
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upconverting nanocomposites dispersed in urea-containing acrylics
D. L.
Inglefield
Jr.
a,
T. R.
Merritt†
b,
B. A.
Magill†
b,
T. E.
Long
a and
G. A.
Khodaparast
*b
aDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
bDepartment of Physics, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: khoda@vt.edu
First published on 8th May 2015
Abstract
Lanthanide-doped upconverting nanoparticles (UCNPs) have the ability to convert low energy photons into high energy photons, making this material appealing for a variety of scientific pursuits, from solar energy conversion to bioimaging. A combination of polymers and nanocomposites increases the utility of these upconverting nanoparticles allowing nanoparticles to be added to any device compatible with polymer coatings. Here, trifluoroacetate salt decomposition enables Er/Yb doped NaYF4 upconverting nanoparticle synthesis. The subsequent deposition of a silica nanoshell yields polar silica-coated upconverting nanoparticles, enabling composite formation with polar urea-containing methacrylic polymers. Hydrogen bonding between urea groups in the polymer and the silica-coated nanoparticles allowed for dispersion of the upconverting nanoparticles to form upconverting composite films. These films exhibit desirable upconversion comparable to the nanoparticles dispersed in methanol. Urea-containing polymers are promising candidates for matrices in nanocomposites formed with polar silica nanoparticles due to favorable polymer–nanoparticle interactions. This architecture is superior to urea-methacrylate homopolymers, since the central low glass transition temperature block will provide critical ductility to the film, thus rendering the film to be durable for optical applications.
1 Introduction
Anti-Stokes emission processes, where high-energy photons are generated from low-energy pump photons, have attracted a great deal of attention due to their inherent importance and applicability to various research fields, including those involving solid state lasers,1–3 optical storage,4 near infrared quantum counters,1 photovoltaics,5–7 bioimaging and biotherapeutics.8–12 For instance, this energy upconversion process has been used for harvesting subbandgap photons to improve solar cell conversion efficiencies;5,7 in addition, upconversion can be utilized in contrast agents for advanced biological labeling and imaging applications due to the avoidance of autofluorescence and deep detection ranges in biological tissues,9 both of which are direct results of the near-infrared excitation spectra and large anti-Stokes shifts of these materials.10While various anti-Stokes mechanisms exist, such as the widely employed processes of simultaneous two photon absorption (TPA) and second harmonic generation (SHG), upconversion based on energy transfer conversion (ETU) remains an appealing alternative for the generation of anti-Stokes emission.1,13,14 The former processes generate upconverted emission through virtual intermediate excited states, a mechanism reliant on high-density excitation, a requirement typically fulfilled by ultrafast lasers.13,14 In contrast, ETU involves unspecified non-radiative energy transfer between real, long-lived intermediate excited states of neighboring lanthanide ions; as a result, the requirement of high-density excitation is largely waived for ETU and, as a consequence, high conversion efficiency is allowed without the need for intense coherent excitation sources.13,14
In particular, lanthanide doped glasses are attractive ETU systems due to their ability to possess emission wavelengths that spam the entire visible region; this feature is achieved through the judicious choice of the activator ion.14,15 The most efficient species of these upconversion materials are fluoride glasses co-doped with the sensitizer–activator pair of Yb3+ and Er3+, namely hexagonal NaYF4:Yb,Er,16 which exhibits upconversion quantum yields of up to 3% in bulk glasses.17 This maximal quantum yield is attributable to the high absorption cross-section of Yb3+ and the spectral overlap of Yb3+ solitary optical transition with the 4I15/2 → 4I11/2 transition of erbium, as well as the low optical phonon energies of hexagonal NaYF4, which results in the considerable suppression of multiphonon relaxation processes.1,16,18
Although few studies investigate the use of UCNPs in polymer composites, the initial results appear to be promising.17,19 These upconverting composites represent materials with possible applications in light curable polymers and solar energy technology. For example, upconversion offers the potential for increasing the efficiency of photovoltaic cells through converting light below the optical band gap to light within the photovoltaic cells' optical band gap. Here we present our initial results toward the goal of utilizing urea-containing polymers in upconverting nanocomposites.
Urea-containing polymers appear to be particularly promising for applications using silica and metal coated UCNPs, due to the high levels of hydrogen bonding between urea groups with silica and the well-known urea–metal interaction.20–23 The ABA triblock copolymer architecture is a well-recognized sequence for the formation of mechanically ductile films and coatings. This architecture is superior to urea-methacrylate homopolymers, since the central low glass transition temperature (Tg) block will provide critical ductility to the film, thus rendering the film to be durable for optical applications. Most importantly, the nanoscale phase separated block copolymer morphology provides a template for nanoparticle organization, locating the nanoparticles in specific sequences containing the urea sites for coordination and creating new compositional options for macromolecular composites whose architecture is controlled by polymer chemistry.24–26
In addition to using these nanocomposites as an upconverting coating for solar cells, these composites could also prove useful for fabricating hybrid nanocomposite structures where upconverting particles are suspended near a metal surface27,28 or other metal nanoparticles,7,29 in self-aligned chains or layers where the particle density and orientation are controlled by an external chemical matrix similar to what has been done with semiconductor nanoparticles26,30,31 or in planar structures constructed using nano-imprint lithography.28 These hybrid structures take advantage of the surface plasmon resonance in the nearby metal to enhance either the emission or absorption by the UCNPs in the hybrid material.
2 Materials
Dimethylformamide (DMF), methanol, and hexane were purchased from fisher scientific and used as received. HPLC grade water was obtained from Spectrum Chemical and used as received. 2-Isocyanatoethyl methacrylate (2-ICMA, >98%, stabilized with butylated hydroxytoluene) was purchased from TCI America and used as received. Hexylamine (99%) was obtained from Sigma Aldrich and distilled before use. Azobisisobutyronitrile (AIBN, 98%) was purchased from Sigma Aldrich and recrystallized twice from methanol before use. Anhydrous chloroform, neutral alumina (activated), oleic acid (90%), 1-octadecene (90%), sodium nitrate (≥99%), potassium nitrate (≥99%), IGEPAL CO-520 (average Mn 441), ammonium hydroxide (28 wt% ammonia in water, 99.99%), sodium trifluoroacetate (98%) and tetraethyl orthosilicate (TEOS, ≥99%) were obtained from Sigma Aldrich and used as received. Erbium trifluoroacetate (99.9%), yttrium trifluoroacetate (99.9%), and ytterbium trifluoroacetate (99.9%) were obtained from GFS Chemicals and used as received.2.1 Urea methacrylate monomer synthesis
For a typical synthesis of urea-containing methacrylate monomers from amine reactants (hexylamine, propylamine, or aniline): a 100 mL, round-bottom, flask equipped with a magnetic stir bar was charged with one equivalent of liquid amine at 20 wt% in chloroform and purged with argon for 10 min. One equivalent of 2-ICMA was added dropwise with stirring at 0 °C. The reaction was allowed to stir for 4 h at 10 °C, and then warmed slowly to room temperature and stirred for an additional 20 h. Chloroform was removed under reduced pressure, yielding a white solid in quantitative yield. For the reaction of methylamine with 2-ICMA, a 2 M solution of methylamine in THF was used. One equivalent of methylamine in THF was charged to a 100 mL, round-bottom, flask equipped with a magnetic stirrer and purged with argon for 10 min. One equivalent of 2-ICMA was added dropwise with stirring at 0 °C. The reaction was allowed to stir for 4 h at 0 °C, and then warmed slowly to room temperature and stirred for an additional 20 h. THF was removed under reduced pressure, yielding a white solid in quantitative yield. The monomers were redissolved at 10 wt% in chloroform and passed through a neutral alumina column to remove inhibitors; then chloroform was removed under reduced pressure and the product was dried in vacuo at 25 °C for 12 h. The purity and structure of the monomers were confirmed by 1H NMR spectroscopy, with the compositions listed in Table 1. The aromatic derivative 2-(3-phenylureido)ethyl methacrylate (ArUrMA), and the hexyl derivative 2-(3-hexylureido)ethyl methacrylate (HUrMA) were selected for triblock copolymer synthesis based on their physical properties and solubility.2.2 Free radical polymerization of urea methacrylate monomers
For a typical free radical polymerization of urea methacrylate monomers from urea methacrylate homopolymers and copolymers: a 25 mL, round-bottom, flask equipped with a magnetic stirrer was charged with 0.5 g of urea methacrylate monomer dissolved at 5 wt% in DMF (10.6 mL) with 0.1 mol% of AIBN relative to the monomer. The mixture was sparged with argon for 25 min, and reacted at 65 °C for 24 h. The polymers were isolated by precipitation and dried in vacuo at 65 °C for 12 h. The isolated polymer yields and molecular weights were determined by 0.05 M LiBr DMF SEC using a dn/dc estimate of 0.007.2.3 Free radical polymerization of 2-EHMA
For a typical free radical polymerization of 2-EHMA: a 25 mL, round-bottom, flask equipped with a magnetic stirrer was charged with 0.5 g of 2-EHMA dissolved at 5 wt% in dioxane (9.7 mL) with 0.1 mol% of AIBN relative to 2-EHMA. The mixture was sparged with argon for 25 min, and reacted at 65 °C for 24 h. The polymer was isolated by dialysis against 1 L of THF three times for 12 h, and dried in vacuo at 65 °C for 12 h. The isolated polymer yield was 80%. The molecular weight as determined by THF SEC and offline determined dn/dc was 132![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with a polydispersity index (PDI) of 2.07.
000 g mol−1 with a polydispersity index (PDI) of 2.07.
2.4 Difunctional chain transfer agent (CTA) synthesis
Carbodiimide coupling synthesized a difunctional RAFT chain transfer agent using commercially available CDP, according to an adapted literature procedure.32 In a representative synthesis, a 50 mL, round-bottom, flask equipped with a magnetic stirrer was charged with CDP (1.12 g, 2.77 mmol), dissolved in 12 mL of anhydrous DCM and stirred at 0 °C. A solution of hexamethylenediamine (0.107 g, 0.92 mmol) and catalytic DMAP (20 mg) dissolved in 4 mL of anhydrous DCM was added to the CDP solution. Upon the addition of hexamethylenediamine and DMAP, DCC (0.635 g, 3.1 mmol) was immediately added. The reaction was allowed to stir under an argon atmosphere for 4 h at 0 °C, and then warmed slowly to room temperature and stirred for an additional 20 h. The solution was filtered to remove dicyclohexylurea precipitates and washed three times with the saturated sodium bicarbonate solution. The resultant organic layer was dried over MgSO4 and concentrated under reduced pressure. The difunctional CTA was purified using a silica gel column with 80:20 (v:v%) hexane:ethyl acetate to elute impurities. The product was then eluted using 50:50 (v:v%) ethyl acetate:methanol and the solvent was removed under reduced pressure. The product was dried for 16 h in vacuo at 25 °C to obtain the yellow-orange solid of 1,6-bis(4-cyano-4-[(dodecylsulfanylthiocarbanyl)sulfanyl] pentanoic acid)-hexane diamide (dCDP, 70% yield). MS-TOF confirmed the purity of dCDP (m/z = 887.45 g mol−1).
2.5 RAFT polymerization of 2-EHMA difunctional macroCTA
In a representative synthesis, a 500 mL, round-bottom, flask equipped with a magnetic stirrer was charged with 2-EHMA (20 g, 100.8 mmol), dCDP (112 mg, 126.2 mol), and V-501 (17.7 mg, 63.1 mol) dissolved in 202 mL dioxane. The solution was sparged with argon for 50 min, and then reacted at 65 °C for given time intervals. The macroCTA was purified by dialysis against THF three times for 12 h. The solvent was removed under reduced pressure, and the polymer was dried in vacuo for 16 h at 65 °C. The molecular weight and PDI for given reaction times were determined by THF SEC using an offline determined dn/dc for absolute molecular weight determination. A reaction time of 10 h yielded a macroCTA with an Mn of 48900 g mol−1 and a PDI of 1.16 in 28% isolated yield, which was used for the synthesis of triblock copolymers.
2.6 RAFT polymerization of ABA triblock copolymers
In a typical polymerization, a 25 mL, round-bottom, flask equipped with a magnetic stirrer was charged with ArUrMA (157 mg, 630 mol), HUrMA (648 mg, 2.52 mmol), macroCTA (287 mg, 5.87 mol), and V-501 (1.36 mg, 4.85 mol) dissolved in 6.32 mL of 65:35 (v:v%) dioxane:DMF. The solution was sparged with argon for 25 min and then reacted at 65 °C for given time intervals. The triblock copolymers were precipitated into 4:1 MeOH:H2O and dried in vacuo at 65 °C for 16 h. Comparing the integration at δ3.8 which corresponds to the methylene protons adjacent to the ester present in all repeating units with the integration at 7.5 which corresponds to the aromatic protons closest to the urea linkage on the ArUrMA repeating units estimated molecular weights by 1H NMR spectroscopy. This integration comparison combined with the molecular weight of the macroCTA as determined by THF SEC allowed for the estimation of triblock Mn. The triblocks are referred to as the wt% of the outerblocks incorporated as estimated by 1H NMR spectroscopy with the naming convention UrMA-wt%. Triblock copolymer films were cast from 100 mg mL−1 THF solutions into Teflon molds, dried at 25 °C for 48 h, and then dried in vacuo at 25 °C for 16 h. For annealing, the films were further dried in vacuo at 100 °C for 16 h.
2.7 Upconverting nanoparticle synthesis
Er/Yb doped NaYF4 upconverting nanoparticles with a silica shell nanoshell were achieved in a two part synthesis shown in Scheme 3. First, upconverting cores were synthesized by the high temperature decomposition of trifluoroacetate salts to yield rare earth doped NaYF4 nanoparticles (UCNPs) using an adapted literature procedure,33 where the time of reaction determines the nanoparticle size and morphology (initially, small spherical particles are formed, as reaction times increase the particles grow in size and transition from spherical to hexagonal morphologies takes place). In our synthesis of targeting spherical nanoparticles with diameters of 20–30 nm, a 500 mL, three-necked, round-bottom, flask equipped with a magnetic stirrer was charged with sodium trifluoroacetate (0.66 g), yttrium trifluoroacetate (1.87 g), ytterbium trifluoroacetate (560 mg), and erbium trifluoroacetate (56 mg) dissolved in a mixture of 22.5 mL of oleic acid and 22.5 mL of 1-octadecene. The mixture was stirred under vacuum at 100 °C for 45 min, forming a transparent yellow solution. Then the reaction flask was purged with argon for 5 minutes, and placed in a molten salt bath (1:1 NaNO3:KNO3 weight) stabilized at 342 °C. The solution was refluxed for 25 min under constant argon flow and vigorous stirring, after which the nanoparticle growth was halted by removing the flask from the molten salt bath and immediately adding 15 mL octadecene to cool the reaction mixture. After the mixture reached room temperature, 200 mL of ethanol was added to precipitate the UCNPs. The UCNPs were isolated by centrifugation at 4000 rpm for 30 min, and then redispersed in 30 mL of cyclohexane followed by sonication for 30 min. The final concentration of UCNPs in cyclohexane was 48 mg mL−1. Deposition of a silica shell on the UCNPs was achieved by base catalyzed hydrolysis of TEOS adapted from Priyam et al.12 In a typical synthesis, a 50 mL, round-bottom, flask equipped with a magnetic stirrer was charged with 6 mL of the aforementioned UCNP solution in cyclohexane, 18 mL of cyclohexane, and 1.2 mL of CO-520, and the mixture was stirred for 20 min. Then, 252 L of ammonia solution (28 wt%) was added and the mixture was sonicated for 20 min. 189 L of TEOS was added and the mixture was stirred at 25 °C at 400 rpm for 45 h. UCNP containing silica shells (UCNP@SiO2) were precipitated by adding acetone to the reaction mixture, filtered, and washed two times with 50:50 ethanol:water. UCNP@SiO2 was dried in vacuo for 16 h at 25 °C.
2.8 Formation of the UCNP@SiO2 composite
Urea-containing methacrylic polymers were combined with UCNP@SiO2 to form upconverting composites according to the following method. Dry UCNP@SiO2 (25 mg) was dispersed in MeOH (1 mL) by sonication for 1 h. The urea-containing methacrylic polymer (100 mg, 80:20 HUrMA:ArUrMA molar ratio) was dissolved in 1 mL of MeOH and added to the UCNP@SiO2 dispersion in MeOH and the mixture was stirred for 5 min. The mixture was sonicated for 20 minutes before casting into a polytetrafluoroethylene mold, and dried at 25 °C for 16 h then at 65 °C for 8 h to ensure complete solvent removal.
3 Methods
1H nuclear magnetic resonance (NMR) spectroscopy was performed on a Varian Unity 400 MHz NMR spectrometer using 64 scans. Mass spectrometry was performed on an Agilent 6220 Accurate Mass TOF LC-MS system. Differential scanning calorimetry (DSC) was performed using a TA Instruments Q 2000 differential scanning calorimeter with a heating rate of 10 °C min−1, and all reported values are from the second heat treatment of a heat/cool/heat cycle using a cooling rate of 5 °C min−1. Thermogravimetric analysis (TGA) was performed using a TA Instruments Q500 thermogravimetric analyzer with a heating rate of 10 °C min−1 under a nitrogen atmosphere. Dynamic mechanical analysis (DMA) was performed on a TA Instruments Q800 dynamic mechanical analyzer in tension mode using a heating rate of 3 °C min−1 and an amplitude of 15 μm. Sonication was performed using a Bransonic 2510R-MTH sonicator bath. Size exclusion chromatography (SEC) of urea-containing methacrylic homopolymers was performed using a Waters size exclusion chromatograph equipped with an auto sampler, three 5 μm PLgel Mixed-C columns, a Waters 2410 refractive index detector operating at 880 nm, and a Wyatt Technologies miniDAWN multi-angle laser light scattering detector operating at 690 nm. The urea-containing methacrylic homopolymer samples were run at 50 °C in 0.01 M LiBr DMF solution with a flow rate of 1 mL min−1. THF SEC was peformed using a Waters 515 HPLC pump equipped with a Waters 717 plus auto sampler, a Wyatt Technology miniDawn MALLS detector and a Waters 2414 refractive index detector with a flow rate of 1 mL min−1. Offline dn/dc values for free radical poly(2-EHMA) and poly(2-EHMA) macroCTA polymers were determined using an Optilab T-rEX refractometer (λ = 658 nm) to provide absolute Mw values in SEC analysis. Atomic force microscopy (AFM) analysis of the triblock copolymer morphology was performed on a Veeco Multimode AFM in the tapping mode using 42 N m−1 spring constant tips and a set point ratio of 0.62.Spectral characterization methods were performed using a pulsed Ti:Sapphire laser, as the excitation source, (Coherent Chameleon Ultra II) operating at 980 nm with a pulse duration of 140 fs and a repetition rate of 80 MHz. The average excitation power was controlled using an acousto-optical modulator and set to 30 mW for all measurements. A 40 mm aspheric lens was used to both excite and collect emission from hexane or water UCNP dispersions contained in 1 cm-thick cells. With the aid of an 800 nm edge long-pass dichroic beam splitter, the photoluminescence (PL) was separated from the co-aligned excitation–emission beam. After this spectral separation, the PL was delivered, through a 100 m wide slit, to a 0.55 m focal length spectrometer (HORIBA Jobin Yvon iHR550) equipped with a 600 grooves per mm, 500 nm blaze grating, and measured using a liquid-nitrogen cooled charged-coupled device (CCD) (HORIBA Jobin Yvon Symphony II).
Lifetimes for our UCNPs were found using the Chameleon ultra fast laser to excite UCNP cores at 980 nm through the 20× objective lens of the Zeiss LSM 510. The light was chopped at 5 Hz using a mechanical chopper to provide a signal that varied over a time interval longer than the expected PL lifetime and within the range of time that our detector could resolve. The light was gathered through the same objective, the laser beam was removed via a pair of dichroic filters and the CCD in the Zeiss LSM 510 was used to measure the photoluminescence as a function of time.
For PL measurements, UCNP cores were measured in cyclohexane and UCNP@SiO2 were measured in methanol. Solvents with similar solubility were chosen to disperse the nanoparticles for optical measurements. Methanol was used in place of water because of water's absorbance in the IR range. The weight percentage of the UCNP cores in cyclohexane and UCNP@SiO2 in methanol was kept constant at 16 mg mL−1.
4 Results and discussion
The facile reaction of amines with 2-ICMA achieved the synthesis of urea-containing methacrylic monomers in high purity and quantitative yield (Scheme 1). However, alumina separation of inhibitors reduced the overall yield. Free radical polymerization using AIBN as an initiator yielded urea-containing methacrylic polymers. Table 1 summarizes the overall yields and polymer properties. The urea-containing methacrylic polymers are extremely brittle regardless of the Tg, presumably due to the high degree of hydrogen bonding in the polymers. All of the polymers possess a sufficiently high molecular weight for characterization of physical properties, and begin to degrade in TGA at temperatures greater than 220 °C. The Tg of the urea-containing methacrylate polymers logically follows changes in the polymer side-chain structure. The aromatic derivative has the highest Tg and increasing the alkyl side-chain length decreases the Tg, presumably due to the increasing free volume in the polymer. The high density of urea groups makes these polymers effective for complexing metal ions and nanoparticles due to the well documented urea–metal interaction.21–23 Incorporation into ABA triblock copolymers with the low Tg polymer poly(2-EHMA) increases the physical usefulness of these polymers. | ||
| Scheme 1 Synthesis of urea-containing methacrylic monomers and homopolymers. | ||
| Fed molar ratio of ArUrMA to HUrMA | Obtained ratio by 1H NMR | M n (g mol−1) | M w (g mol−1) | PDI | T g (°C) |
|---|---|---|---|---|---|
| 100:0 |
100:0 |
150000 |
171000 |
1.14 | 128 |
| 80:20 |
78:22 |
162600 |
185400 |
1.14 | 114 |
| 60:40 |
58:42 |
145900 |
166800 |
1.14 | 106 |
| 40:60 |
39:61 |
123300 |
150300 |
1.21 | 91 |
| 20:80 |
20:80 |
140600 |
174400 |
1.24 | 86 |
| 0:100 |
0:100 |
255200 |
315700 |
1.23 | 77 |
Based on the physical properties and solubility of poly(HUrMA), and the high Tg and sustainability of poly(ArUrMA) for TEM analysis, a copolymer of these monomers was targeted for use in RAFT triblock synthesis. To ensure the random polymerization behavior and miscibility of these monomers, a series of copolymers was prepared using free radical polymerization (Scheme 2). Table 2 summarizes the properties of these copolymers. The copolymers are of sufficiently high molecular weight, and the molar ratio of each monomer charged in the polymerization is very close to the molar ratio in the resultant polymer as determined by 1H NMR spectroscopy, upon comparing the integrations of HUrMA urea protons with ArUrMA urea protons. The copolymers closely follow the Fox equation:
 | (1) |
:20 HUrMA:ArUrMA copolymer composition was selected due to its relatively high Tg while retaining THF solubility for film casting.
 | ||
| Scheme 2 Synthesis of urea-containing triblock copolymers. | ||
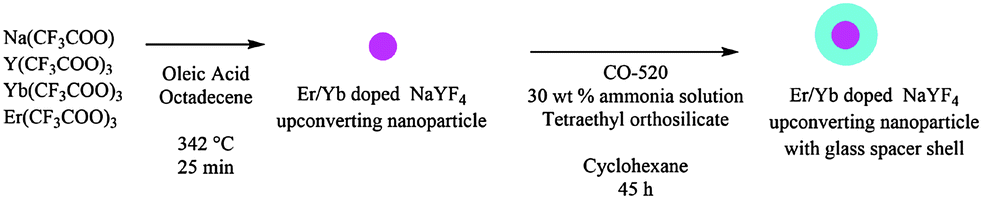 | ||
| Scheme 3 Synthesis of upconverting nanoparticles with the silica shell. | ||
| Polymers | M n (g mol−1) | wt% outer blocks | T g (°C) UrMa | T d5% (°C) |
|---|---|---|---|---|
| Free radical poly(2-EHMA) | 132100 |
— | — | 270 |
| MacroCTA | 49000 |
0 | — | 182 |
| UrMA-12 | 55900 |
12 | — | 178 |
| UrMA-41 | 82200 |
41 | 84 | 184 |
| UrMA-45 | 82900 |
45 | 67 | 184 |
| UrMA-51 | 100000 |
51 | 67 | 190 |
| UrMA-56 | 112000 |
56 | 79 | 196 |
| UrMA-73 | 179000 |
— | 73 | 201 |
| Free radical 80:20 poly(HUrMA-co-ArUrMA) |
140600 |
0 | 86 | 224 |
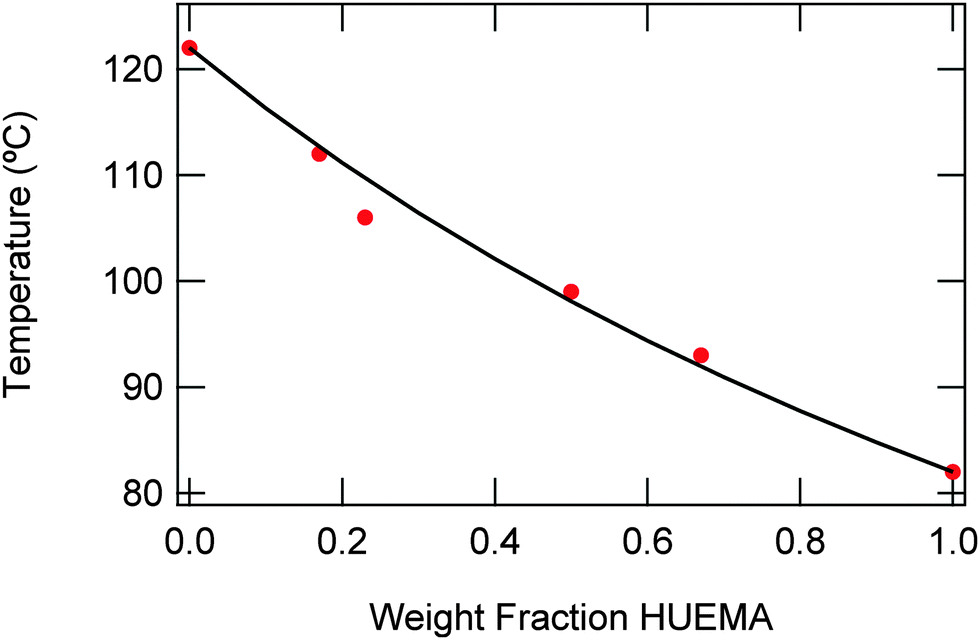 | ||
| Fig. 1 Fit of urea-containing copolymers to the Fox equation; dots indicate measured values while the line indicates the expected value from theory. | ||
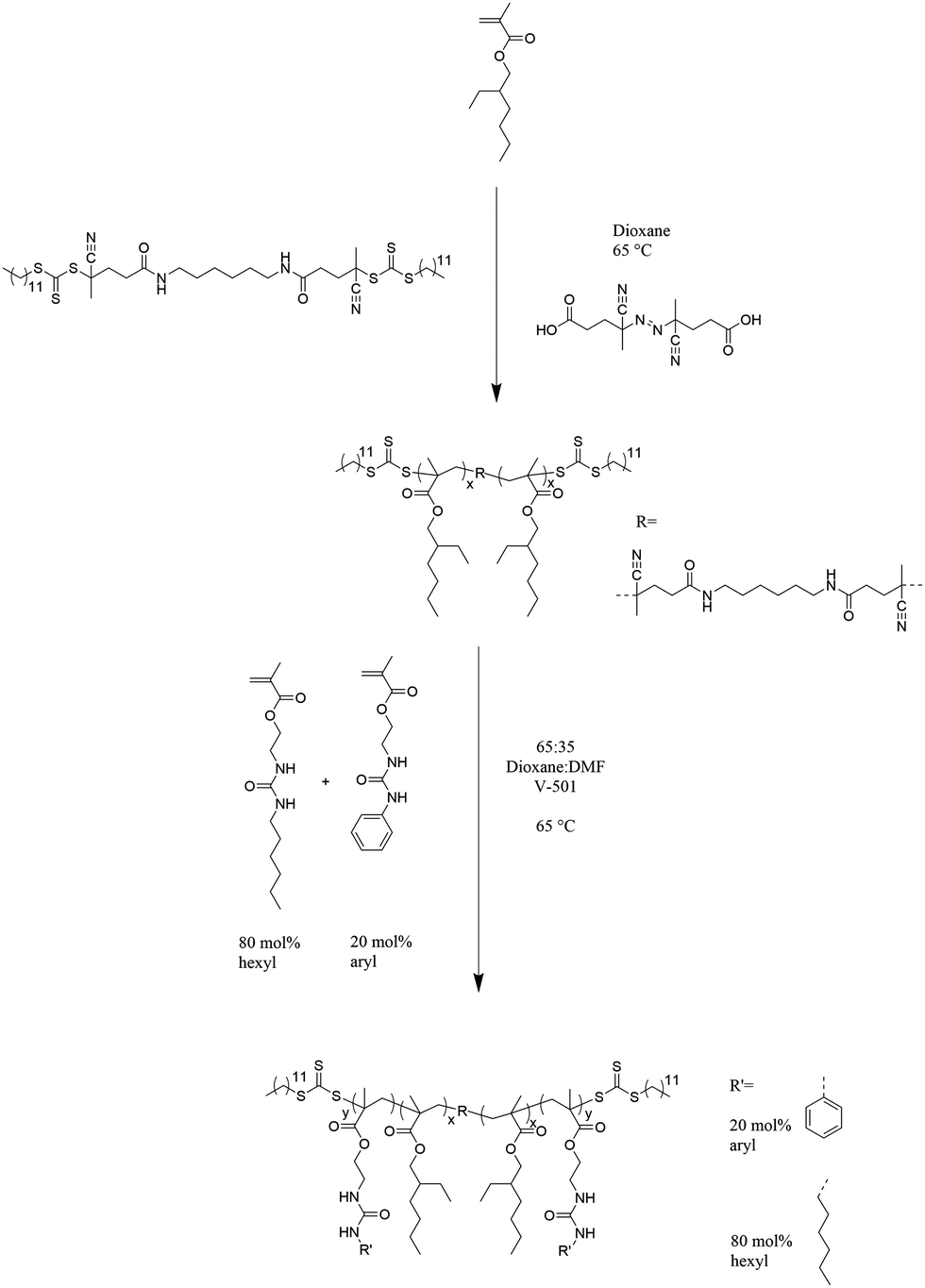
Polymerization of 2-EHMA in the presence of dCDP in dioxane using V-501 as an initiator (Scheme 2) enabled the controlled growth of difunctional poly(2-EHMA) macro CTA. This RAFT polymerization provided near linear molecular weight growth over time and acceptably low PDIs at the longer reaction times according to THF SEC and offline determined dn/dc giving absolute Mw values. Dialysis ensured complete removal of residual monomers, and purification of this polymer by precipitation from dioxane into cold methanol proves to be difficult because of the melting temperature of dioxanes. The molecular weight of 49000 g mol−1 obtained from a 10 h polymerization served as the macroCTA for subsequent triblock synthesis due to its high molecular weight and low PDI.
Chain extension of the difunctional poly(2-EHMA) macroCTA using an 80:20 molar ratio of HUrMA:ArUrMA yielded urea-containing ABA triblock copolymers as shown in Scheme 2. Because of the complex solubility of the obtained triblocks with a non-polar central block and polar outer blocks, a 65:35 dioxane:DMF (v:v%) solvent mixture was used to ensure solvation of the macroCTA and final triblocks at the polymerization temperature. Precipitation into 4:1 methanol:water (v:v%) purified the triblock copolymers. SEC could not reliably determine molecular weights because of the complex solubility. Therefore, the triblock copolymer molecular weight was determined from the known molecular weight of the macro CTA by 1H NMR spectroscopy comparing the integration of ester adjacent methylene protons (present in all repeating units) to the urea adjacent aromatic protons (present in the ArUrMA repeating units). This calculation assumed 20 mol% incorporation of ArUrMA into the outer blocks, which is a reasonable assumption from the incorporation in poly(HUrMA-co-ArUrMA) conventional free-radical copolymers (Table 2). The molecular weights are summarized in Table 1 and the polymerization behavior displays a nearly linear molecular weight growth over time, indicating good control of the reaction. However, PDIs could not be determined without SEC data. Each triblock composition retained THF solubility which was used to cast films.
DSC and TGA investigated the thermal properties of the ABA triblock copolymers and free radical controls of each block, which are summarized in Table 2. The Td5% of 270 °C of the free radical poly(2-EHMA) is significantly higher than the Td5% of the poly(2-EHMA) macroCTA at 184 °C because of the trithiocarbonate functionality that begins degrading around this temperature, greatly reducing the thermal stability of the macroCTA. Increasing the incorporation of UrMA outer blocks increases the Td5%, due to dilution of the trithiocarbonate endgroups. The Td5% of the poly(UrMA) free radical control polymer at 224 °C is significantly lower than the free radical poly(2-EHMA), likely due to the urea group reducing the thermal stability. DSC analysis reveals the presence of poly(UrMA) phases in triblocks with significant outer block incorporation, however with slightly depressed Tgs compared to the poly(UrMA) free radical control which is likely the consequence of some phase mixing during the DSC experiment. However, the Tg of poly(2-EHMA) was not observable under these DSC conditions, even in the case of poly(2-EHMA) homopolymers. The literature value of Tg of poly(2-EHMA) is about −13 °C.35 The lack of an observable transition here in DSC is likely due to the broad nature of this transition, which was further elucidated in DMA.
DMA and AFM further investigated the phase separated morphology and mechanical properties of the triblock copolymers. Although the triblocks were fairly brittle which limited their mechanical testing, the triblocks with lower incorporations of outer blocks were evaluated by DMA. DMA of annealed films revealed a more defined rubbery plateau and an increase in the rubbery plateau storage modulus with an increase in outer block incorporation, as shown in Fig. 2a. The UrMA-12 triblock copolymer did not contain enough outer blocks to form a rubbery plateau region, and its behavior resembled that of the poly(2-EHMA) free radical control, which contains a broad Tg transition around −20 °C and did not flow until 40 °C. The triblocks with a defined rubbery plateau also showed this broad poly(2-EHMA) transition around −20 °C, and flowed around 100 °C after going through the Tg of poly(UrMA). The tan delta in Fig. 2b clearly shows the nature of these transitions, with a very broad poly(2-EHMA) transition and flow after going through the Tg of poly(UrMA).
 | ||
| Fig. 2 DMA of triblock copolymers: (a) storage modulus. (b) Tan delta. Copolymers are labeled according to the wt% of outer block incorporation. | ||
AFM revealed the phase separated surface morphology of the triblock copolymers. AFM imaged both the unannealed film, as shown in Fig. 3, and the annealed film (100 °C in vacuo for 16 h) as shown in Fig. 4. In the unannealed films, the surface morphology follows a logical trend with increasing wt% of outer block incorporation. The hard domains formed by the poly(UrMA) outer blocks grow in size, starting as small spherical domains and growing into lamella-like structures lacking long-range order, and eventually forming large domains with inclusions of soft segments formed by the poly(2-EHMA) inner block. Annealing significantly changes the surface morphology, serving to coalesce the hard domains, yielding larger soft domains on the surface of the film. Although different in appearance compared to the unannealed films, the annealed films also follow a visible trend with increasing outer block incorporation. Unfortunately, limited contrast in small-angle X-ray scattering (SAXS) prevented SAXS analysis of the bulk morphology.
 | ||
| Fig. 3 AFM of unannealed triblock copolymers (copolymers are labeled according to the wt% of outer block incorporation). | ||
 | ||
| Fig. 4 AFM of annealed triblock copolymers (copolymers are labeled according to the wt% of outer block incorporation). | ||
The morphology and the particle size of the bare and silica capped NaYF4:Yb,Er nanocrystals were characterized by TEM studies. Fig. 5 shows a bright-field TEM micrograph of a typical sample. The dimensions, where r1 indicates the radius of the UCNP, r2 indicates the outer radius of the silica shell, and the shell thickness, from examination of a small (<50) sample set of UCNPs, given by r1 − r2, were determined to be between 10–15 nm.
 | ||
| Fig. 5 TEM of upconverting nanoparticles: (a) Er/Yb doped NaYF4 core. (b) Er/Yb doped NaYF4 core with the silica shell. | ||
The emission from NaYF4:Yb,Er is the result of a two photon absorption process mediated by the Yb3+ sensitizers in an energy transfer upconversion scheme.1,13 Upon the absorption of a 980 nm (1.27 eV) photon, the Yb3+ ion is elevated from the ground state to the excited state of 2F5/2. The Yb3+ ion relaxes back down to the ground state, transferring the energy of the excited state to the adjacent Er3+ ion, which, in turn, promotes the Er3+ from the ground state 4I15/2 to the 4I11/2 level. The absorption of a second photon populates the 4F7/2 level, resulting from either a second energy transfer or by the direct excitation of the Er3+ ion. The Er3+ ion can then nonradiatively relax to the 2H11/2 and 4S3/2 levels from which it radiatively relaxes, resulting in the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 green emission bands, centered at 520 and 540 nm, respectively. Alternatively, the Er3+ can further nonradiatively relax from these levels to the 4F9/2 level, ultimately producing the 4F9/2 → 4I15/2 red emission band centered at 660 nm. In addition, there is an alternative optical route that can lead to this red emission. While in the 4I11/2 state, the Er3+ ion can decay to the 4I13/2 state and are then subsequently promoted to the 4F9/2 state through an energy transfer from the excited Yb3+ ions. Once again, radiative relaxation from this level produces the 4F9/2 → 4I15/2 red emission.
Fig. 6 shows the emission spectra of bare UCNP cores in cyclohexane solution and UCNP@SiO2 dispersed in triblock co-polymers, respectively. Both spectra feature two strong emission bands at 545 and 660 nm (2H11/2 → 4I15/2 and 4F9/2 → 4I15/2), respectively, a weaker emission band at 520 nm (2H11/2 → 4I15/2), and a small artifact at 700 nm from the laser. While the inclusion of the silica shell did not alter the peak positions or spectral shapes of these bands, it did result in an increase in Rr/g (Ratio of red to green) from 0.92 for the bare cores to 1.28 for the Si coated cores, the intensity ratio between the 540 nm-centered green emission and the 680 nm-centered red emission, opposite to our expectation that the Si shell would decrease Rr/g. The main expectation from the deposition of a silica shell onto the UCNP was the depletion of proximate surface quenching centers and their associated high vibrational quanta.36 This would affect the phonon-assisted relaxations in the Er3+activator and, hence, alter the relative populations of the excited states. Due to the greater reliance of the red upconversion pathways on nonradiative relaxations, particularly in the 4I11/2 → 4I13/2 transition, the value of Rr/g was expected to decrease in this case.33,37 The increase in Rr/g we report may be due to the corpulent silica shell layer that is well beyond the thicknesses used in previous work, 5–20 nm.12,17,36 In contrast to their thinner counterparts, thick silica shells readily absorb the pumped photons as well as the emitted upconverted photons, detrimentally affecting the luminescence yield; in addition, an amorphous silica shell suffers from considerable lattice mismatch with the UCNP core, producing interface defects that give rise to additional phonon modes36 which, in turn, may selectively promote the red emission and lead to an increase in Rr/g reducing the effect of proximate surface quenching from the silica shell.
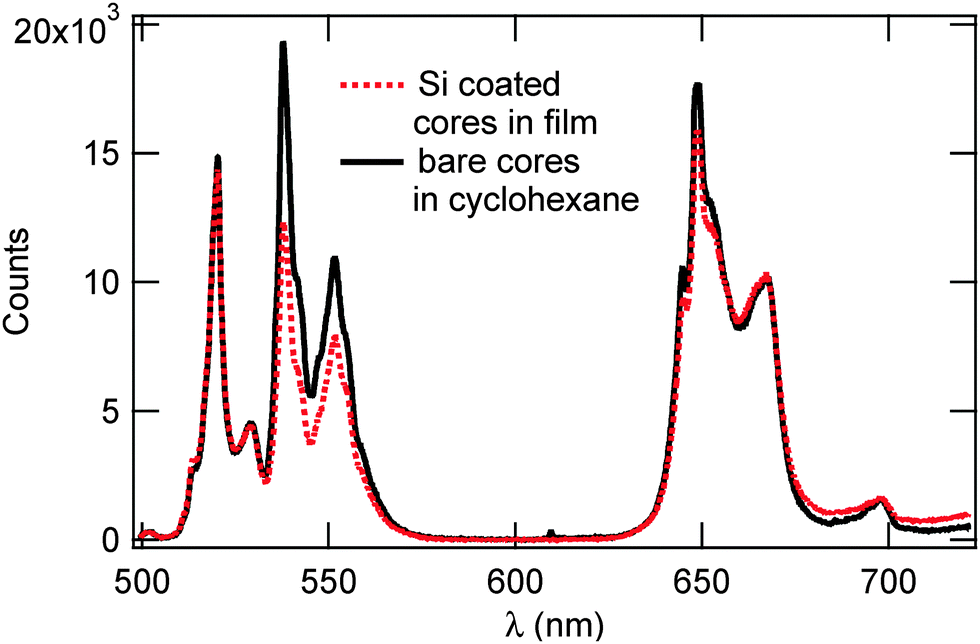 | ||
| Fig. 6 Photoluminescene spectra of bare UCNP cores in solution versus Si coated cores in tri-block polymers. Both spectra were acquired using 980 nm excitation with an average power of 300 mW. | ||
Direct comparison of the upconversion from cores in solution to cores in polymers is difficult due to the difference in transmittivity between the solution and air, the density of the cores in the solution versus in the polymer, as well as the polymer being easily focused on in comparison to the cores in solution. Thus, to evaluate the effect of the polymer on upconversion we compared the overall lineshape of the emission before and after incorporation of the cores into the polymer. This comparison is shown in Fig. 7, and we observed that the incorporation of the UCNPs into the polymer did not change the overall shape or the location of the emission peaks of the UCNP's PL. The incorporation of the cores into the polymer did cause a reduction in the intensity of two of the green emission peaks, in comparison to the red emission peaks, resulting in a Rr/g = 1.094 for the cores in triblock polymers and a decrease from 1.28 observed for Si coated cores in solvent. This suggests that the addition of the polymer resulted in an increase in the surface quenching of the green emission.
 | ||
| Fig. 7 Comparison of the optical emission properties of glass coated UCNPs in tri-block co-polymer and homo-polymer composites. For both spectra the excitation wavelength was 980 nm with the average power of 300 mW. | ||
In Fig. 7, we compare the PL spectra of UCNPs in a tri-block copolymer versus UCNPs embedded in a homo-polymer when excited with the same intensity of 980 nm light. Compared to the triblock copolymer, the Rr/g ratio decreased for the particles in the homo-polymer to an Rr/g = 0.753. The overall shape of both emissions is the same and the emission peaks are located at the same wavelengths. In the homo-polymer the highest energy green peak is also the largest, similar to the cores in the triblock polymer. The greater decrease in Rr/g, as well as the overall smaller emission intensity, observed for the cores in the homo-polymer suggests that our tri-block polymer affects the PL spectra less than the homo-polymer.
In upconverting nanoparticles the emission lifetime can be related directly to the quantum efficiency. As such, we compared the fluorescence lifetimes of our polymer–UCNP composites to lifetimes of polymer encapsulated NaYF4 UCNPs in previous studies.17,38 This allows us to evaluate the quantum efficiency of our UCNPs after incorporation into the copolymer and determine whether our triblock polymers are suitable for constructing polymer–UCNP composites.
To measure the UCNP lifetimes we modulated the laser before the microscope using an optical chopper at 2 krpm, and then the fluorescence lifetimes were found by an exponential fit of the decay of the PL signal that occurs after the laser beam was blocked using the chopper. For the green (red) decay time we used a dichroic filter to remove the red (green) portion of the PL. The decay time of the full PL as well as the decay times of the green and the red portion of the PL are shown in Table 3. The life times of our polymer embedded UCNPs are longer than similar UCNPs with surfaces modified by phosphate ligands (76 μs)17 but they are shorter by a factor of 8–10 of similar bare UCNPs in solution.38
| Samples | Lifetimes (μs) |
|---|---|
| Bare cores in the homo-polymer | 98.5 ± 8.6 |
| Silica coated cores in the tri-block copolymer | 99.6 ± 5.8 |
| Silica coated cores in the tri-block copolymer (red emission) | 112.5 ± 9.3 |
| Silica coated cores in the tri-block copolymer (green emission) | 98.5 ± 9.4 |
5 Conclusion
In summary, we have presented a study regarding the integration of upconverting NaYF4:Yb,Er nanocrystals into a uniform nanocomposite. The raw material for the composite is a urea-containing triblock copolymer. Precursors for these nanocomposites, bare UCNPs and silica capped UCNPs were synthesized and subjected to structural and spectral analysis. While both nanoparticles produced the expected photoluminescence, the addition of a thick silica capping layer resulted in the redistribution of the primary emission bands in favor of the red emission, a possible consequence of interface defects between the host matrix and the amorphous silica layer. When incorporated into polymer, the quantum efficiency of our nanoparticles, determined by the PL lifetimes, decreased by about a factor of 10 when compared to ideally formed crystals without a silica capping layer. In contrast to other examples of similar UCNPs coated with a polymer,17 our UCNPs show 34% longer PL lifetimes and as such have a higher quantum efficiency.Acknowledgements
This work was supported by NSF-Career Award DMR-0846834 and Institute of Critical Technology and Applied Sciences (ICTAS) at Virginia Tech.References
- F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed.
- D. Finlayson and B. Sinclair, Advances in Lasers and Applications, The Institute of Physics, London, 1999 Search PubMed.
- J. W. Kim, J. I. Mackenzie and W. A. Clarkson, Opt. Express, 2009, 17, 11935–11943 CrossRef CAS.
- G. Wang, Q. Peng and Y. Li, Acc. Chem. Res., 2011, 44, 322–332 CrossRef CAS PubMed.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- J. Goldschmidt, S. Fischer, H. Steinkemper, F. Hallermann, G. von Plessen, K. Kramer, D. Biner and M. Hermle, IEEE Journal of Photovoltaics, 2012, 2, 134–140 CrossRef.
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1 CrossRef CAS.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- B. E. Cohen, Nature, 2010, 467, 407–408 CrossRef CAS PubMed.
- P. Yuan, Y. H. Lee, M. K. Gnanasammandhan, Z. Guan, Y. Zhang and Q.-H. Xu, Nanoscale, 2012, 4, 5132–5137 RSC.
- A. Priyam, N. M. Idris and Y. Zhang, J. Mater. Chem., 2012, 22, 960–965 RSC.
- H. Dong, L.-D. Sun and C.-H. Yan, Nanoscale, 2013, 5, 5703–5714 RSC.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642–5643 CrossRef CAS PubMed.
- H. Schäfer, P. Ptacek, K. Kömpe and M. Haase, Chem. Mater., 2007, 19, 1396–1400 CrossRef.
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- S. Schietinger, T. Aichele, H.-Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134–138 CrossRef CAS PubMed.
- H. Na, J. S. Jeong, H. J. Chang, H. Y. Kim, K. Woo, K. Lim, K. A. Mkhoyan and H. S. Jang, Nanoscale, 2014, 6, 7461–7468 RSC.
- A. Mandal, S. Sekar, K. M. Seeni Meera, A. Mukherjee, T. P. Sastry and A. B. Mandal, Phys. Chem. Chem. Phys., 2014, 16, 20175–20183 RSC.
- S. A. Sadeek and M. S. Refat, J. Coord. Chem., 2005, 58, 1727–1734 CrossRef CAS PubMed.
- N. Nishat, S. Ahmad and R. Tansir Ahamad, J. Appl. Polym. Sci., 2006, 100, 928–936 CrossRef CAS PubMed.
- R. B. Penland, S. Mizushima, C. Curran and J. V. Quagliano, J. Am. Chem. Soc., 1957, 79, 1575–1578 CrossRef CAS.
- J.-F. Lutz, B. Sumerlin and K. Matyjaszewski, Macromol. Rapid Commun., 2014, 35, 122 CrossRef CAS PubMed.
- T. Pakula, K. Koynov, H. Boerner, J. Huang, H. il Lee, J. Pietrasik, B. Sumerlin and K. Matyjaszewski, Polymer, 2011, 52, 2576–2583 CrossRef CAS PubMed.
- S. Maria, A. S. Susha, M. Sommer, D. V. Talapin, A. L. Rogach and M. Thelakkat, Macromolecules, 2008, 41, 6081–6088 CrossRef CAS.
- K. Aslan, Z. Leonenko, J. Lakowicz and C. Geddes, J. Fluoresc., 2005, 15, 643–654 CrossRef CAS PubMed.
- M. Saboktakin, X. Ye, S. J. Oh, S.-H. Hong, A. T. Fafarman, U. K. Chettiar, N. Engheta, C. B. Murray and C. R. Kagan, ACS Nano, 2012, 6, 8758–8766 CrossRef CAS PubMed.
- P. Kannan, F. A. Rahim, X. Teng, R. Chen, H. Sun, L. Huang and D.-H. Kim, RSC Adv., 2013, 3, 7718–7721 RSC.
- J. R. Kumpfer and S. J. Rowan, ACS Macro Lett., 2012, 1, 882–887 CrossRef CAS.
- S. Padalkar, J. Capadona, S. Rowan, C. Weder, R. Moon and L. Stanciu, J. Mater. Sci., 2011, 46, 5672–5679 CrossRef CAS.
- M. H. Allen, S. T. Hemp, M. Zhang, M. Zhang, A. E. Smith, R. B. Moore and T. E. Long, Polym. Chem., 2013, 4, 2333–2341 RSC.
- X. Ye, J. E. Collins, Y. Kang, J. Chen, D. T. N. Chen, A. G. Yodh and C. B. Murray, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22430–22435 CrossRef CAS PubMed.
- M. Lejars, A. Margaillan and C. Bressy, Polym. Chem., 2014, 5, 2109–2117 RSC.
- C. L. Elkins, T. Park, M. G. McKee and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4618–4631 CrossRef CAS PubMed.
- D. Xie, H. Peng, S. Huang and F. You, J. Nanomater., 2013, 2013, 10 Search PubMed.
- H.-X. Mai, Y.-W. Zhang, L.-D. Sun and C.-H. Yan, J. Phys. Chem. C, 2007, 111, 13721–13729 CAS.
- W. Deng, L. Sudheendra, J. Zhao, J. Fu, D. Jin, I. M. Kennedy and E. M. Goldys, Nanotechnology, 2011, 22, 325604 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |