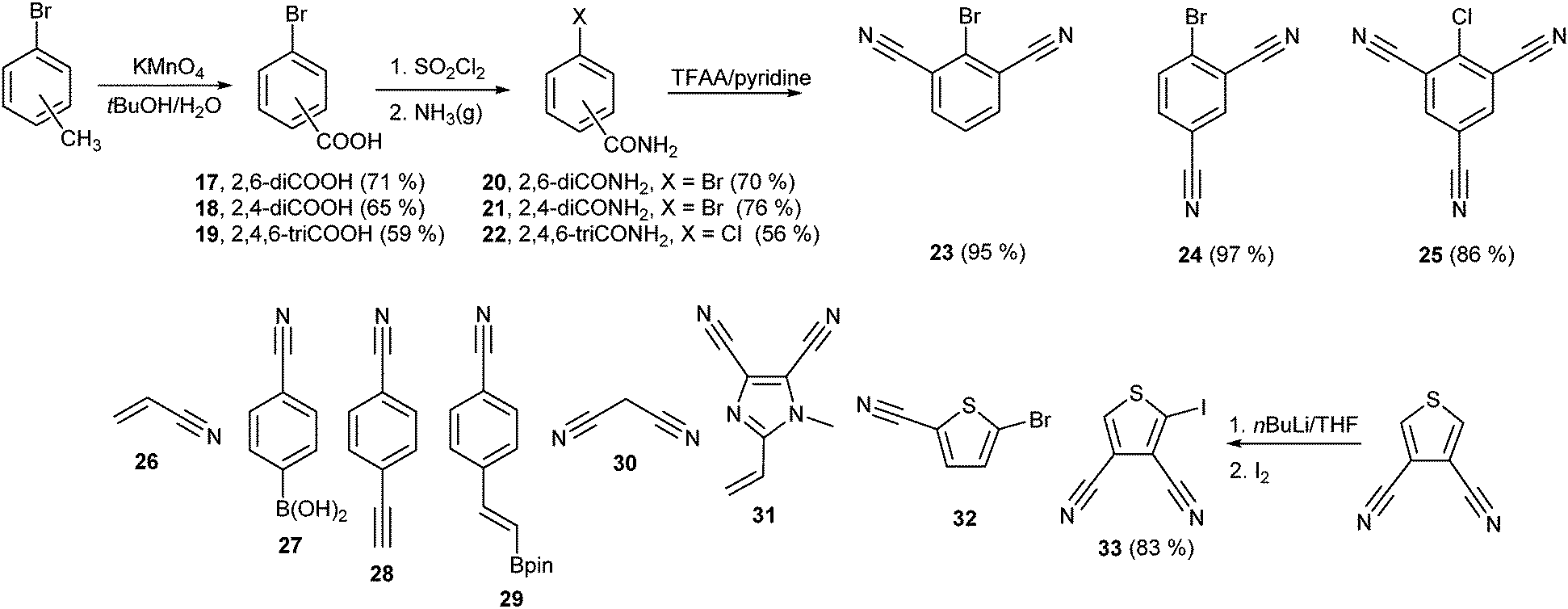 | ||
| Scheme 2 Structure and selected synthesis of cyano-substituted acceptor moieties 23–33. | ||
The synthetic details and spectral characterization of precursors and intermediates 14–25 are given in the ESI.† Target tripodal push-pull molecules 1–12 were obtained by a combination of the aforementioned A- and D-precursors, mainly in terms of cross-coupling reactions as shown in Scheme 3. Threefold cyanation27 of triiodotriphenylamine proved to be tedious and, therefore, chromophore 1 was synthesized from trialdehyde 35 (ref. 4g) by a one-pot reaction with hydroxylamine and subsequent Beckmann rearrangement.28 Chromophore 2 was prepared by Heck–Mizoroki cross-coupling reaction of 13 and acrylonitrile 26 with a similar outcome as described by Tian et al.29 The biphenyl π-linker in chromophore 3 has been generated by the Suzuki–Miyaura reaction between 13 and boronic acid 27 (76%).
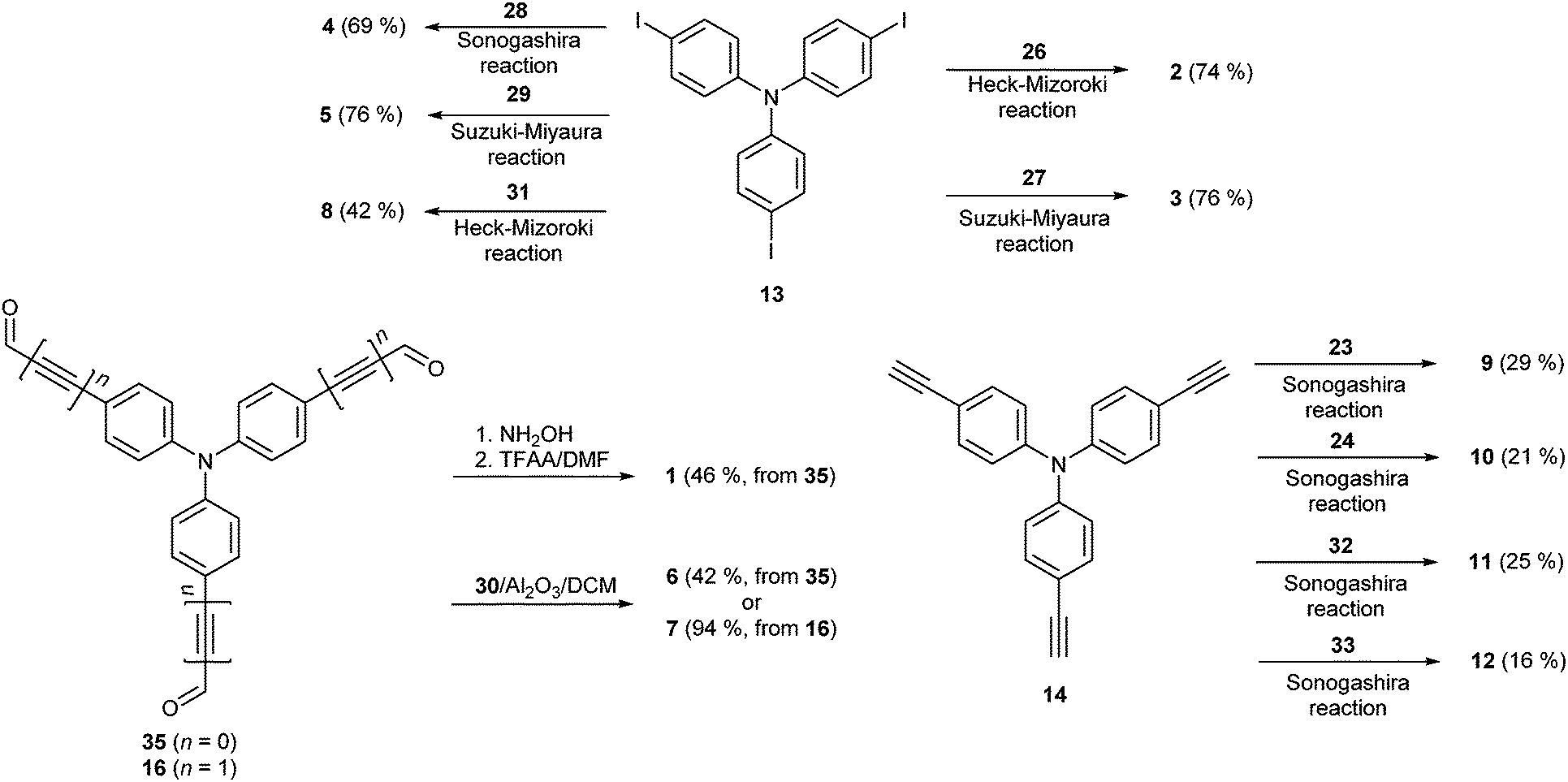 | ||
| Scheme 3 Combination of A- and D-parts leading to tripodal push-pull molecules 1–12. | ||
A Sonogashira reaction of 13 and terminal acetylene 28 provided chromophore 4 in generally lower yield than reported by Ogilby et al. (69 vs. 94%).5d The synthesis of chromophore 5 has been described by Cho et al. via a Horner–Wadsworth–Emmons reaction.21b However, the Suzuki–Miyaura cross-coupling on 13 using boronic acid pinacol ester 29 delivered 5 in a slightly improved yield of 76%. Al2O3-catalyzed Knoevenagel condensation30 of aldehydes 35/16 and malononitrile 30 afforded smoothly target chromophores 6 and 7 with dicyanovinyl (DCV) moieties in the yields of 42 and 94%. Chromophore 6 has also been previously synthesized by the pyridine/acetic acid-catalyzed condensation in a similar yield.11q,31 Target chromophore 8 bearing peripheral dicyanoimidazole (DCI) acceptor moieties was prepared by Heck–Mizoroki olefination of 13 and vinazene 31. Although carried out under optimized conditions (dioxane, elevated temperature and Pd(0) precatalyst), a Sonogashira reaction of triacetylene 14 and halogenated dinitriles 23 and 24 afforded chromophores 9 and 10 only in modest yields. This reaction was complicated by the predominant formation of one- and two-fold cross-coupling products which were tedious to separate and to suppress even in the case of an excess of 23 and 24. Moreover, a similar Sonogashira reaction with trinitrile 25 proved to be unsuccessful at all, which is most likely caused by the presence of chloro instead of bromo substituents (see above). Cross-coupling reactions with thiophene analogues 32 and 33 afforded chromophores 11 and 12 in similar modest yields of 25 and 16%, respectively.
Thermal properties
Thermal behaviour of compounds 1–12 was studied by differential scanning calorimetry (DSC). Fig. 3 shows thermograms of representative compounds 2 and 9 while Table 1 lists the measured melting temperatures (Tm) and temperatures of thermal decompositions (Td).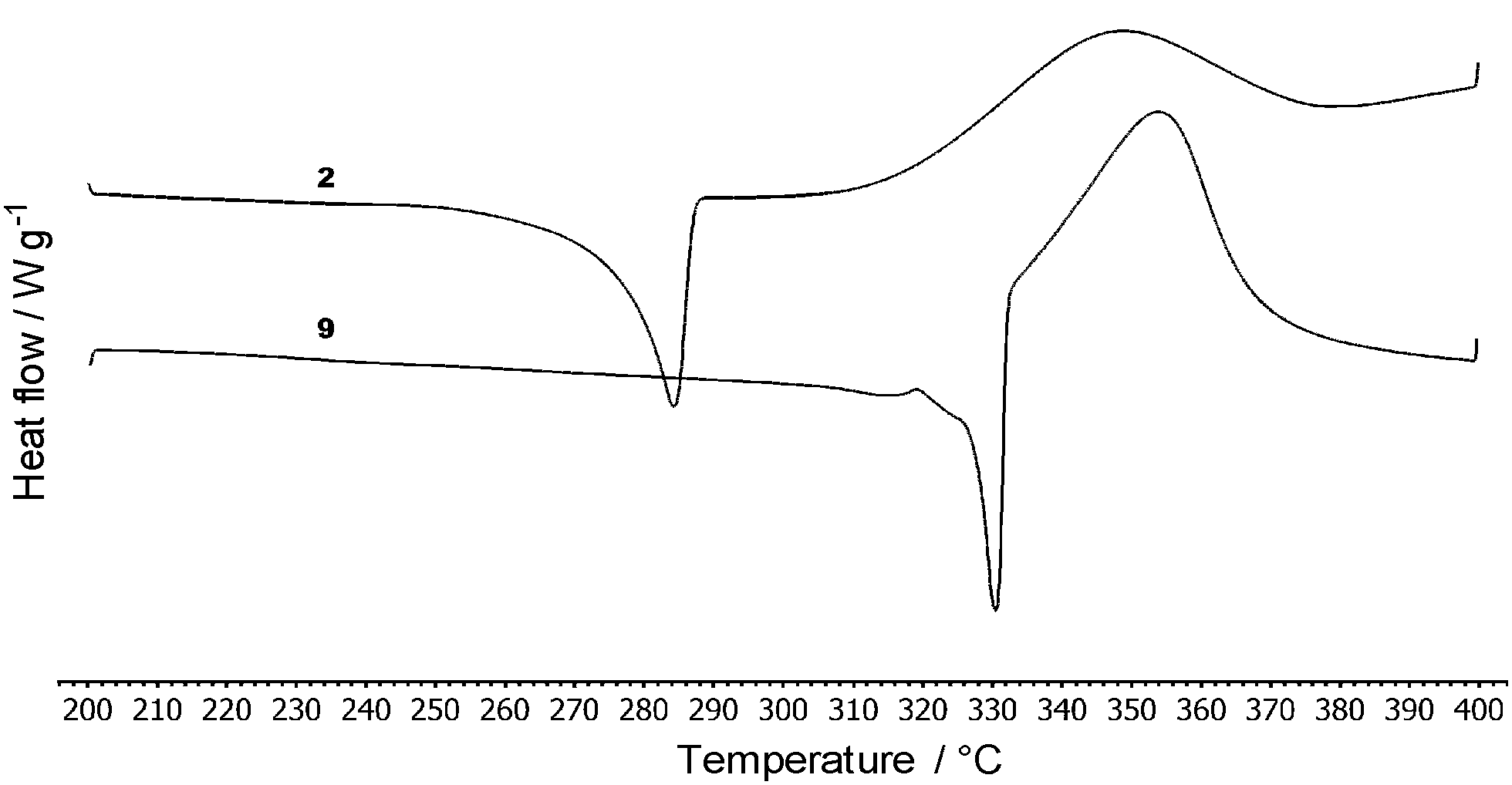 | ||
| Fig. 3 Representative DSC curves of compounds 2 and 9 determined with a scanning rate of 3 °C min−1 under N2. | ||
| Comp. | T m (°C) | T d (°C) | E 1/2(ox1) (V) | E 1/2(red1) (V) | ΔEc (V) | E HOMO (eV) | E LUMO (eV) | λ Amax (nm eV−1) | λ Fmax (nm eV−1) | Φ F (%) | Stokes shift (cm−1) | δ 2PA (GM) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a T m = melting point (the point of intersection of a baseline before the thermal effect with a tangent). b T d = thermal decomposition (pyrolysis in a N2 atmosphere). c E 1/2(ox1) and E1/2(red1) are half-wave potentials of the first oxidation and reduction, respectively; all potentials are given vs. SCE; ΔE = E1/2(ox1) − E1/2(red1). d Electrode surface inhibition. e −EHOMO/LUMO = E1/2(ox1/red1) + 4.429 (ref. 32). f Measured in THF. g The wavelength of the maximum 2PA cross section is shown in parenthesis (measured in THF). | ||||||||||||
| 1 | 343 | — | 1.49 | −2.01 | 3.50 | −5.92 | −2.42 | 339/3.66 | 377/3.29 | 0.62 | 2970 | — |
| 2 | 275 | 317 | 1.12 | −1.74 | 2.86 | −5.55 | −2.69 | 394/3.15 | 460/2.70 | 0.48 | 3640 | 260 (770) |
| 3 | 329 | 340 | 1.02 | −2.02 | 3.04 | −5.45 | −2.41 | 372/3.33 | 448/2.77 | 0.64 | 4560 | 566 (760) |
| 4 | 217 | — | 1.04 | −1.85 | 2.89 | −5.47 | −2.58 | 387/3.20 | 458/2.71 | 0.59 | 4010 | 785 (780) |
| 5 | 291 | 477 | 0.85 | −1.80 | 2.65 | −5.28 | −2.63 | 403/3.08 | 510/2.43 | 0.51 | 5210 | 1100 (810) |
| 6 | 145 | 322 | 1.41 | —d | — | −5.84 | — | 453/2.74 | 549/2.26 | 0.29 | 3860 | 757 (830) |
| 7 | 84 | 176 | 1.29 | —d | — | −5.72 | — | 470/2.64 | 611/2.03 | 0.29 | 4910 | 667 (810) |
| 8 | 188 | 366 | 0.97 | −1.66 | 2.63 | −5.40 | −2.77 | 413/3.00 | 497/2.49 | 0.59 | 4090 | 620 (810) |
| 9 | 327 | 333 | 1.16 | −1.55 | 2.71 | −5.59 | −2.88 | 420/2.95 | 533/2.33 | 0.69 | 5050 | 852 (810) |
| 10 | — | 282 | 1.15 | −1.44 | 2.59 | −5.58 | −2.99 | 414/3.00 | 499/2.48 | 0.55 | 4126 | 631 (810) |
| 11 | 191 | 240 | 1.09 | −1.45 | 2.54 | −5.52 | −2.98 | 397/3.12 | 475/2.61 | 0.46 | 4140 | 148 (750) |
| 12 | 198 | 252 | 1.17 | −1.41 | 2.58 | −5.60 | −3.02 | 412/3.01 | 516/2.40 | 0.49 | 4890 | 375 (830) |
The measured melting points of 1–12 range from 84 to 343 °C. The temperature of decomposition was estimated within the range of 176–477 °C. Compound 1 provided a very sharp peak of melting/crystallization at 343/263 °C and does not undergo decomposition until its boiling point (436 °C). Thermal behaviour of compounds 2, 3, 5–9 and 11–12 is very similar with the melting point peak followed by a subsequent decomposition at a higher temperature. Compound 4 showed a wide enantiotropic solid–solid transition (37–82 °C), melting point at 217 °C and surprisingly no decomposition peak. However, the sample was clearly carbonized after heating up to 500 °C. For compound 10, thermal decomposition preceded the melting point. As can be seen from the whole series (Table 1), DCV-terminated tripodal compounds 6 and 7 showed the lowest melting points and temperatures of thermal decompositions. TPA derivatives substituted with cyanoaromates 3–5 and 9–10 resisted thermal decomposition above 300 °C. Heterocyclic acceptor moieties such as DCI and (di)cyanothiophene as in 8 and 11–12 bring also noticeable thermal stability up to 366 °C, especially for the imidazole-terminated compound 8. The measured DSC data of compounds 1–12 reflect their different molecular structure (varied peripheral acceptors and π-linkers). The following relations can be deduced from the DSC measurements:
• The melting temperatures within the most evaluated subseries of compounds 1–5 are mostly affected by an insertion of multiple bonds. In general, chromophores bearing triple bond proved less stable than the corresponding olefinic derivatives (e.g.4vs.3 and 5vs.3).
• This can be further demonstrated by the DCV acceptor linked to the TPA core via an acetylenic spacer in 7, which lowered the thermal stability very significantly when compared to 6 (ΔTm = 61 °C, ΔTd = 146 °C).
• Chromophores bearing benzene-derived acceptors (e.g.4 or 9/10) are generally more thermally stable than the corresponding thiophene derivatives (e.g.11 and 12).
• Chromophore 8 with the imidazole-based acceptor (DCI) showed the highest thermal resistance among all heterocyclic compounds (8, 11–12) with Td = 366 °C.
Electrochemistry
Electrochemical measurements of chromophores 1–12 were carried out in acetonitrile containing 0.1 M Bu4NPF6 in a three electrode cell by cyclic voltammetry (CV), rotating disk voltammetry (RDV) and polarography. The working electrode was a platinum disk (2 mm in diameter) for CV and RDV experiments. A saturated calomel electrode (SCE) separated by a bridge filled with a supporting electrolyte and a Pt wire were used as reference and auxiliary electrodes. The acquired data are summarized in Table 1, while representative CV diagrams of compounds 5, 7–9 and 11 are given in the ESI.†The measured half-wave potentials of the first oxidation and reduction (E1/2(ox1) and E1/2(red1)) of chromophores 1–12 were found within the range of 0.85 to 1.49 and −1.41 to −2.02 V, respectively. The first reduction and oxidation are one-electron processes, followed by subsequent oxidations and reductions, and are obviously a function of the peripheral acceptor and the π-linker (Table 1). Whereas the first oxidation can be attributed to the central TPA donor, the first reduction is mostly localized on the peripheral CN groups and the adjacent π-system. The first oxidation potentials of chromophores 1–5 bearing monocyano-substituted acceptor moieties steadily decreased from 1.49 to 0.85 V as a function of the π-system elongation. The first reduction potentials showed similar trends with exception for 3 in which nonplanar biphenyl π-linker allows only reduced ICT. The difference between the first oxidation and reduction potentials (electrochemical gap, ΔE) represents a straightforward way for the evaluation of the ICT. When going from 1 (ΔE = 3.50 V) to 2 (ΔE = 2.86 V), the electrochemical gap decreased by 0.64 V, as a consequence of the insertion of an additional olefinic subunit. However, further π-system extension as in 3 resulted in an increased gap of 3.04 V. This demonstrates “ICT transparency” of the olefinic unit in comparison to the aromatic 1,4-phenyl moiety.33 Further extension and planarization of the π-linker by acetylenic and olefinic subunits (3vs.4 and 5) lowered the electrochemical gap up to 2.65 V. Dicyano-substituted (hetero)aromatic moieties attached to the TPA core affected both the first oxidation and the reduction potentials (8–12, Table 1). The evaluation of the ICT based on the overall electrochemical gap ΔE implies that these (hetero)aromates impart a stronger resonance effect than aromates (ΔE of 2.71–2.54 vs. 3.50–2.65 V for 1–5vs.8–12). Positioning of the CN groups along the (hetero)aromatic scaffold plays also a significant role as can be seen for instance on chromophores 9 and 10. The latter, 2,4-dicyano-substituted chromophore showed a reduced gap by 0.12 V. The lowest electrochemical gap (ΔE = 2.58 V) was measured for chromophore 11 bearing a cyano-acceptor based on polarizable thiophene with the cyano group attached at the resonant and most extended C5 position (as compared to the dicyano acceptor in 12). A comparison of chromophores 4 and 11 also allows comparison of 1,4-phenylene and 2,5-thienylene π-systems.34 With very similar oxidation potentials (1.04 and 1.09 V), the latter showed significantly lowered first reduction potential (−1.45 vs. −1.85 V) as well as the electrochemical gap (ΔE = 2.89 vs. 2.54 V).
One photon absorption and emission
Target chromophores are coloured molecules, their colours range from yellowish to red (see the ESI†). Optical properties of 1–12 were investigated by electronic absorption and emission spectra; the longest-wavelength absorption/emission maxima λAmax/λFmax, the fluorescence quantum yields ΦF and the Stokes shifts are summarized in Table 1. Fig. 4 shows absorption and emission spectra of representative chromophores 5, 7–9 and 11.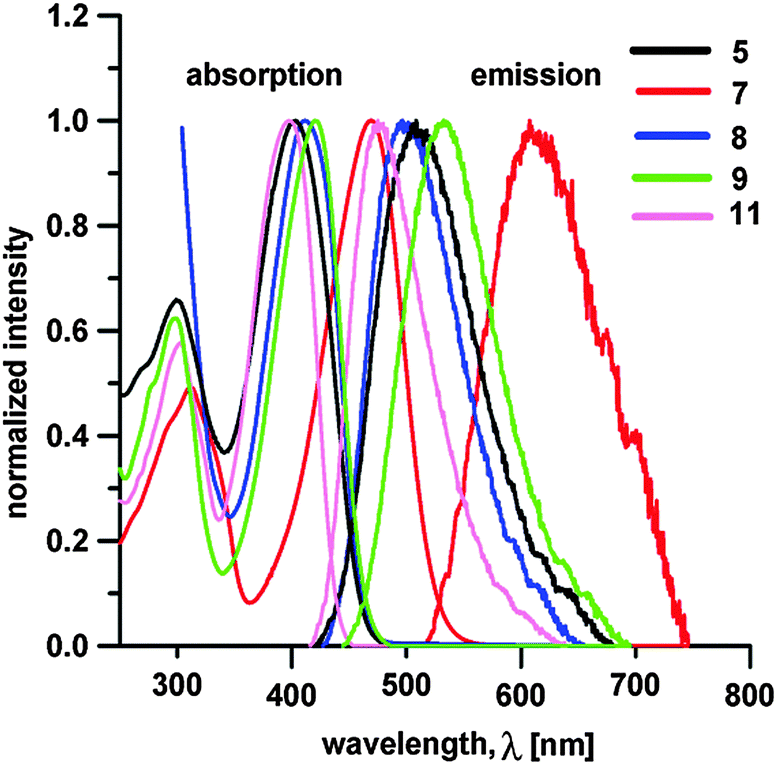 | ||
| Fig. 4 Representative absorption (left) and fluorescence (right) spectra of chromophores 5, 7–9 and 11 measured in THF. | ||
Whereas the absorption maxima range from 339 to 470 nm, the emission maxima are red-shifted to 377–611 nm. The absorption spectra are dominated by one intense CT-band whose position is clearly dependent on the nature of the appended cyano-acceptors and the π-system. When considering the monocyano-substituted series of molecules 1–5, the λAmax values showed a very similar trend as seen for the electrochemical gaps ΔE. Namely, extension of the π-system by olefinic (1vs.2) or aromatic units (3vs. 2), planarization (4/5vs.3) and further elongation by acetylenic subunit (4) affected the absorption maxima within the range of 339 to 403 nm. It should be noted that the longest-wavelength absorption maxima of 1–12 correlate very tightly with the measured electrochemical gap ΔE (see the ESI†). Chromophores 6 and 7 bearing strong electron DCV acceptors showed the most bathochromically shifted CT-bands with λAmax at 453 and 470 nm, respectively. In contrast to 1–5, chromophores 8–10 and 12 exhibited red-shifted absorption maxima up to 412–420 as a result of the appended dicyano-substituted (hetero)aromatic acceptor moieties. However, the differences in λAmax are just within the range of 8 nm, which implies that, in contrast to electrochemical gap, the central (hetero)aromatic π-system and positioning of the CN groups do not affect the absorption properties very significantly.
The fluorescence spectra exhibited one single band with λFmax appearing at 377–611 nm. In general, its position is affected by the same structural features as the aforementioned absorption bands. Both positions of the longest-wavelength absorption and fluorescence maxima showed tight correlations (see the ESI†). The quantum yields lie within the range of 0.29 to 0.69. The lowest quantum yields were measured for chromophores 6 and 7 with strongly electron withdrawing DCV moieties. The weaker (di)cyano acceptors in 1–5 and 8–12 lead generally to blue-shifted emission with a high quantum yield. This is in accordance with our previous observations on linear and X-shaped push-pull molecules with strong or weak ICT.20g,h,33a
Two photon absorption
The 2PA properties of tripodal chromophores 1–12 were studied by means of two-photon excited fluorescence spectroscopy within the spectral range from 750 to 850 nm. The advantage of this method is that low concentrations are used and that the order of the non-linear effect is monitored by detecting the intensity of the emitted fluorescence as a function of excitation power. In the case of two-photon excitation, the emitted fluorescence is dependent on the square of the excitation laser power. This was checked in all experiments to confirm the 2PA process. The excitation power was in all cases kept below 20 mW in order to minimize thermal effects and in most cases the 2PA cross-sections were measured within the range of 10–20 mW. The samples were 10−4 M solutions of the dyes in THF. The contribution of the solvent was taken into account by subtracting the intensity of scattered light, obtained by a cuvette containing the solvent only, from the measurements. The two-photon absorption cross-sections were determined by using Rhodamine B in methanol (10−4 M concentration), as reference.The 2PA spectra of all compounds, i.e. the 2PA cross-sections (δ2PA) as a function of excitation wavelength, are shown in Fig. 5. For a better comparison of the 2PA properties, target chromophores 1–12 were split into four groups according to the peripheral acceptor moieties. Whereas the first group includes chromophores 2–5 with monocyano acceptors, chromophores 6 and 7 in the second group possess DCV acceptors. The third and fourth group of molecules consist of dicyano aromatic (9 and 10) and (di)cyano heteroaromatic acceptor moieties (8, 11–12), respectively. In all cases, good 2PA properties were observed with the 2PA cross-section exceeding 1000 GM for compound 5. In general, chromophores 4 and 5 with monocyano acceptors displayed a larger 2PA cross section than the other tripodal molecules in the whole series. Moreover, these two chromophores also exhibited good fluorescence quantum yields (>0.50). The 2PA results are summarized in Table 1.
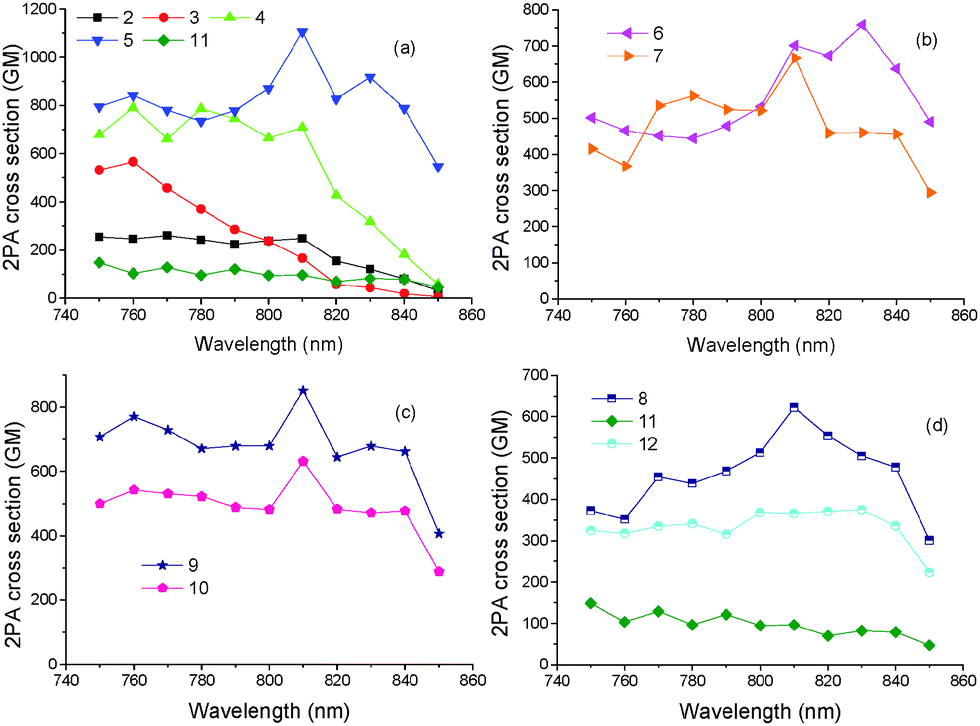 | ||
| Fig. 5 Two-photon absorption cross-sections, δ2PA, of (a) 2, 3, 4, 5 and 11 (b) 6 and 7 (c) 9 and 10 and (d) 8, 11, 12. All compounds have been measured in THF solutions at 10−4 M concentration. | ||
Fig. 5a compares the 2PA spectra of mono-cyano terminated chromophores 2–5 and 11. Compound 1 showed negligible 2PA cross-section within the investigated spectral area. It was observed that by increasing the length of the π-linker, i.e. passing from 2 to 4 and 5, the 2PA cross sections increased. In addition, it is also obvious that upon changing the acetylenic π-conjugated bridge by an olefinic one (4vs.5), the 2PA cross-sections significantly increased up to 1100 GM (at 810 nm) for 5 while this is 785 GM (at 780 nm) for 4. Since the fluorescence quantum yields of 4 and 5 are similar (Table 1), the 2PA action cross-section product Φ × δTPA is again higher for 5 than for 4. This renders molecule 5 as a promising candidate for up-converted lasing or two-photon imaging. A replacement of the 1,4-phenylene moiety in 4 with 2,5-thienylene one (11) led to more than seven-times decrease of the 2PA cross sections (Table 1, 1100 vs. 148 GM). Hence, heteroaromatic polarizable moiety based on thiophene in 11 did not bring the expected higher 2PA activity. Another conclusion drawn from Fig. 5a is that the peaks of the 2PA spectra appear at approximately twice the wavelengths of the one photon absorption spectra. This is in accordance with the two-photon allowed states for octupolar molecules. Specifically, the 2PA spectrum of 4 is blue-shifted compared to 5 while that of 3 is blue shifted compared to 2 as in the case of their one-photon absorption spectra.
The 2PA spectra of chromophores 6 and 7 bearing the strongest DCV acceptor are shown in Fig. 5b. Chromophore 6 exhibited a large 2PA cross-section of 760 GM at 830 nm. Interestingly, an extension of the π-conjugated path by an additional acetylene subunit as in 7 did not lead to enhanced 2PA cross-sections. The fluorescence quantum yields of 6 and 7 are relatively low making the Φ × δ2PA values significantly smaller than those of compounds 4 and 5. However, it should be noted that, according to their one-photon absorption spectra, the maximum of the 2PA spectra of 6 and 7 is expected at wavelengths close to 900 nm, which are not covered by our experiment. On the other hand, chromophore 6 with a DCV acceptor exhibited much larger 2PA cross-section than structurally related 2, which possesses only one cyano group.
Fig. 5c presents the 2PA cross-sections of 9 and 10 bearing dicyano aromatic acceptors with different orientation of the appended CN groups. The position of the cyano groups played significant role as 9 exhibited an approximately 1.35-times larger 2PA cross-sections compared to 10. Moreover, the fluorescence quantum yield is higher for 9 (0.69) compared to 10 (0.55), which provides 9 a larger Φ × δ2PA value. A comparison of the structurally related chromophores 10 and 4, with di- and monocyano aromatic acceptor moieties, reveals that the second cyano group has broadened and red-shifted the 2PA spectra. This is in accordance with one photon absorption spectra, but, on the other hand, it also causes a decrease of the 2PA response.
Finally, the 2PA cross-sections of chromophores 8, 11 and 12 with (di)cyano-heteroaromatic acceptors are compared in Fig. 5d. Chromophores 11 and 12 with thiophene acceptors exhibited relatively small 2PA cross-section values compared to their analogues 4 and 10 having a phenyl ring. Thus, a replacement of 1,4-phenylene by 2,5-thienylene moieties coupled either to cyano or dicyano groups is not a useful strategy towards increasing the 2PA activity in TPA-derived tripodal chromophores. This is in contrast to general trends observed for second order NLO chromophores.34 However, an addition of the second cyano group into 11 leads to 12 with more than three-times higher 2PA cross-section without affecting the florescence quantum yield. This makes the thiophene substituted chromophores 11 and 12 extremely sensitive to the number of cyano groups. On the other hand, 2PA cross-sections measured for the analogous chromophores 4 and 10 with 1,4-phenylene parent moieties showed less sensitivity. Chromophore 8 bearing three peripheral DCI acceptor units exhibited a relative large 2PA cross-section of 620 GM at 810 nm accompanied by a good quantum yield of 0.59. Although, a straightforward comparison of the 2PA properties of the herein studied compounds with literature data is not easy because of various experimental approaches used (sample concentration, solvent used, pulse duration, laser power, spectral range etc.), chromophores 4 and 5 are found to exhibit similar or better 2PA activity compared with other tri-branched compounds with D(–π–A)3 arrangement and similar molecular weight.11b,h
Quantum chemical calculations
The spatial and electronic properties of the parent TPA and chromophores 1–12 were investigated using the Gaussian W09 package35 at the DFT level. The initial geometries of molecules 1–12 were estimated using the PM3 method implemented in ArgusLab36 and these were subsequently optimized using the DFT B3LYP/6-311++G(2d,p) method. The energies of the HOMO and LUMO, their differences and ground state dipole moments were calculated with the DFT B3LYP/6-311++G(2d,p) and are summarized in Table 2.| Comp. | Symmetry group | E HOMO (eV) | E LUMO (eV) | ΔE (eV) | μ (D) |
|---|---|---|---|---|---|
| a Calculated at the DFT B3LYP/6-311++G(2d,p) level. | |||||
| TPA | D 3 | −5.29 | −0.90 | 4.39 | 0.024 |
| 1 | D 3 | −6.53 | −2.53 | 4.00 | 0.000 |
| 2 | C 3 | −6.14 | −2.90 | 3.24 | 0.227 |
| 3 | D 3 | −5.86 | −2.39 | 3.47 | 0.002 |
| 4 | D 3 | −5.80 | −2.67 | 3.13 | 0.001 |
| 5 | C 3 | −5.62 | −2.75 | 2.87 | 0.221 |
| 6 | C 3 | −6.76 | −3.80 | 2.94 | 5.675 |
| 7 | C 3 | −6.49 | −3.81 | 2.68 | 7.199 |
| 8 | C 3 | −6.30 | −3.01 | 3.29 | 7.603 |
| 9 | D 3 | −5.78 | −2.84 | 2.94 | 0.035 |
| 10 | C 3 | −6.05 | −3.17 | 2.88 | 6.034 |
| 11 | C 3 | −5.81 | −2.82 | 2.99 | 5.199 |
| 12 | C 3 | −6.07 | −3.13 | 2.94 | 11.914 |
The calculated energies of the HOMO/LUMO of 1–12 range from −6.76/−3.81 to −5.62/−2.39 eV and are obviously a function of the structure of the particular chromophore. Both energies correlate tightly with the experimental data obtained by the electrochemical measurements (Table 1). Hence, the used DFT method can be considered as a reasonable tool for the description of electronic properties of 1–12 (see the ESI†). When going from parent TPA to molecule 1, the HOMO–LUMO gap decreased from 4.39 to 4.00 eV as a result of attaching three cyano groups and generating a push-pull system. Further extension of the π-system within the series 1–5 affected the HOMO and LUMO levels as well as the gap in very similar way as observed by the electrochemistry. The lowest HOMO–LUMO gap within this subseries has been calculated for 5 bearing a cyano-substituted styryl linker. Further attachment of an additional CN group (4vs.9 and 10) resulted in drop of the calculated gap from 3.13 to 2.94 and 2.88 eV. DCV acceptors in chromophores 6–7 impart also strong ICT and reduce the gaps up to 2.94 and 2.68 eV, respectively. A replacement of the terminal 1,4-phenylene moiety in 4 by 2,5-thienylene one in 11 slightly decreases the HOMO–LUMO gap from 3.13 to 2.99 eV in a similar way as observed by CV measurements. In general, both experimental and calculated HOMO–LUMO gaps correlate tightly as shown in the ESI.†
The calculated ground state dipole moments are almost zero for molecules having the D3 group of symmetry, whereas noticeable values were obtained for molecules 6–8 and 10–12 having branched and unsymmetrical acceptor moieties (DCV, DCI, 2,4-dicyanobenzene and 5(2,4)-(di)cyanothiophene).
The HOMO and LUMO localizations in representative chromophores 5, 7–9 and 11 are shown in Fig. 6. For complete listing see the ESI.† As expected, the HOMO is localized on the central amino donor and adjacent alternating positions of the TPA, whereas the LUMO is spread over the peripheral cyano acceptor and the adjacent π-linker. Fig. 6 shows a clear charge separation and thus confirms the ICT character of chromophores 1–12. As can be seen, the LUMO is predominantly localized over one or two particular branche(s), while the third branch is occupied by the LUMO+1. The HOMO as well the HOMO−1 remained on the central amino donor. This is a common feature of tripodal molecules based on TPA.23b,c,29
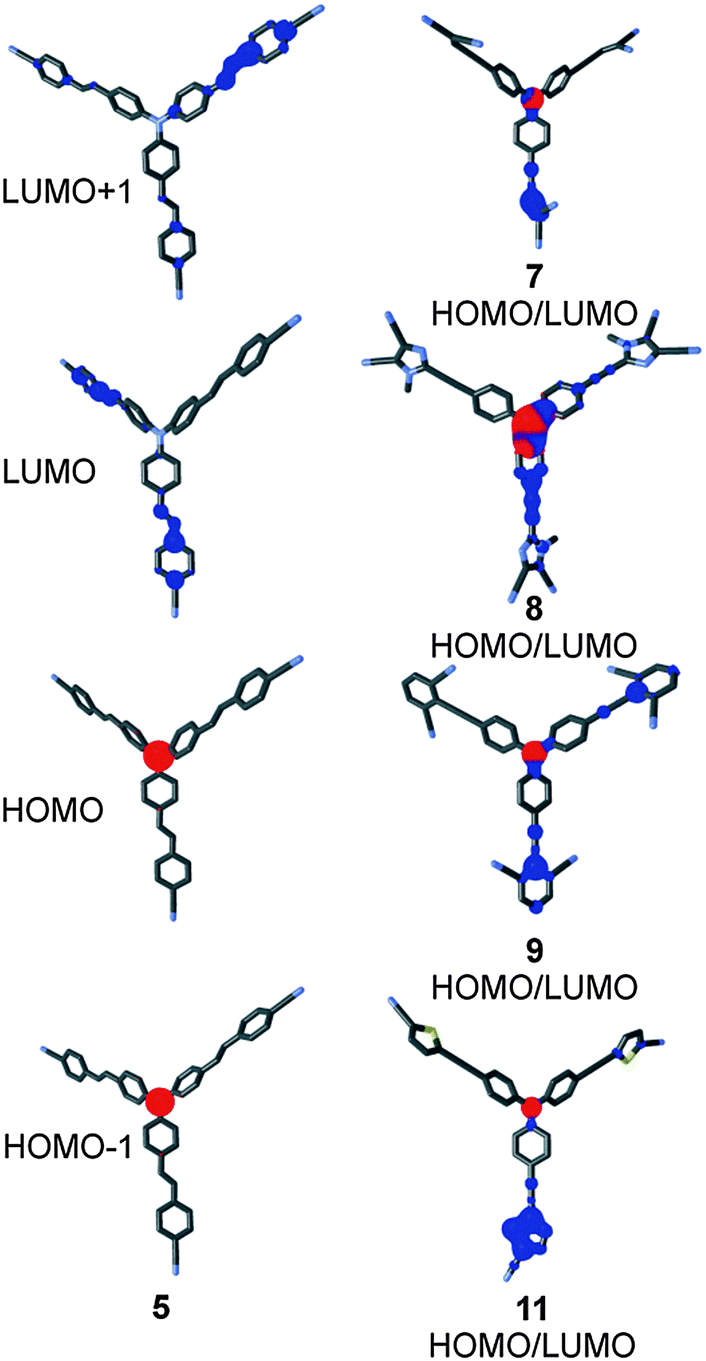 | ||
| Fig. 6 HOMO/HOMO−1 (red) and LUMO/LUMO+1 (blue) localizations in 5 and HOMO/LUMO mix in 7–9 and 11. | ||
Conclusions
A series of twelve tripodal D(–π–A)3 molecules based on triphenylamine has been designed and synthesized. The investigated push-pull systems possess one central amino donor connected via a π-linker to three peripheral cyano acceptors. Fine tuning of the properties has been achieved either by modulation of the acceptors as well as by the systematic elongation of the π-system. The synthesis has been carried out in a modular manner by connecting donor and acceptor parts via cross-coupling reactions. Target chromophores were further studied by differential scanning calorimetry, electrochemistry, one and two photon absorption and emission spectra and also quantum chemical calculations. Thorough structure–property relationships were subsequently evaluated.Compounds 1–12 showed melting point and subsequent thermal decomposition within the range of 84–343 and 176–477 °C. These values indicate relatively high thermal resistance of TPA derived tripodal molecules.
The electrochemical properties (HOMO and LUMO levels and gap) were mainly affected by the type of peripheral acceptor, length and composition of the π-linker as well as its planarity. Using these structural changes as a tool, the electrochemical HOMO–LUMO gap could be finely tuned by almost 1 V (3.50 to 2.54 V).
One photon absorption and emission properties studied by electronic absorption/emission spectra revealed significant CT-bands appearing between 339/377 and 470/611 nm. Hence, optical tuning could be carried out within the range of 130/230 nm. Quantum yields ranging from 0.29–0.69 were measured for 1–12.
The two photon absorption properties of 1–12 were measured by two-photon excited fluorescence spectroscopy within the spectral range 750 to 850 nm. A critical assessment has been carried out within/across the particular series of chromophores in order to evaluate structure–property relationships. Extension of the π-system and replacement of acetylenic by more polarizable olefinic units led to a significant increase of 2PA activity. Hence, chromophore 5 bearing a cyano substituted styryl linker showed the most enhanced NLO activity within the whole series. An attachment of aromatic acceptors bearing two CN groups (9 and 10) as well as strongly withdrawing DCV units in 6 and 7 did not lead to increased δTPA coefficients compared to 5. A similar conclusion can be made for heterocyclic cyano acceptors such as DCI and 5(2,4)-(di)cyanothiophene.
HOMO and LUMO levels calculated using the DFT and their differences correlate tightly with the linear properties measured by electrochemistry and absorption/emission spectra. Considering all the aforementioned structural features of 1–12, we believe that this work could be treated as a useful guide in designing/tuning tripodal push-pull molecules with a central donor and peripheral acceptors.
Experimental
Materials and methods
For detailed description of used materials and methods used in this work see the ESI.† Synthesis and full spectral characterization of target chromophores 1–5 is given in the ESI.† Two general methods were used for the synthesis of chromophores 6–7 and 9–12.General method for Knoevenagel condensation (chromophores 6 and 7)
Triphenylamine-4,4′,4′′-triscarboxaldehyde 35 (165 mg, 0.5 mmol) or aldehyde 16 (201 mg, 0.5 mmol) and malononitrile 30 (150 mg, 2.27 mmol) were dissolved in DCM (20 ml) and alumina (930 mg, 8.9 mmol, Brockmann activity II–III) was added. The reaction mixture was stirred for 4 h and monitored by TLC (SiO2, DCM/EtOAc 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Anhydrous Na2SO4 (350 mg, 2.46 mmol) can eventually be added to accelerate the reaction. The resulting coloured suspension was filtered and the filtrate was concentrated in vacuo. The crude products were purified by column chromatography (SiO2, DCM/EtOAc 10:1 or DCM) to afford 6 or 7.
:1) Mp 145 °C. IR (HATR) νmax/cm−1 3087, 2220 (CN), 1559, 1496, 1269, 1186, 827. 1H-NMR: δH (400 MHz; CD2Cl2) 7.25 (6H, d, 3J 8.8, CHAr), 7.73 (3H, s, CH
:1). Anhydrous Na2SO4 (350 mg, 2.46 mmol) can eventually be added to accelerate the reaction. The resulting coloured suspension was filtered and the filtrate was concentrated in vacuo. The crude products were purified by column chromatography (SiO2, DCM/EtOAc 10:1 or DCM) to afford 6 or 7.
:1) Mp 145 °C. IR (HATR) νmax/cm−1 3087, 2220 (CN), 1559, 1496, 1269, 1186, 827. 1H-NMR: δH (400 MHz; CD2Cl2) 7.25 (6H, d, 3J 8.8, CHAr), 7.73 (3H, s, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 7.90 (6H, d, 3J 8.8, CHAr). 13C-NMR: δC (100 MHz; CD2Cl2) 81.6, 113.6, 114.7, 125.4, 128.2, 133.3, 150.7, 158.5. HR-MALDI-MS (DHB): calcd for C30H15N7 (M+) 473.1384 found 473.1385.
C), 7.56 (6H, d, 3J 9.0, CHAr). 13C-NMR: δC NMR (100 MHz; CD2Cl2): 86.2, 93.0, 111.4, 112.47, 114.9, 115.6, 124.5, 135.0, 140.8, 148.5. HR-MALDI-MS (DHB): calcd for C36H15N7 (M+) 545.1384 found 545.1386.
:3) to afford 8 as orange solid. Yield (144 mg, 42%). Rf 0.4 (SiO2; EtOAc/hexane 1:3). Mp 188 °C. IR (HATR), νmax/cm−1 2294, 2231 (CN), 1592, 1506, 1324, 1285, 1179, 972, 818. 1H-NMR: δH (400 MHz; CD2Cl2) 3.83 (9H, s, CH3), 6.77 (6H, d, 3J 16.0, CHCH), 7.16 (6H, d, 3J 8.5, CHAr), 7.54 (6H, d, 3J 8.5, CHAr), 7.81 (6H, d, 3J 16.0, CHCH). 13C-NMR: δC (100 MHz; CD2Cl2) 33.2, 109.1, 109.5, 112.8, 113.3, 122.8, 125.0, 129.5, 130.9, 139.7, 148.6, 151.3. HR-MALDI-MS (DCTB): calcd for C42H27N13 (M+) 713.2507 found 713.2526.
C), 7.90 (6H, d, 3J 8.8, CHAr). 13C-NMR: δC (100 MHz; CD2Cl2) 81.6, 113.6, 114.7, 125.4, 128.2, 133.3, 150.7, 158.5. HR-MALDI-MS (DHB): calcd for C30H15N7 (M+) 473.1384 found 473.1385.
C), 7.56 (6H, d, 3J 9.0, CHAr). 13C-NMR: δC NMR (100 MHz; CD2Cl2): 86.2, 93.0, 111.4, 112.47, 114.9, 115.6, 124.5, 135.0, 140.8, 148.5. HR-MALDI-MS (DHB): calcd for C36H15N7 (M+) 545.1384 found 545.1386.
:3) to afford 8 as orange solid. Yield (144 mg, 42%). Rf 0.4 (SiO2; EtOAc/hexane 1:3). Mp 188 °C. IR (HATR), νmax/cm−1 2294, 2231 (CN), 1592, 1506, 1324, 1285, 1179, 972, 818. 1H-NMR: δH (400 MHz; CD2Cl2) 3.83 (9H, s, CH3), 6.77 (6H, d, 3J 16.0, CHCH), 7.16 (6H, d, 3J 8.5, CHAr), 7.54 (6H, d, 3J 8.5, CHAr), 7.81 (6H, d, 3J 16.0, CHCH). 13C-NMR: δC (100 MHz; CD2Cl2) 33.2, 109.1, 109.5, 112.8, 113.3, 122.8, 125.0, 129.5, 130.9, 139.7, 148.6, 151.3. HR-MALDI-MS (DCTB): calcd for C42H27N13 (M+) 713.2507 found 713.2526.
General method of Sonogashira cross-coupling (chromophores 9–12)
Tris(4-ethynylphenyl)amine 14 (317 mg, 1.00 mmol) and the corresponding halogen derivative 23, 24, 32 or 33 (3.6 mmol) were dissolved in dry 1,4-dioxane (40 ml) and triethylamine (10 ml, 71.8 mmol). Argon was bubbled through the mixture for 10 min, whereupon Pd(PPh3)4 (116 mg, 0.10 mmol) and CuI (38 mg, 0.2 mmol) were added and the reaction mixture was heated to 92 °C for 8 h. The reaction mixture was quenched with saturated solution of NH4Cl (150 ml) and extracted with DCM (2 × 150 ml). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, EtOAc/hexane 2:3 or EtOAc/hexane 3:4) to afford target chromophores 9–12.
:3). Mp 327 °C (dec.). IR (HATR), νmax/cm−1 2921, 2206 (CN), 1593, 1504, 1320, 1289, 1145, 836, 801, 736. 1H-NMR: δH (400 MHz; CD2Cl2) 7.19 (6H, d, 3J 8.8, CHAr), 7.52 (3H, t, 3J 8.0, CHAr), 7.64 (6H, d, 3J 8.8, CHAr), 7.90 (6H, d, 3J 8.0, CHAr). 13C-NMR: δC (100 MHz; CD2Cl2) 83.4, 103.5, 116.2, 116.5, 124.5, 128.3, 131.2, 134.3, 136.4 148.1, 149.5. HR-MALDI-MS (DHB): calcd for C48H21N7 (M+) 695.1853 found 695.1852.
:3). Mp 282 °C (dec.). IR (HATR), νmax/cm−1 2922, 2204 (CN), 1593, 1503, 1320, 1277, 1145, 836, 800, 735. 1H-NMR: δH (400 MHz; CD2Cl2) 7.24 (6H, d, 3J 8.8, CHAr), 7.66 (6H, d, 3J 8.8, CHAr), 7.81 (3H, d, 3J 8.4, CHAr), 7.92 (3H, dd, 3J 8.4, 4J 1.2, CHAr), 8.04 (3H, d, 4J 1.2, CHAr). 13C-NMR: δC (100 MHz; CD2Cl2) 85.7, 101.1, 112.5, 116.4, 116.7, 116.8, 117.2, 124.9, 132.0, 133.2, 134.3, 136.1, 136.5, 148.4. HR-MALDI-MS (DHB): calcd for C48H21N7 (M+) 695.1853 found 695.1872.
:4). Mp 191 °C. IR (HATR), νmax/cm−1 2925, 2208 (CN), 1593, 1504, 1320, 1291, 834, 808, 737. 1H-NMR: δH (400 MHz; CD2Cl2) 7.18 (6H, d, 3J 8.8, CHAr), 7.31 (3H, d, 3J 4.0, Th), 7.55 (6H, d, 3J 8.8, CHAr), 7.62 (3H, d, 3J 4.0, Th). 13C-NMR: δC (100 MHz; CD2Cl2) 81.2, 97.0, 110.4, 114.1, 117.1, 124.8, 131.2, 132.0, 133.6, 138.0, 147.9. HR-MALDI-MS (DHB): calcd for C39H18N4S3 (M+) 638.0688 found 638.0705.
:4). Mp 198 °C (dec.). IR (HATR), νmax/cm−1 2848, 2236 (CN), 1592, 1472, 1377, 1169, 831, 728, 718. 1H-NMR: δH (500 MHz; CD2Cl2) 7.19 (6H, d, 3J 9.0, CHAr), 7.59 (6H, d, 3J 9.0, CHAr), 7.99 (3H, s, Th). 13C-NMR: δC (100 MHz; CD2Cl2) 78.2, 103.1, 111.8, 111.9, 112.3, 114.9, 115.8, 124.6, 133.7, 135.5, 136.6, 148.0. HR-MALDI-MS (DHB): calcd for C42H27N13 (M+) 713.2507 found 713.2532.
Acknowledgements
This research was supported by the Czech Science Foundation (13-01061S).Notes and references
- (a) L. Guo and M. S. Wong, Adv. Mater., 2014, 26, 5400 CrossRef CAS PubMed; (b) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS PubMed; (c) S. R. Marder, Chem. Commun., 2006, 131 RSC.
- (a) Special issue on “Organic electronics and optoelectronics”, ed. S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923; (b) Special issue on “Materials for electronics”, ed. R. D. Miller and E. A. Chandross, Chem. Rev., 2010, 110, 1; (c) Special issue on “Molecular electronics – from a visionary concept towards reality”, ed. M. Mayor, Chimia, 2010, 64, 348.
- (a) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed; (b) F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641 CrossRef CAS PubMed.
- (a) S. Yao and K. D. Belfield, Eur. J. Org. Chem., 2012, 3199 CrossRef CAS PubMed; (b) H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863 CrossRef CAS PubMed; (c) C. Le Droumaguet, A. Sourdon, E. Genin, O. Mongin and M. Blanchard-Desce, Chem. – Asian J., 2013, 8, 2984 CrossRef CAS PubMed; (d) B. Dumat, G. Bordeau, E. Faurel-Paul, F. Mahuteau-Betzer, N. Saettel, G. Metge, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Am. Chem. Soc., 2013, 135, 12697 CrossRef CAS PubMed; (e) S. Picard, E. Genin, G. Clermont, V. Hugues, O. Mongin and M. Blanchard-Desce, New J. Chem., 2013, 37, 3899 RSC; (f) L. Zhu, W. Lv, S. Liu, H. Yan, Q. Zhao and W. Huang, Chem. Commun., 2013, 49, 10638 RSC; (g) G. Wang, X. Zhang, J. Geng, K. Li, D. Ding, K.-Y. Pu, L. Cai, Y-H. Lai and B. Liu, Chem. – Eur. J., 2012, 18, 9705 CrossRef CAS PubMed; (h) Z. Yang, N. Zhao, Y. Sun, F. Miao, Y. Liu, X. Liu, Y. Zhang, W. Ai, G. Song, X. Schen, X. Yu, J. Sun and W.-Y. Wong, Chem. Commun., 2012, 48, 3442 RSC; (i) X. Zhang, X. Ren, Q.-H. Xu, K. P. Loh and Z.-K. Chen, Org. Lett., 2009, 11, 1257 CrossRef CAS PubMed; (j) G. Wang, X. Zhang, J. Geng, K. Li, D. Ding, K.-Y. Pu, L. Cai, Y.-H. Lai and B. Liu, Chem. – Eur. J., 2012, 18, 9705 CrossRef CAS PubMed; (k) G. Wang, K.-Y. Pu, X. Zhang, K. Li, L. Wang, L. Cai, D. Ding, Y.-H. Lai and B. Liu, Chem. Mater., 2011, 23, 4428 CrossRef CAS; (l) O. P. Varnavski, J. C. Ostrowski, L. Sukhomlinova, R. J. Twieg, G. C. Bazan and T. Goodson III, J. Am. Chem. Soc., 2002, 124, 1736 CrossRef CAS PubMed; (m) W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CAS.
- (a) F. Hammerer, G. Garcia, S. Chen, F. Poyer, S. Achelle, C. Fiorini-Debuisschert, M.-P. Teulade-Fichou and P. Maillard, J. Org. Chem., 2014, 79, 1406 CrossRef CAS PubMed; (b) S. Achelle, P. Couleaud, P. Baldeck, M. P. Teulade-Fichou and P. Maillard, Eur. J. Org. Chem., 2011, 1271 CrossRef CAS PubMed; (c) M. Gary-Bobo, Y. Mir, C. Rouxel, D. Brevet, I. Basile, M. Maynadier, O. Vaillant, O. Mognin, M. Blanchard-Desce, A. Morère, M. Garcia, J.-O. Durand and L. Raehm, Angew. Chem., Int. Ed., 2011, 50, 11425 CrossRef CAS PubMed; (d) S. P. McIlroy, E. Cló, L. Nikolajsen, P. K. Frederiksen, C. B. Nielsen, K. V. Mikkelsen, K. V. Gotheft and P. O. Ogilby, J. Org. Chem., 2005, 70, 1134 CrossRef CAS PubMed; (e) J. R. Starkey, A. K. Rebane, M. A. Drobizhev, F. Meng, A. Gong, A. Elliott, K. McInnerney and C. W. Spangler, Clin. Cancer Res., 2008, 14, 6564 CrossRef CAS PubMed.
- (a) A. Abbotto, L. Beverina, R. Bozio, S. Bradamante, C. Ferrante, G. A. Pagani and R. Signorini, Adv. Mater., 2000, 12, 1963 CrossRef CAS; (b) F. Hao, X. Zhang, Y. Tian, H. Zhou, L. Li, J. Wu, S. Zhang, J. Yang, B. Jin, X. Tao, G. Zhou and M. Jiang, J. Mater. Chem., 2009, 19, 9163 RSC.
- (a) D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843 CAS; (b) B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I.-Y. Sandy Lee, D. McCord-Maughon, J. Qin, H. Röckel, M. Rumi, X.-L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS; (c) J. H. Strickler and W. W. Webb, Opt. Lett., 1991, 16, 1780 CrossRef CAS.
- (a) W. Zhou, S. M. Kuebler, K. L. Braun, T. Yu, J. K. Cammack, C. K. Ober, J. W. Perry and S. R. Marder, Science, 2002, 296, 1106 CrossRef CAS PubMed; (b) L. Kelemen, P. Ormos and G. Vizsnyiczai, J. Eur. Opt. Soc.-Rapid, 2011, 6, 11029 CrossRef; (c) M. Jin, J. Xie, J.-P. Malval, A. Spangenberg, O. Soppera, D.-L. Versace, T. Leclerc, H. Pan, D. Wan, H. Pu, P. Baldeck, O. Poizat and S. Knopf, J. Mater. Chem. C, 2014, 2, 7201 RSC.
- (a) F. Bureš, RSC Adv., 2014, 4, 58826 RSC; (b) P. D. Jarowski and Y. Mo, Chem. – Eur. J., 2014, 20, 17214 CrossRef CAS PubMed; (c) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef CAS PubMed; (d) J. Y. Lee, K. S. Kim and B. J. Mhin, J. Chem. Phys., 2001, 115, 9484 CrossRef CAS PubMed; (e) F. Bureš, O. Pytela, M. Kivala and F. Diederich, J. Phys. Org. Chem., 2011, 24, 274 CrossRef PubMed.
- (a) F. A. Martin, C. Baudequin, C. Fiol-Petit, M. Darabantu, Y. Ramondec and N. Plé, Tetrahedron, 2014, 70, 2546 CrossRef CAS PubMed; (b) Y. Jiang, Y. Wang, J. Hua, J. Tang, B. Li, S. Qian and H. Tian, Chem. Commun., 2010, 46, 4689 RSC; (c) Y. Jiang, Y. Wang, B. Wang, J. Yang, N. He, S. Qian and J. Hua, Chem. – Asian J., 2011, 6, 157 CrossRef CAS PubMed; (d) S. Zeng, X. Ouyang, H. Zeng, W. Ji and Z. Ge, Dyes Pigm., 2012, 94, 290 CrossRef CAS PubMed.
- (a) C. Rouxel, C. Le Droumaguet, Y. Macé, S. Clift, O. Mongin, E. Magnier and M. Blanchard-Desce, Chem. – Eur. J., 2012, 18, 12487 CrossRef CAS PubMed; (b) L. Porrès, O. Mongin, C. Katan, M. Charlot, T. Pons, J. Mertz and M. Blanchard-Desce, Org. Lett., 2004, 6, 47 CrossRef PubMed; (c) C. Le Droumaguet, O. Mongin, M. H. V. Werts and M. Blanchard-Desce, Chem. Commun., 2005, 2802 RSC; (d) J. Wu, Y. Zhao, X. Li, M. Shi, F. Wu and X. Fang, New J. Chem., 2006, 30, 1098 RSC; (e) Y.-P. Tian, L. Li, J.-Z. Zhang, J.-X. Yang, H.-P. Zhou, J.-Y. Wu, P.-P. Sun, L.-M. Tao, Y.-H. Guo, C.-K. Wang, H. Xing, W.-H. Huang, X.-T. Tao and M.-H. Jiang, J. Mater. Chem., 2007, 17, 3646 RSC; (f) O. Mongin, L. Porrès, C. Katan, T. Pons, J. Mertz and M. Blanchard-Desce, Tetrahedron Lett., 2003, 44, 8121 CrossRef CAS PubMed; (g) C. Huang, M. M. Sartin, N. Siegel, M. Cozzuol, Y. Zhang, J. M. Hales, S. Barlow, J. W. Perry and S. R. Marder, J. Mater. Chem., 2011, 21, 16119 RSC; (h) J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsoon, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem. – Eur. J., 2009, 15, 198 CrossRef CAS PubMed; (i) Y.-X. Yan, X.-T. Tao, Y.-H. Sun, C.-K. Wang, G.-B. Xu, J.-X. Yang, Y. Ren, X. Zhao, Y.-Z. Wu, X.-Q. Yu and M.-H. Jiang, J. Mater. Chem., 2004, 14, 2995 RSC; (j) A. Bhaskar, G. Ramakrishna, Z. Lu, R. Twieg, J. M. Hales, D. J. Hagan, E. Van Stryland and T. Goodson III, J. Am. Chem. Soc., 2006, 128, 11840 CrossRef CAS PubMed; (k) H. Xiao, C. Mei, B. Li, N. Ding, Y. Zhang and T. Wei, Dyes Pigm., 2013, 99, 1051 CrossRef CAS PubMed; (l) M. Parent, O. Mongin, K. Kamada, C. Katan and M. Blanchard-Desce, Chem. Commun., 2005, 2029 RSC; (m) D. W. Brousmiche, J. M. Serin, J. M. J. Fréchet, G. S. He, T.-C. Lin, S. J. Chung and P. P. Prasad, J. Am. Chem. Soc., 2003, 125, 1448 CrossRef CAS PubMed; (n) S. J. Chung, K.-S. Kim, T.-C. Lin, G. S. He, J. Swiatkiewicz and P. P. Prasad, J. Phys. Chem. B, 1999, 103, 10741 CrossRef CAS; (o) V. Hrobáriková, P. Hrobárik, P. Gajdoš, I. Fitilis, M. Fakis, P. Persephonis and P. Zahradník, J. Org. Chem., 2010, 75, 3053 CrossRef PubMed; (p) G. Bordeau, R. Lartia, G. Metge, C. Fliorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Am. Chem. Soc., 2008, 130, 16836 CrossRef CAS PubMed; (q) R. Lartia, C. Allain, G. Bordeau, F. Schmidt, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Org. Chem., 2008, 73, 1732 CrossRef CAS PubMed; (r) S.-I. Kato, T. Matsumoto, M. Shigeiwa, H. Gorohmaru, S. Maeda, T. Ishi-i and S. Mataka, Chem. – Eur. J., 2006, 12, 2303 CrossRef CAS PubMed; (s) Y. Jiang, Y. Wang, J. Yang, J. Hua, B. Wang, S. Qian and H. Tian, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1830 CrossRef CAS PubMed; (t) J.-W. Seo, S. Y. Jang, D. Kim and H.-J. Kim, Tetrahedron, 2008, 64, 2733 CrossRef CAS PubMed.
- (a) B. Xu, H. Fang, F. Chen, H. Lu, J. He, Y. Li, Q. Chen, H. Sun and W. Tian, New J. Chem., 2009, 33, 2457 RSC; (b) T.-C. Lin, W.-L. Lin, C.-M. Wang and C.-W. Fu, Eur. J. Org. Chem., 2011, 912 CrossRef CAS PubMed; (c) Z.-Q. Yan, B. Xu, Y.-J. Dong, W.-J. Tian and A.-W. Li, Dyes Pigm., 2011, 90, 269 CrossRef CAS PubMed; (d) P. Wei, X. Bi, Z. Wu and Z. Xu, Org. Lett., 2005, 7, 3199 CrossRef CAS PubMed.
- (a) Y. Qian and M. Luo, Dyes Pigm., 2014, 101, 240 CrossRef CAS PubMed; (b) L. Guo, K. F. Li, M. S. Wong and K. W. Cheah, Chem. Commun., 2013, 49, 3579 RSC; (c) T.-C. Lin, Q. Zheng, C.-Y. Chen, G. S. He, W.-J. Huang, A. I. Ryasnyanskiy and P. N. Prasad, Chem. Commun., 2008, 389 RSC.
- (a) T. Kengthanomma, P. Thamyongkit, J. Gasiorowski, A. M. Ramil and N. S. Sariciftci, J. Mater. Chem. A, 2013, 1, 10524 RSC; (b) J. Zhang, J. Yu, C. He, D. Deng, Z.-G. Zhang, M. Zhang, Z. Li and Y. Li, Org. Electron., 2012, 13, 166 CrossRef CAS PubMed.
- E. Gagnon, T. Maris and J. D. Wuest, Org. Lett., 2010, 12, 404 CrossRef CAS PubMed.
- (a) K. Melánová, D. Cvejn, F. Bureš, V. Zima, J. Svoboda, L. Beneš, T. Mikysek, O. Pytela and P. Knotek, Dalton Trans., 2014, 43, 10462 RSC; (b) J. F. Eubank, F. Nouar, R. Luebke, A. J. Cairns, L. Wojtas, M. Alkordi, T. Bosquet, M. R. Hight, J. Eckert, J. P. Embs, P. A. Georgiev and M. Eddaoudi, Angew. Chem., Int. Ed., 2012, 51, 10099 CrossRef CAS PubMed; (c) D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186 CrossRef CAS PubMed.
- (a) N. Niammont, N. Kimpitak, G. Tumcharern, P. Rashatasakhon and M. Sukwattanasinitt, RSC Adv., 2013, 3, 25215 RSC; (b) N. Niammont, W. Siripornnoppakhun, P. Rashatasakhon and M. Sukwattanasinitt, Org. Lett., 2009, 11, 2768 CrossRef PubMed; (c) K. Vongnam, T. Vilaivan, M. Sukwattanasinitt and P. Rashatasakhon, J. Fluoresc., 2014, 24, 197 CrossRef CAS PubMed.
- S. Körsten and G. J. Mohr, Chem. – Eur. J., 2011, 17, 969 CrossRef PubMed.
- (a) N. Niammont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel and M. Sukwattanasinitt, Chem. Commun., 2013, 49, 780 RSC; (b) Z. Zhao, J. Liu, J. W. Y. Lam, C. Y. K. Chan, H. Qiu and B. Z. Tang, Dyes Pigm., 2011, 91, 258 CrossRef CAS PubMed.
-
(a) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed;
(b) M. Kivala and F. Diederich, Pure Appl. Chem., 2008, 80, 411 CrossRef CAS;
(c) J. Kulhánek and F. Bureš, Beilstein J. Org. Chem., 2012, 8, 25 CrossRef PubMed;
(d) N. N. P. Moonen, R. Gist, C. Boudon, J.-P. Gisselbrecht, P. Seiler, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Org. Biomol. Chem., 2003, 1, 2032 RSC;
(e) F. Bureš, W. B. Schweizer, J. C. May, C. Boudon, J.-P. Gisselbrecht, M. Gross, I. Biaggio and F. Diederich, Chem. – Eur. J., 2007, 13, 5378 CrossRef PubMed;
(f) F. Bureš, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, M. Gross and F. Diederich, Eur. J. Org. Chem., 2008, 994 CrossRef PubMed;
(g) F. Bureš, H. Čermáková, J. Kulhánek, M. Ludwig, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růži
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ka, Eur. J. Org. Chem., 2012, 529 CrossRef PubMed;
(h) L. Dokládalová, F. Bureš, W. Kuznik, I. V. Kityk, A. Wojciechowski, T. Mikysek, N. Almonasy, M. Ramaiyan, Z. Padělková, J. Kulhánek and M. Ludwig, Org. Biomol. Chem., 2014, 12, 5517 RSC.
ka, Eur. J. Org. Chem., 2012, 529 CrossRef PubMed;
(h) L. Dokládalová, F. Bureš, W. Kuznik, I. V. Kityk, A. Wojciechowski, T. Mikysek, N. Almonasy, M. Ramaiyan, Z. Padělková, J. Kulhánek and M. Ludwig, Org. Biomol. Chem., 2014, 12, 5517 RSC. - (a) J. Yoo, S. K. Yang, M.-Y. Jeong, H. C. Ahn, S.-J. Jeon and B. R. Cho, Org. Lett., 2003, 5, 645 CrossRef CAS PubMed; (b) W. J. Yang, D. Y. Kim, C. H. Kim, M.-Y. Jeong, S. K. Lee, S.-J. Jeon and B. R. Cho, Org. Lett., 2004, 6, 1389 CrossRef CAS PubMed; (c) W. J. Yang, D. Y. Kim, M.-Y. Jeong, H. M. Kim, S.-J. Jeon and B. R. Cho, Chem. Commun., 2003, 2618 RSC; (d) W. J. Jang, D. Y. Kim, M.-Y. Jeong, H. M. Kim, Y. K. Lee, X. Fang, S.-J. Jeon and B. R. Cho, Chem. – Eur. J., 2005, 11, 4191 CrossRef PubMed; (e) B. J. Zhang and S.-J. Jeon, Chem. Phys. Lett., 2003, 377, 210 CrossRef CAS; (f) B. Wang, Y. Wang, J. Hua, Y. Jiang, J. Huang, S. Qian and H. Tian, Chem. – Eur. J., 2011, 17, 2647 CrossRef CAS PubMed; (g) E. Ripaud, Y. Olivier, P. Leriche, J. Cornil and J. Roncali, J. Phys. Chem. B, 2011, 115, 9379 CrossRef CAS PubMed.
- (a) J. Zhang, D. Deng, C. He, Y. He, M. Zhang, Z.-G. Zhang, Z. Zhang and Y. Li, Chem. Mater., 2011, 23, 817 CrossRef CAS; (b) J. Jia, Y. Zhang, P. Xue, P. Zhang, X. Zhao, B. Liu and R. Lu, Dyes Pigm., 2013, 96, 407 CrossRef CAS PubMed.
- (a) Z. Fang, R. D. Webster, M. Samoc and Y.-H. Lai, RSC Adv., 2013, 3, 17914 RSC; (b) M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zaleśny, W. Bartkowiak, H. Ångren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 CrossRef PubMed; (c) P. Hrobárik, V. Hrobáriková, I. Sigmundová, P. Zahradník, M. Fakis, I. Polyzos and P. Persephonis, J. Org. Chem., 2011, 76, 8726 CrossRef PubMed.
- J. Kulhánek, F. Bureš, T. Mikysek, J. Ludvík and O. Pytela, Dyes Pigm., 2011, 90, 48 CrossRef PubMed.
- D. M. Johnson and P. G. Rasmussen, Macromolecules, 2000, 33, 8597 CrossRef CAS.
- J.-Y. Balandier, F. Quist, C. Amato, S. Sergueev, Y. Geerts, J. Cornil and S. Bouza-Kraoui, WO 2011/02712, 2011.
- J. Ni, K.-J. Wei, Y. Liu, X.-C. Huang and D. Li, Cryst. Growth Des., 2010, 10, 3964 CAS.
- H. M. S. Kumar, B. V. S. Reddy, P. T. Reddy and J. S. Yadav, Synthesis, 1999, 586 CrossRef CAS PubMed.
- Y.-P. Tian, L. Li, Y.-Z. Zhang, J.-X. Yang, H. Zhou, J. Wu, P. Sun, L. Tao, Y. Guo, C.-K. Wang, H. Xing, W. Huang, X.-T. Tao and M.-H. Jiang, J. Mater. Chem., 2007, 17, 3646 RSC.
- (a) F. Texier-Boullet and A. Foucaud, Tetrahedron Lett., 1982, 23, 4927 CrossRef CAS; (b) M. Klikar, F. Bureš, O. Pytela, T. Mikysek, Z. Padělková, A. Barsella, K. Dorkenoo and S. Achelle, New J. Chem., 2013, 37, 4230 RSC.
- C. Lambert, W. Gaschler, E. Schmälzlin, K. Meerholz and C. Bräuchle, J. Chem. Soc., Perkin Trans. 2, 1999, 577 RSC.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
-
(a) J. Kulhánek, F. Bureš, O. Pytela, T. Mikysek, J. Ludvík and A. Růžika, Dyes Pigm., 2010, 85, 57 CrossRef PubMed;
(b) M.-B. S. Kirketerp, M. Å. Petersen, M. Wanko, H. Zettergren, A. Rubio, M. B. Nielsen and S. B. Nielsen, ChemPhysChem, 2010, 11, 2495 CrossRef CAS PubMed.
- J. Kulhánek, F. Bureš, J. Opršal, W. Kuznik, T. Mikysek and A. Růžika, Asian J. Org. Chem., 2013, 2, 422 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc, Wallingford CT, 2013 Search PubMed.
- ArgusLab 4.0, Mark A. Thompson, Planaria Software LLC, Seattle, WA. http://www.arguslab.com.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of chromophores 1–5 and all intermediates 13–25, electrochemical and optical data, correlations and NMR spectra. See DOI: 10.1039/c5tc01293g |
| This journal is © The Royal Society of Chemistry 2015 |