
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The band structure of graphene oxide examined using photoluminescence spectroscopy†
H. F.
Liang
*ab,
C. T. G.
Smith
b,
C. A.
Mills
bc and
S. R. P.
Silva
*b
aXi'an Technological University, BOX 277, No. 95 Jinhua North Road, Xi'an City, 710032, P. R. China. E-mail: hfliang2004@gmail.com
bAdvanced Technology Institute, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: s.silva@surrey.ac.uk
cAdvanced Coatings Group, Surface Engineering Department, Tata Steel Research Development and Technology, Swinden Technology Centre, Rotherham, S60 3AR, UK
First published on 16th November 2015
Abstract
Photoluminescence (PL) spectra have been used to elucidate the band structure of graphene oxide (GO) reduced in aqueous solution. The GO reduction is measured in situ via the identification of four PL peaks produced from GO solutions with different concentrations. Using corresponding UV-visible and photoluminescence excitation (PLE) spectroscopy, and on progressing from high energy to low energy transitions, the four PL peaks are identified as σ–σ* and π–π* transitions, a π band tail due to oxygen localized states, and a π band tail due to trapped water, respectively. The labeling of the band structure has been used to challenge the prevailing assignation of the low energy transitions, reported in the literature, to molecular σ–σ* and π–π* transitions alone.
1. Introduction
Due to its attractive electrical and optical properties,1–3 graphene oxide (GO) has potential for applications in electronics4,5 and optoelectronics,6–8 and in particular photodetectors. Special attention has been focused on GO's complex band structure but, despite this, significant differences in experiment and theoretical band structures have been reported.9–14 This has also been mirrored in the experimental band structure obtained from photoluminescence (PL), where notable differences;15–24 and even contradictory values for physical properties, such as band gap and resistivity have been reported.25,26To date, researchers have typically observed PL peaks for GO located in the blue (around 430 nm [2.88 eV]) and red regions (around 580 nm [2.14 eV]) of the visible spectrum.16–18,20 The low energy PL peak has been attributed to microstructure and defect states induced by oxygen.16,17,19–21 With reduction this peak blue-shifts and even disappears,16,20 with a new peak appearing in the blue region of the spectrum due to recovery of the sp2 carbon network.16,19–21
However, the band gap of as-prepared GO has been determined to be approx. 3.5 eV, suggesting that the corresponding PL peak should be positioned at 354 nm which is somewhat different from that previously reported experimentally.27 The GO band gap and resistivity are also found to decrease with reduction25–27 which should cause the PL peak to red-shift, opposite to the reported PL results.16,20 The reduction has been confirmed as being due to the removal of oxygen groups27 with a consequent conversion of sp3 carbons to sp2.
Here, PL experiments have been completed which allow us to form a clearer understanding of the as-produced GO band structure and the effects on the band structure during reduction. Four PL peaks, at 4.04 eV and 3.44 eV, a broad peak between 3.31 and 2.82 eV, and at 2.14 eV, have been identified for as-produced GO, assigned to σ–σ* and π–π* transitions, a π band tail due to oxygen localized states (including lone electron pair transition), and a π band tail due to trapped water, respectively. Relative intensity variations of these peaks are then examined with respect to the reduction process.
2. Experimental
2.1. Aqueous GO solution preparation
GO was synthesized from graphite using the Hummers and Offeman method.28 The as-synthesized GO was suspended in de-ionized water using a sonic bath, resulting in aqueous solutions with concentrations from 0.01 to 0.5 mg ml−1. Part of the resulting GO solution was reduced by a hydrothermal method using a water bath (95 °C) for different time periods. Alternatively, GO solutions with different pH values were produced by adding sodium hydroxide (NaOH, 0.1 mol L−1) and hydrochloric acid (HCL, 5% aqueous solution) respectively. Aqueous GO solutions (0.1 mg ml−1) were also prepared containing vitamin C (0.05 mg ml−1). After standing for 96 hours at room temperature, the solution color changed from brown to black, and sediment could be observed at the bottom of the bottle. Aliquots of the supernatant were then decanted for PL measurement.For comparison, GO power was directly reduced by vacuum thermal annealing at temperatures of 50 and 200 °C, respectively. The reduced graphene oxide was then suspended in de-ionized water for PL testing.
2.2. GO characterization
Ultraviolet-visible (UV-vis) absorption spectra (185–1200 nm [1.03–6.70 eV], 0.5 nm resolution) were acquired using UV-vis spectroscopy (Varian Cary 5000 UV-vis-NIR absorption spectrometer, Agilent Technologies, USA), utilizing quartz cuvettes with optical path lengths of 10 mm. PL and Photoluminescence excitation (PLE) spectra were obtained at different excitation/measurement wavelengths (1.0 nm resolution) using a spectrophotometer (Varian Cary Eclipse Fluorescence Spectrophotometer, Agilent, USA), where a xenon lamp was used to excite the GO sample and a red-sensitive photomultiplier tube was used for detection. The excitation and emission slit widths were both 10 nm, and the detector applied voltage was 700 V.XPS (X-ray photoelectron spectroscopy, K-Alpha, Thermo Fisher Scientific) was undertaken using Aluminium Kα X-rays, with beam energy, beam spot size and beam current density of 1486.60 eV, 400 μm and 4.778 A cm−2, respectively, resulting in a spectral resolution of approx. 0.5 eV. The XPS data were collected under scanning mode and analyzed in Thermo Scientific Advantage 4.51 software, respectively. A peak fitting process was performed where the linear background was first deducted from the original XPS data, after which the spectra were aligned using the C–C bond energy (284.6 eV), and then fitted using a Gauss peak.
Thermogravimetric analysis (TGA) was carried out under an ambient air environment at temperatures from ambient to 900 °C, with 5 °C measurement intervals (Q500 TGA, TA Instruments, USA). Raman spectra (Renishaw Systems 2000 Raman microscope) were recorded using a green laser (514 nm, 30 mW) at 2 cm−1 resolution.
3. Results and discussion
3.1. PL, PLE and absorption spectra
Fig. 1 shows the PL (5.39 eV excitation energy) and PLE (3.18 eV measurement energy) spectra for as-prepared GO solution, which are superimposed with the corresponding absorption spectrum. Four PL peaks can be observed at 4.04 and 3.44 eV, a broad peak between 3.31 and 2.82 eV (including a shoulder at 2.95 eV), and at 2.14 eV. Excitation energy-dependent PL experiments (ESI,† Fig. S1 and S2) also confirm the presence of these four peaks. Two corresponding peaks can be seen in the PLE spectrum, a shoulder peak at 5.39, and 4.20 eV. A third peak may be inferred by the increasing slope of the curve up to 6.20 eV. The fourth peak could not be seen due to a very low PL efficiency. The peak observed at 3.61 eV is the Raman peak for water. The absorption curve also reveals three peaks in similar positions to those found in the PLE spectrum. Finally, a long absorption tail from 3.88 eV to low energies can be observed. The PL spectrum is divided into two sections corresponding to PL measurement using different GO solution concentrations – below 2.85 eV using 0.03 mg ml−1 solution, and above 2.85 eV nm using 0.3 mg ml−1 solution respectively. The graph shows the recorded data with no normalization required to align the intensity of the plots recorded above and below 2.85 eV. | ||
| Fig. 1 PL (black triangles, excitation energy = 5.39 eV), PLE (red squares, measurement energy = 3.18 eV) and absorption spectrum (blue circles) of as-prepared GO from aqueous solution. GO solutions of two different concentrations (0.03 and 0.3 mg ml−1) were examined to obtain PL peaks in different regions of the ultraviolet/visible spectrum, below and above 2.85 eV respectively. Absorption and PLE were acquired using 0.03 mg ml−1 solution. | ||
Generally, previous work has only reported two PL peaks, whereas here we report on four peaks. Fig. 2 shows the variation of the fourth PL peak (2.14 eV) with increasing solution concentration. The peak is seen to effectively disappear for GO solution concentrations below 0.03 mg ml−1. At higher concentrations, the peak at 1 mg ml−1 can be seen to be lower intensity than that of the 0.5 and 0.25 mg ml−1 concentrations, and displays a slight red-shift. This suggests that the optimum concentration for observing the 2.14 eV peak in PL is about 0.5 mg ml−1, which gives a good balance between the appropriate absorption for excitation and scattering for emission.
 | ||
| Fig. 2 PL intensity (excitation energy = 3.444 eV) in the red region of the visible spectrum for different aqueous GO solution concentrations (0.015–1.0 mg ml−1). | ||
However, it is impossible to observe the other PL peaks (at 4.04 eV, 3.44 eV, and a broad peak between 3.31 eV and 2.82 eV) using this high concentration solution. Typically, to observe these high energy peaks, the concentration must be decreased (Fig. 3), and an effective PL signal can usually be achieved by tuning the solution absorbance excitation wavelength to approx. 0.1 a.u. Similarly, as-produced GO displays an increase in absorption with wavelength, as shown in Fig. 4. Thus, in order to resolve all the peaks in the PL spectrum, the different solution concentrations were applied. Previously, only the two low energy PL peaks were observed due to the use of higher solution concentrations.20,21,24
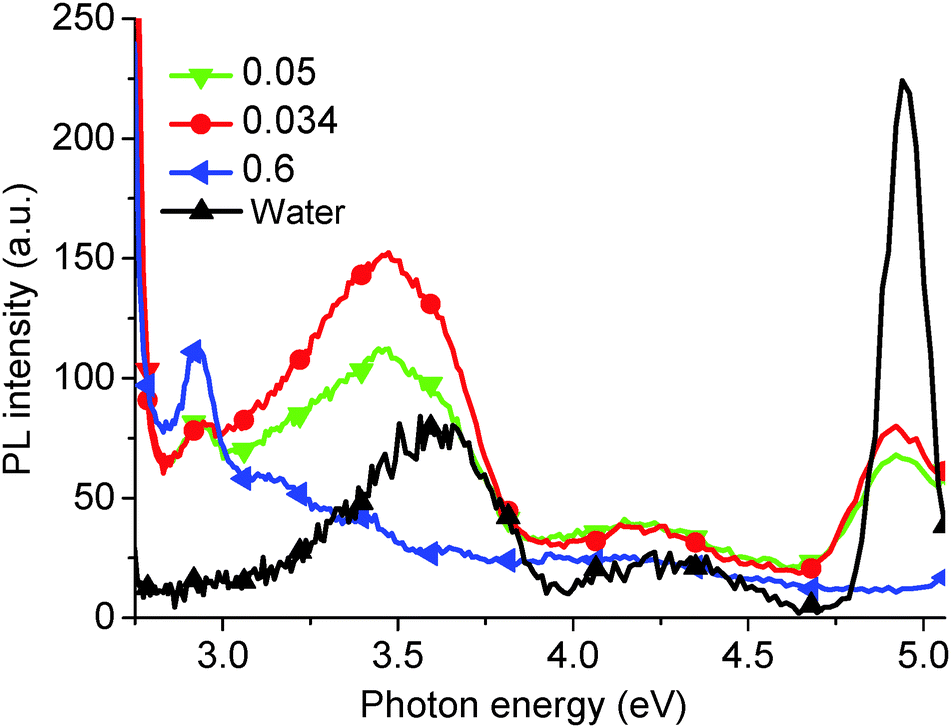 | ||
| Fig. 3 PL spectra (excitation energy = 5.39 eV) for increasing GO solution concentrations (0.034, 0.05, 0.6 mg ml−1) after hydrothermal reduction for 5 min. For comparison, the PL spectrum of water is given. | ||
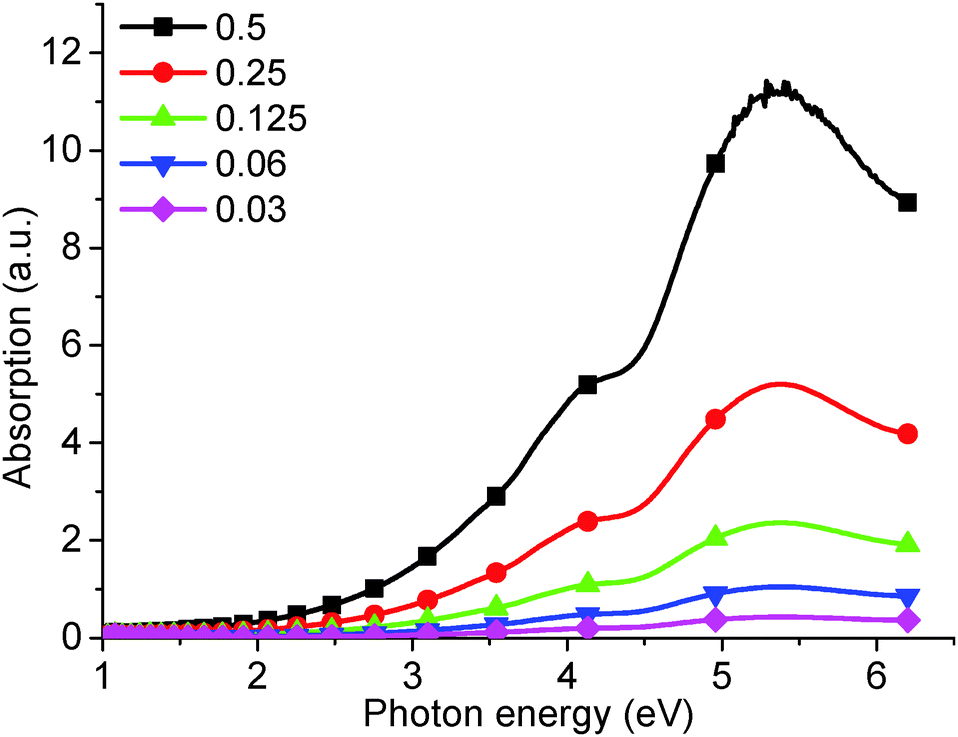 | ||
| Fig. 4 UV-visible absorption of as-produced GO from increasingly concentrated aqueous solution: 0.03, 0.06, 0.125, 0.25, 0.5 mg ml−1. | ||
The absorption peak at 5.39 eV (Fig. 4) has been confirmed as being due to the π–π* band transition.19 The calculated band gap, taken from the slope of the absorption curve, is about 3.5 eV.27 This is in close agreement with the 3.44 eV PL peak observed in Fig. 1, which is consequently assigned to the π–π* band transition.
The UV-visible peak at 6.53 eV (Fig. 1) is reported for the first time in our experiments and is determined to be due to the σ–σ* transition, similar to that seen in diamond-like carbon/amorphous carbon films.29,30 Attaching oxygen to graphene will convert a planar sp2 carbon to sp3,31,32 and produces a corresponding absorption peak at higher energy than the π–π* band transition. The PL peak at 4.31 eV (Fig. 1), at a higher energy than the corresponding π–π* transition (3.44 eV), was therefore assigned to a σ–σ* transition. This has been fully discussed using multiple analytical methods on α-C thin films.33
The absorption shoulder at 4.20 eV (Fig. 4), corresponding to the 2.95 eV PL peak (Fig. 1), is determined to be due to a transition from the oxygen lone electron pair to the carbon π band.34 Finally, the long absorption tail was assigned to the presence of localized states on the oxygen group and trapped water,19,34 and corresponds to the broad PL peak between 3.31 eV and 2.82 eV and the peak at 2.14 eV, respectively. We show that the PL peak, at 2.14 eV, decreases correspondingly as trapped water is removed; similarly, the broad peak decreases concomitant with the removal of the oxygen. A new peak produced at 2.82 eV after an extensive reduction is shown to be ascribed to states induced by the recovery of micro-sized sp2 carbon sites.
Consequently, comparing the three curves (PL, PLE and absorption), we confirm that the PL peaks at 4.04 eV, 3.44 eV, the broad peak between 3.31 and 2.82 eV including a 2.95 eV peak, and the 2.14 eV peak are due to σ–σ* and π–π* transitions, a π band tail due to oxygen localized states (including long electron pair transition), and a π band tail due to trapped water, respectively. A summary of peak assignations is given in Table 1.
| Peak position (eV) | Transition | ||
|---|---|---|---|
| PL | PLE | UV-visible | |
| 4.05 | (6.20) | 6.53 | σ–σ* |
| 3.44 | 5.39 | 5.39 | π–π* |
| Broad peak between 3.31 and 2.82 eV including the 2.95 eV peak | 4.20 | Long absorption tail (including the shoulder at 4.20 eV) | Oxygen localized states including lone electron pair-π band |
| 2.39 | None | Long absorption tail | Trapped water localized states |
3.2. Hydrothermal reduction
In order to support our assignations, PL measurements were carried out on GO solutions lightly reduced using a hydrothermal reduction method (Fig. 5). At high energies (Fig. 5a), the peak at 4.04 eV increased up to 120 min reduction time, and then decreased at 240 min. The peak at 3.44 eV reached a maximum after 5 min annealing, then decreased, and finally, the broad peak between 3.31 and 2.82 eV decreased after reduction. At lower energies (Fig. 5b), the peak at 2.39 eV decreased with annealing time but did not display any blue shift, observed previously in the literature.16,19,20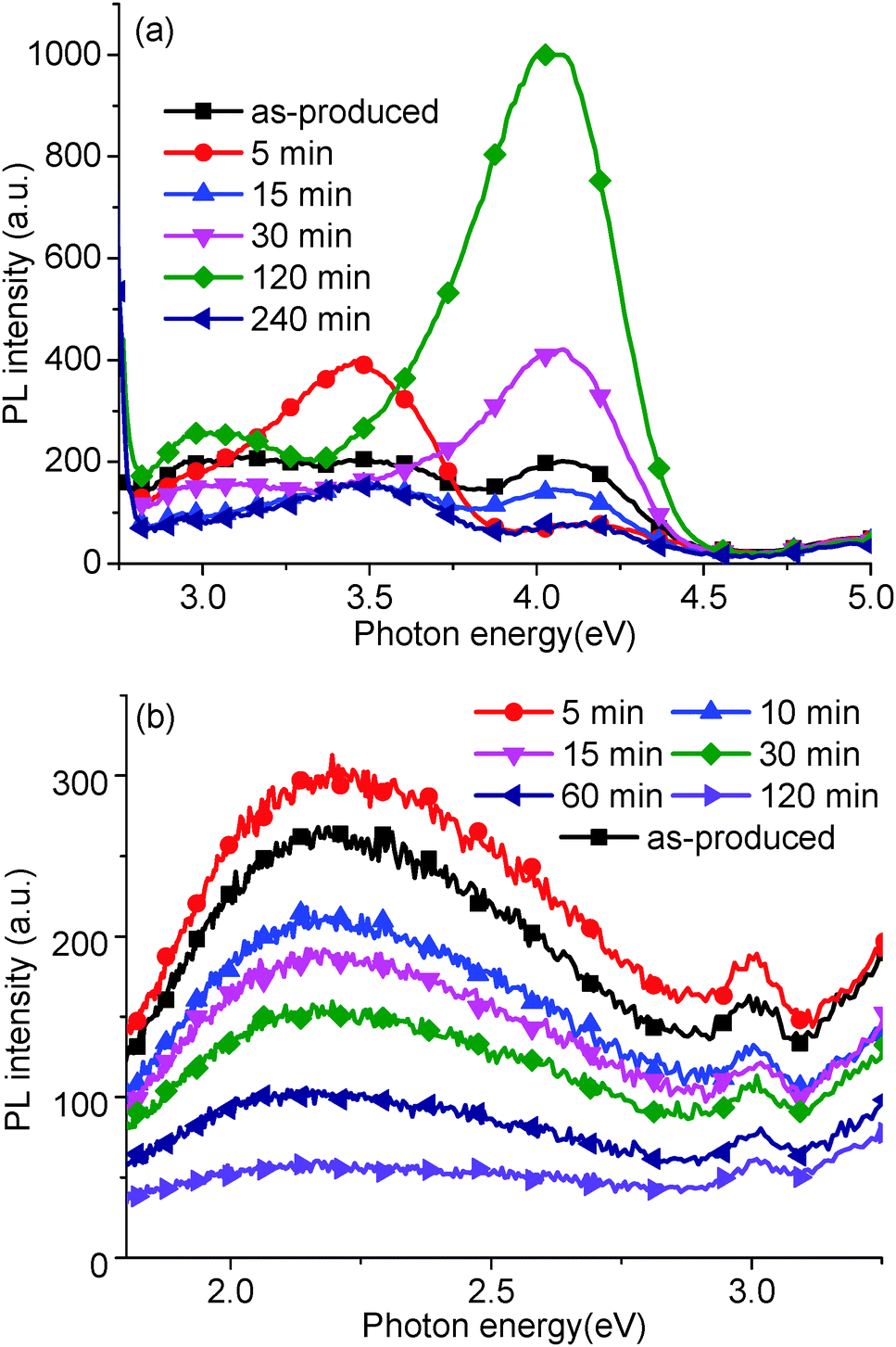 | ||
| Fig. 5 PL spectrum, recorded using (a) 5.39 eV and (b) 3.44 eV excitation energies, of as-produced GO and after hydrothermal reduction at 95 °C with different processing times: 5, 10, 15, 30, 60, 120 and 240 min. | ||
These peak variations relate to the relative sp2 and sp3 carbon hybridization induced by the removal of oxygen groups and trapped water. With gentle reduction, the oxygen groups have been removed gradually, causing a concurrent increase in sp2 carbon content. Thus, the density of states would decrease in the σ band in comparison to the increase in the π band states. Based on this, the peak at 4.04 eV, corresponding to the σ–σ transition, should decrease with GO reduction. In fact, in our experiments, the peak at 4.04 eV initially increases (up to 120 min). To explain this, we assume that water trapped in the GO structure initially plays a key part in the structure determined by PL.
At 95 °C, trapped water would slowly be removed from the GO structure (Section 3.4 addresses this point further), simultaneously the peaks at 4.04 eV and 2.14 eV increase and rapidly decrease respectively. Thus, trapped water could quench PL in the ultraviolet and contribute to the PL peak at 2.14 eV. Previous work has suggested that water can be trapped by hydrogen bonding to hydroxyl and epoxide groups, which will affect the C–C bond angle and the length of C–C base plane.32,35 A possible reason for the quenching of the 4.04 eV peak is that the hydrogen bonding between the trapped water and a GO oxygen group could change the bond angle and length of sp3 carbons on the GO. After 120 min, the peaks at 4.04 eV, 3.44 eV, and the broad peak between 3.31 and 2.82 eV all decreased, resulting in the recovery of sp2 carbons on the reduced GO, and indicated by the dark colour of the GO solution.
The corresponding UV-visible absorption curves (Fig. 6) produced results in agreement with the PL measurements. The absorption peaks at 6.53 and 5.39 eV for the reduced GO produced a similar trend in intensity to that of the 4.04 and 3.44 eV PL peaks, respectively. The absorption shoulder at 4.20 eV disappears at reduction times greater than 120 min, which corresponds to the decreasing PL shoulder at 2.95 eV (see Fig. 5). In addition, the slight red-shift of the 5.39 eV peak and increasing long absorption tail indicate the recovery of sp2 carbon moieties and the removal of oxygen groups. However, the PL peak at 3.44 eV did not increase in intensity, which was in agreement with published results,15,19 possibly due to a low PL efficiency of reduced GO.
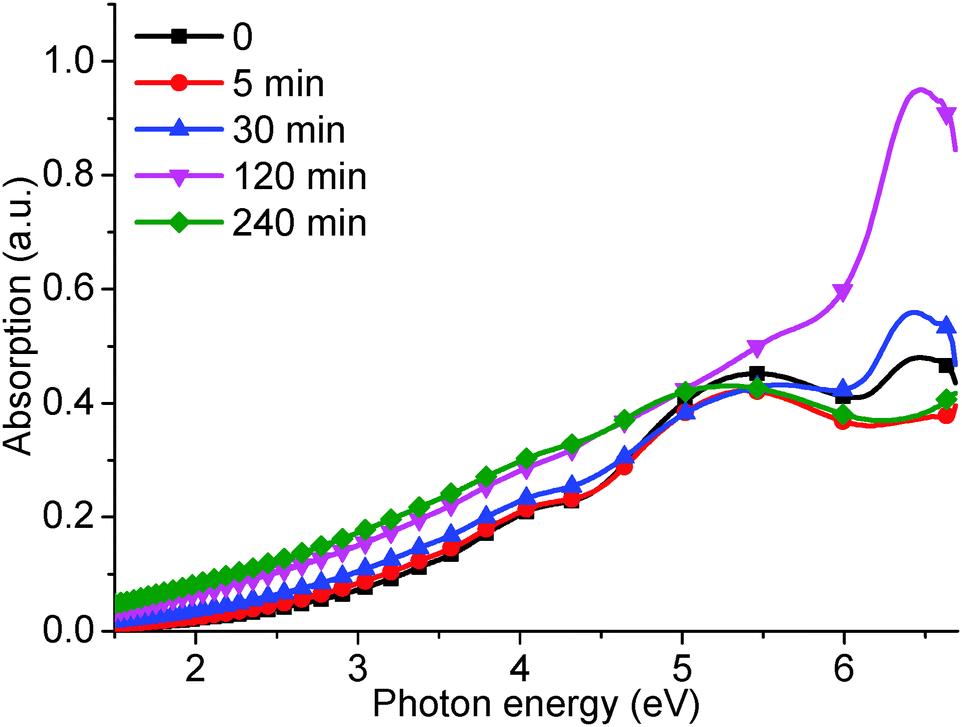 | ||
| Fig. 6 UV-visible absorption curves for as-produced GO and subjected to different hydrothermal reduction times: 5, 30, 120, and 240 min. | ||
3.3. pH dependent reduction
In order to further support our results, pH-dependent PL experiments were also performed (Fig. 7) which provides another effective method for tuning the GO structure.23,24 Previous work has shown that adding base (NaOH) quenches PL in the red region of the visible spectrum.23,24 Our results corroborate this effect (Fig. 7b), but we also show that the intensities of the high energy 4.04 and 3.44 eV PL peaks are enhanced upon increasing the pH using NaOH (Fig. 7a). | ||
| Fig. 7 (a) PL spectra of GO (excitation wavelength = 5.39 eV) as a function of pH at pH 1, 4, 9, 11, and 13, and (b) PL spectra of GO (excitation wavelength = 3.44 eV) as a function of pH, when subjected to acid (blue diamonds) and base (purple down triangles) conditions, compared to pristine GO (black circles). | ||
Obviously, in this case the structure of the GO has been altered, with –OH and –COOH rich GO produced after adding alkali and acid, respectively.24 Adding NaOH provided abundant –OH groups which attack the C–C base plane and open C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds to produce C–O bonds, which consequently affects the carbon bond angle and length.36 This results in the conversion of sp2 carbon atoms to sp3,31 causing an increase in the 4.04 eV peak. The addition of the dissociated Na+ could potentially dehydrate the GO, removing the trapped water and causing the reduction of the 2.14 eV peak.
C bonds to produce C–O bonds, which consequently affects the carbon bond angle and length.36 This results in the conversion of sp2 carbon atoms to sp3,31 causing an increase in the 4.04 eV peak. The addition of the dissociated Na+ could potentially dehydrate the GO, removing the trapped water and causing the reduction of the 2.14 eV peak.
Inversely, adding acid removes existing hydroxyl (–OH) groups attached to the GO carbon base plane, resulting in the production of sp2 carbon atoms from their sp3 analogues and the subsequent decrease in the 4.04 eV peak. Alteration of the GO structure in this manner produces PL spectra similar to those associated with the hydrothermal reduction process above, and further suggests that the PL peaks at different wavelengths are due to the corresponding assigned transitions.
3.4. Evidence for trapped water in the GO structure
Experiments were undertaken to attempt to prove that the 2.14 eV PL peak was correctly assigned with respect to water trapped in the GO structure. As-synthesized GO powder was annealed in a vacuum chamber (50 °C for 24 h), and then suspended into de-ionized water for PL testing. Fig. 8 shows the PL spectra, excited at a wavelength of 3.67 eV, for a sample annealed at 50 °C compared to PL spectra of as-produced GO and GO which is reduced at 200 °C. When compared to the PL spectra of as-produced GO, the 2.14 eV peak for GO annealed at 50 °C disappears. This peak can be attributed to adsorbed water, because at such a low temperature it is unlikely that chemical changes occur in the structure of the GO and that only trapped water is slowly removed. Upon increasing the annealing temperature to 200 °C, a valley appears in the PL spectrum at 2.88 eV due to the removal of oxygen groups from the GO surface (Fig. 8). A corresponding PL peak (2.88 eV) is observed from the GO solution (Fig. 1). Moreover, the disappearance of the valley at 2.88 eV (Fig. 8) is in agreement with that of our aqueous GO solution (Fig. 1), and additional PL and PLE spectra of the water alone, measured at different energies, demonstrates a red shift in the water Raman peak (ESI,† Fig. S3 and S4).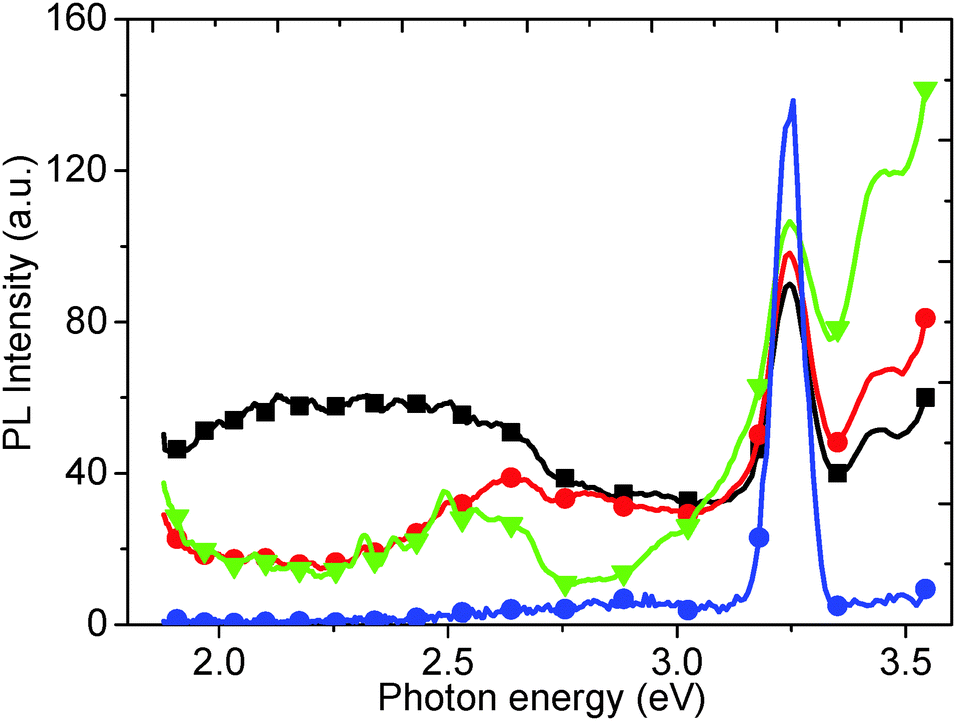 | ||
| Fig. 8 PL spectra of as-produced GO (black squares), and after thermal annealing at 50 °C (red circles) and 200 °C (green down triangles). The PL spectrum for water (blue circles) is also given for comparison. | ||
Removal of trapped water and oxygen functionalities could also be observed by examining the GO samples by TGA (Fig. 9), where four weight loss peaks can be observed at 60, 190, 225 and 600 °C. These peaks were attributed to loss of trapped water, easily removed hydroxyl groups, epoxide removal and less easily removed hydroxyl, and C–C base plane decomposition, respectively.37 The first peak, at 60 °C, decreased after annealing at 50 °C for 24 h (Fig. 9a), due to the loss of most of the trapped water. The first two peaks (60 and 190 °C) were completely removed when annealing at 200 °C (Fig. 9c), which suggests that part of the hydroxyl functionalities are removed along with the trapped water.37 Further increasing the annealing temperature to 250 °C removes more oxygen functionalities, as indicated by the disappearance of the third peak at 225 °C (Fig. 9d). Comparing the differences in the PL spectra in Fig. 8 with the changes observed in the TGA data (Fig. 9), we can confirm that the 2.14 eV peak is related to the presence of trapped water.
 | ||
| Fig. 9 TGA of (a) as-produced GO (initial weight: 13.18 mg), and after reduction at (b) 50 °C (12.47 mg), (c) 200 °C (12.53 mg), and (d) 250 °C (3.11 mg): normalized weight (solid black lines), derivative weight (dashed red lines). | ||
The TGA data is supported by XPS spectra (Fig. 10) which show the relative intensities of the C–C, C–O and CO peaks of the GO as it is reduced. The as-produced GO has a peak intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.76:0.18 for the fitted C–C, C–O, and CO peaks respectively. After gentle reduction at 50 °C, the ratio changes to 1:0.53:0.18 with a noticeable reduction in the C–O peak. Further reduction at 200 °C and 250 °C gives ratios of 1:0.23:0.17 and 1:0.24:0.13 respectively, suggesting a reduction in oxygen moieties on the GO surface and a subsequent increase in sp2 character in the GO.
:0.76:0.18 for the fitted C–C, C–O, and CO peaks respectively. After gentle reduction at 50 °C, the ratio changes to 1:0.53:0.18 with a noticeable reduction in the C–O peak. Further reduction at 200 °C and 250 °C gives ratios of 1:0.23:0.17 and 1:0.24:0.13 respectively, suggesting a reduction in oxygen moieties on the GO surface and a subsequent increase in sp2 character in the GO.
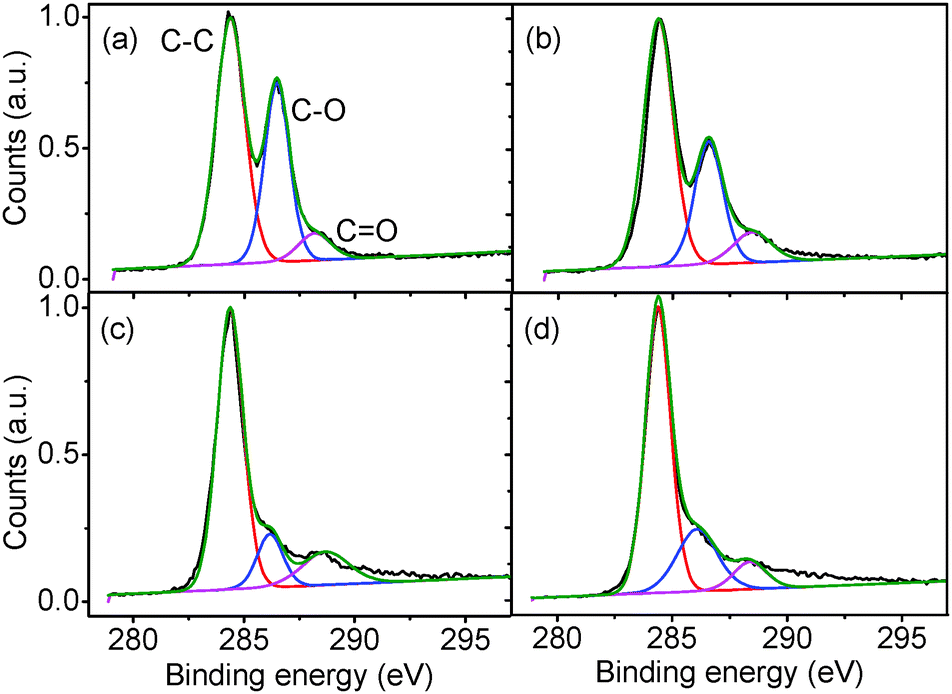 | ||
| Fig. 10 XPS spectra of (a) as-produced GO, and after thermal reduction under (b) 50, (c) 200, and (d) 250 °C: raw data (black line), composite origin fit to spectrum (green line), and individual fits to C–C (red line), C–O (blue line) and CO (purple line) peaks. | ||
3.5. Effect on GO oxygen moieties
The PLE peak (Fig. 1), and corresponding absorption shoulder (Fig. 4), at 4.02 eV disappear when the GO is hydrothermally reduced for 240 min (Fig. 11), corresponding to a disappearance of the PL shoulder at 2.95 eV (Fig. 3 and 5a). Water, trapped in the GO structure, hydrogen bonds to hydroxyl and epoxide defects on the GO,35 which changes the lone electron pair density associated with the GO. With reduction, the trapped water is removed, releasing the lone electron pairs on the GO defect oxygen groups. However, as interstitial water molecules are removed, these defects, present on the surface of the GO molecules, can interact with neighboring GO molecules causing a further redistribution of the oxygen lone pairs. This causes the reduction of the corresponding absorption and PL/PLE peaks to be non-linear, unlike the reduction of the 2.14 eV PL peak, due to the loss of the water molecules. However, with extensive reduction, for 240 min, the PLE peak at 4.20 eV disappears due to the removal of the oxygen groups from the GO surface. In comparison, excess NaOH provides abundant oxygen lone pair electrons from the additional –OH groups, which enhances the PL shoulder at 2.95 eV, as shown in Fig. 7a. | ||
| Fig. 11 PLE intensity (3.18 eV emission wavelength and 0.03 mg ml−1 GO concentration) as a function of hydrothermal reduction time. | ||
3.6. Ascorbic acid reduction
Finally, we have measured the PL of GO reduced using vitamin C (ascorbic acid). Traditionally, GO has been reduced using hydrazine hydrate however, due to the concentration of functional groups on GO that can be reduced, there is a wide variety of possible reducing agents. These range from strong, classical reducing agents, such as sodium borohydride (NaBH4) and lithium aluminium hydride (LiAlH4), to more exotic materials such as green tea leaf extracts, and microorganisms such as E. coli. One of the most interesting reducing agents is Ascorbic acid, also known as vitamin C, as it can reduce to a similar extent to hydrazine, without the inherent toxicity.38 The ascorbic acid is dehydrated upon reduction of the GO,38 but there are questions as to whether the ascorbic acid is consumed, or is then regenerated and participates catalytically in further GO reduction.After reduction with ascorbic acid, the 2.14 eV peak disappears and, in this case, a new peak is formed at 2.82 eV, potentially suggesting that the PL peak shifts from red to blue (Fig. 12). However, we attribute the 2.82 eV peak after reduction to new states, induced by macro sp2 carbon sheet formation during reduction, due to recovery of the sp2 carbon network, rather than from a blue shift of the 2.14 eV peak.21 The shoulder at 2.95 eV is attributed to the water solvent.
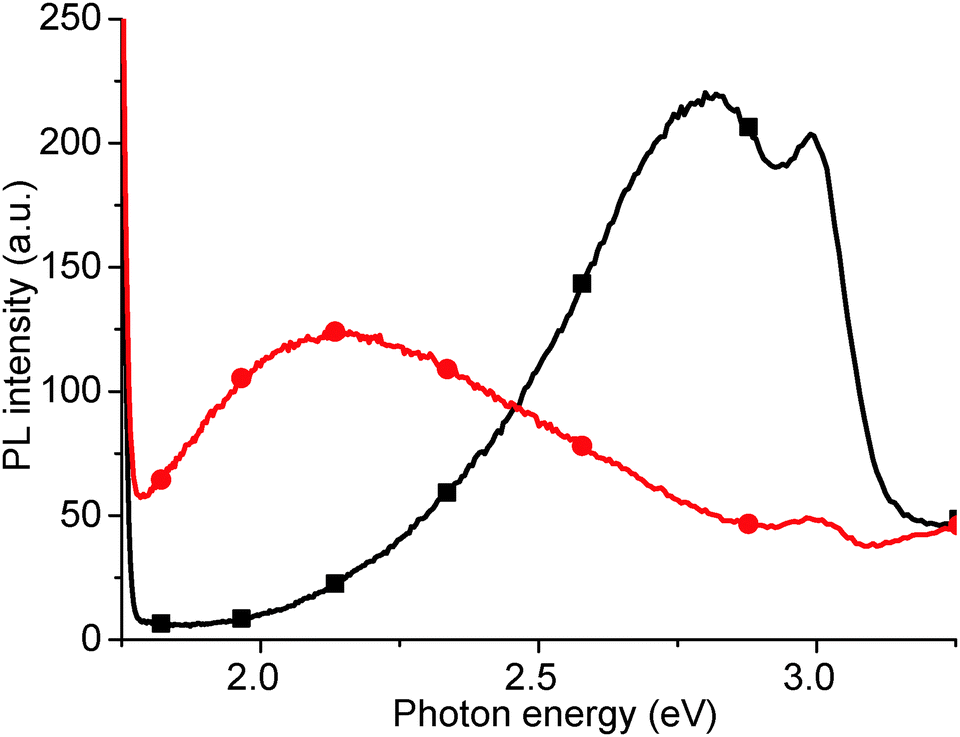 | ||
| Fig. 12 PL spectrum of as-produced GO (red circles), and after room temperature reduction in ascorbic acid solution (black squares). | ||
3.7. Summary
Based on our PL experiments, a complete band structure for the hydrothermal reduction of GO can be postulated (Fig. 13), containing four bands, σ, π, band 1 and band 2 (Fig. 13a), corresponding to PL peaks at 4.04 eV, 3.44 eV, the broad peak between 3.31 and 2.82 eV, and the 2.14 eV peak, respectively, and which are produced by sp3 carbon, sp2 carbon, oxygen states (including lone electron pairs) and trapped water states in π band tail, respectively. Initially during reduction, trapped water is removed, causing the 2.14 eV peak to disappear and the 4.04 and 3.44 eV peaks to change intensity due to variation in the sp3 hybridization content (Fig. 13b). After further reduction, the density of states in the σ band decreases, and the density of states in the π band increases and shift to lower energy (Fig. 13c). A new band tail due to the formation of macro size sheets of sp2 carbon is formed which contributes to the PL peak seen in the ascorbic acid reduction at 2.82 eV.21Fig. 13 also gives illustrative diagrammatic representations of the GO structure as the reduction progresses initially by losing interstitial water molecules (a and b) and then through the reduction of GO to rGO and the reclamation of macro sized sp2 carbon sheets (b and c). | ||
| Fig. 13 Diagrammatic representations of the evolution of the chemical structure and band structure of GO during reduction: (a) as-produced GO with water intercalated GO sheets and high oxygen functionality, (b) GO with reduced intercalated water and oxygen functionality after gentle reduction and (c) reduced GO with minimal oxygen functionality and an increased sp2 character produced under stronger reducing conditions. The chemical structure diagrams show carbon (grey), oxygen (red) and hydrogen (white) atoms, either as part of the GO or as H2O molecules. For comparison in each case, the relevant PL plots are reproduced: (a) as-produced GO (from Fig. 1), (b) hydrothermally reduced at 95 °C for increasing time increments (from Fig. 5b), and (c) hydrothermally reduced at 95 °C for 120 min (from Fig. 5a and b). Included is the experimental scale on the x-axis to give an idea of the positions of the bands in the diagrammatic representation (DoS = density of states). | ||
To support the analysis of the conformational changes occurring, complementary Raman spectra of the thermally reduced GO materials have been completed (ESI,† Fig. S5). With the reduction of the GO, the PL background in the Raman spectra is decreased, which is consistent with our results showing similar PL reduction in the visible spectral range (Fig. 5b and 8). The D/G peak ratios also show a slight increase from 0.846 for as-produced GO to 0.868 for rGO reduced at 200 °C, suggesting an increase in the sp2 character of the rGO.
4. Conclusions
The current explanation of the reduction of GO, typically based on using two PL peaks, does not provide all the information required to explain the typical GO band gap (3.5 eV) or the apparent blue shift of the low energy (2.14 eV) peak. Detailed PL experiments have been completed to understand the band structure of GO, based on the analysis of four PL peaks at 4.04 eV, 3.44 eV, a broad peak between 3.31 and 2.82 eV, and a 2.14 eV peak, explained as being due to transitions of σ–σ*, π–π*, an oxygen states (including the lone electron pair) and a trapped water states in π band tail, respectively. The relative intensity variation of these peaks was studied by increasing the extent of the hydrothermal reduction of the GO, and the PL measurements were supported by UV-visible and Raman spectroscopy, and TGA analysis. Based on our PL results, a complete band structure model has been postulated which outlines the changes in the density of states of the individual bands.Acknowledgements
The authors would like to thank Violeta Doukova, Chris Buxley and John Underwood (all University of Surrey) for assistance in the production of TGA and spectroscopy data, and for technical assistance, respectively. We also acknowledge support from the Chinese Scholarship Council (grant number 201208610014), the Shannxi Youth Science and Technology Funding (grant number 2014KJXX-71), and the European Union 7th Framework Program project SMARTONICS (grant agreement number 310229).Notes and references
- Y. Cao, J. Zhu, J. Xu, J. He, J.-L. Sun, Y. Wang and Z. Zhao, Small, 2014, 10, 2345–2351 CrossRef CAS PubMed.
- H. Chang, Z. Sun, Q. Yuan, F. Ding, X. Tao, F. Yan and Z. Zheng, Adv. Mater., 2010, 22, 4872–4876 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- S. Saxena, T. A. Tyson, S. Shukla, E. Negusse, H. Chen and J. Bai, Appl. Phys. Lett., 2011, 99, 013104 CrossRef.
- P. Zhu, E. Cruz-Silva and V. Meunier, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 085427 CrossRef.
- K. D. G. I. Jayawardena, R. Rhodes, K. K. Gandhi, M. R. R. Prabhath, G. D. M. R. Dabera, M. J. Beliatis, L. J. Rozanski, S. J. Henley and S. R. P. Silva, J. Mater. Chem. A, 2013, 1, 9922–9927 CAS.
- S. Shi, V. Sadhu, R. Moubah, G. Schmerber, Q. Bao and S. R. P. Silva, J. Mater. Chem. C, 2013, 1, 1708–1712 RSC.
- C. T. G. Smith, R. W. Rhodes, M. J. Beliatis, K. D. G. I. Jayawardena, L. J. Rozanski, C. A. Mills and S. R. P. Silva, Appl. Phys. Lett., 2014, 105, 073304 CrossRef.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS PubMed.
- H. Huang, Z. Li, J. She and W. Wang, J. Appl. Phys., 2012, 111, 054317 CrossRef.
- A. Hunt, D. A. Dikin, E. Z. Kurmaev, Y. H. Lee, N. V. Luan, G. S. Chang and A. Moewes, Carbon, 2014, 66, 539–546 CrossRef CAS.
- Y. Luo, K.-Y. Lian, Y.-F. Ji, X.-F. Li, M.-X. Jin and D.-J. Ding, J. Phys. Chem. C, 2013, 117, 6049–6054 Search PubMed.
- S. Zhang, J. Zhou, Q. Wang and P. Jena, J. Phys. Chem. C, 2013, 117, 1064–1070 CAS.
- T. Bansal, A. D. Mohite, H. M. Shah, C. Galande, A. Srivastava, J. B. Jasinski, P. M. Ajayan and B. W. Alphenaar, Carbon, 2012, 50, 808–814 CrossRef CAS.
- Z. Luo, P. M. Vora, E. J. Mele, A. T. C. Johnson and J. M. Kikkawa, Appl. Phys. Lett., 2009, 94, 111909 CrossRef.
- D. A. Sokolov, Y. V. Morozov, M. P. McDonald, F. Vietmeyer, J. H. Hodak and M. Kuno, Nano Lett., 2013, 13, 5777–5784 CrossRef PubMed.
- F. Liu, T. Tang, Q. Feng, M. Li, Y. Liu, N. Tang, W. Zhong and Y. Du, J. Appl. Phys., 2014, 115, 164307 CrossRef.
- T. Sakthivel, V. Gunasekaran and S. J. Kim, Mater. Sci. Semicond. Process., 2014, 19, 174–178 CrossRef CAS.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I.-S. Chen, C.-W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- C.-T. Chien, S.-S. Li, W.-J. Lai, Y.-C. Yeh, H.-A. Chen, I.-S. Chen, L.-C. Chen, K.-H. Chen, T. Nemoto, S. Isoda, M. Chen, T. Fujita, G. Eda, H. Yamaguchi, M. Chhowalla and C.-W. Chen, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef CAS PubMed.
- Z. Gan, S. Xiong, X. Wu, T. Xu, X. Zhu, X. Gan, J. Guo, J. Shen, L. Sun and P. K. Chu, Adv. Opt. Mater., 2013, 1, 926–932 CrossRef.
- J. R. Rani, J. Lim, J. Oh, J.-W. Kim, H. S. Shin, J. H. Kim, S. Lee and S. C. Jun, J. Phys. Chem. C, 2012, 116, 19010–19017 CAS.
- D. Kozawa, Y. Miyauchi, S. Mouri and K. Matsuda, J. Phys. Chem. Lett., 2013, 4, 2035–2040 CrossRef CAS PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2013, 8, 1002–1013 CrossRef PubMed.
- X. Díez-Betriu, S. Álvarez-García, C. Botas, P. Álvarez, J. Sánchez-Marcos, C. Prieto, R. Menéndez and A. de Andrés, J. Mater. Chem. C, 2013, 1, 6905–6912 RSC.
- C. Punckt, F. Muckel, S. Wolff, I. A. Aksay, C. A. Chavarin, G. Bacher and W. Mertin, Appl. Phys. Lett., 2013, 102, 023114 CrossRef.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. L. M. Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- Amorphous Carbon: State of the Art, ed. S. R. P. Silva, J. Roberts and G. A. J. Amaratunga, World Scientific Press, 1998, pp. 1–374 Search PubMed.
- S. R. P. Silva, et al., Amorphous Carbon Thin Films, in Handbook of Thin Films, ed. H. S. Nalwa, Academic Press, 2001, ch. 9, vol. 4, pp. 403–506 Search PubMed.
- D. W. Lee, V. L. De Los Santos, J. W. Seo, L. L. Felix, D. A. Bustamante, J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- Y. Miyajima, Y. Tison, C. E. Giusca, V. Stolojan, H. Watanabe, H. Habuchi, S. J. Henley, J. M. Shannon and S. R. P. Silva, Carbon, 2011, 49, 5229–5238 CrossRef CAS.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS PubMed.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 Search PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc00307e |
| This journal is © The Royal Society of Chemistry 2015 |