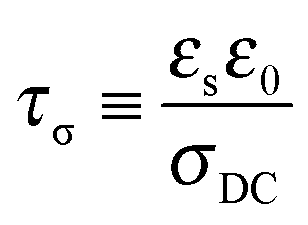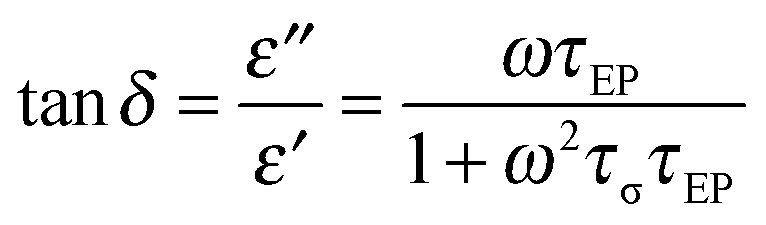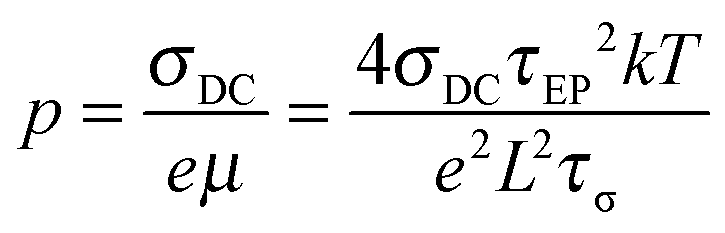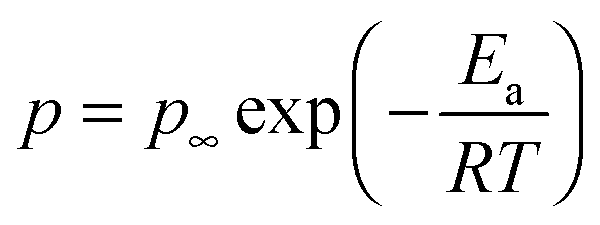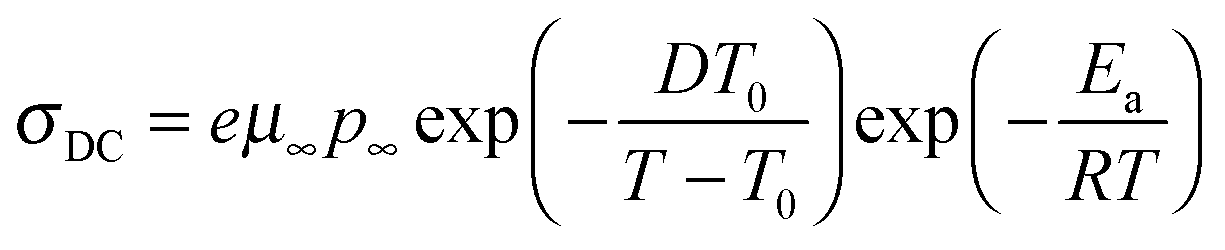Imidazole-containing triblock copolymers with a synergy of ether and imidazolium sites†
Received
17th January 2015
, Accepted 6th March 2015
First published on 6th March 2015
Abstract
Reversible addition–fragmentation chain transfer (RAFT) polymerization enabled the synthesis of well-defined A–BC–A triblock copolymers containing a synergy of pendant ether and imidazolium sites. The soft central BC block comprises low Tg di(ethylene glycol) methyl ether methacrylate (DEGMEMA) and 1-(4-vinylbenzyl) methyl imidazolium units. External polystyrene blocks provide mechanical reinforcement within a nanoscale morphology. Dynamic mechanical analysis (DMA) of the A–BC–A triblock copolymers exhibited a plateau region, which suggested the formation of a microphase-separated morphology. Atomic force microscopy (AFM) and small angle X-ray scattering (SAXS) collectively probed the morphology of the A–BC–A triblock copolymers, revealing long-range order at the nanoscale dimensions. Dielectric relaxation spectroscopy (DRS) examined the ion-transport properties of ionomeric A–BC–A triblock copolymers and random copolymers with different compositions. The role of morphology was demonstrated with block copolymer nanoscale structures providing superior ionic conductivity and mechanical performance compared to random copolymers. Under a 4 V direct current (DC) applied voltage, electromechanical transducers derived from these triblock copolymer membranes with added ionic liquid showed superior actuation performance compared to a benchmark Nafion® membrane, suggesting potential for ionic polymer device applications. This was attributed to optimum modulus, improved ionic conductivity, and microphase-separated morphology of triblock copolymers.
Introduction
The design and synthesis of “smart” macromolecules that respond to external stimuli such as temperature, pH, electrolytes, light, mechanical stress, and applied potential remains an area of intense interest for biological and engineering applications.1–4 In many cases, these macromolecules are designed to mimic behavior of biological polymers and outperform commercially available materials.1 Prior to the development of controlled radical polymerization (CRP) techniques, lack of structural and molecular weight control with broad polydispersity indices (PDI) limited the discovery of well-defined macromolecules with requisite features for designing smart macromolecules for various applications.5–12 CRP facilitated the synthesis of a broad range of triblock copolymers with well-defined architecture and controlled molecular weights.1 Reversible addition–fragmentation chain transfer (RAFT) provides versatility to effectively control molecular weight with judicious chain-transfer agent (CTA) and monomer combinations, solvent, and initiator concentration. RAFT polymerization is currently one of the most versatile CRP synthetic methods capable of affording complex macromolecules with controlled architectures, leading to a tailored range of response to external stimuli. For example, mechanical deformation under applied voltage for electromechanical transducers1 provides potential energy harvesting devices, biomimetic materials, and sensors.13–17 Currently, Nafion® serves as a benchmark material for the electrically stimulated electromechanical transducers due to optimal performance.18 The microphase-separated morphology in Nafion® membranes provides superior performance as an electromechanical transducer. Microphase-separated block copolymers synthesized using CRP techniques present a facile strategy to mimic the semi-crystalline morphology of Nafion® through the inclusion of high modulus domains for mechanical reinforcement and ion-rich soft phases that facilitate ion migration.13,19–25 Thus, recent efforts focus on the synthesis of novel ionomeric block copolymers with the potential for preparing electroactive devices.
The synthesis of a difunctional CTA for a divergent chain growth strategy for triblock copolymers was described in our earlier literature.26 Divergent chain growth maintains thiocarbonylthio groups at the propagating chain end, which results in a polymer main chain that is both thermally and hydrolytically stable. Our group has established suitable conditions for the RAFT polymerization of di(ethylene glycol) methyl ether methacrylate (DEGMEMA) with 1,6-bis(4-cyano-4-(ethylsulfanyl-thiocarbonylsulfanyl)pentanoic acid) hexane diamide (dCEP-NH2) as a CTA.26 Our group has also described ABA triblock copolymers using nitroxide-mediated polymerization (NMP) with polystyrene external blocks and an ionic imidazolium-containing central block. These well-defined triblock copolymers exhibited sufficient modulus and ionic conductivity for electromechanical transducers.2,27 We also reported the synthesis of high molecular weight poly[Sty-b-(nBA-co-DMAEMA)-b-Sty] using RAFT synthesis, and triblock copolymers with tunable properties showed electro-responsiveness under the application of low voltage (2–4 V).3 Elabd, Winey and co-workers recently synthesized block copolymers containing 1-[(2-methacryloyloxy)ethyl]-3-butylimidazolium bis(trifluoromethane sulfonyl)imide (MEBIm-TFSI) and methyl methacrylate (MMA) utilizing RAFT polymerization to understand the influence of phase separation on ionic conductivity.28,29
The self-assembled, nanostructured morphology of block copolymers enabled enhanced conductivity compared to random analogs for solid-state polymer electrolytes. Block copolymers provide a unique opportunity in polymer–ionic liquid mixtures, where a variety of nanoscale, self-assembled morphologies are accessible as a function of composition and thermodynamic incompatibility.30 Elabd and Winey reported a polymerized ionic liquid diblock copolymer exhibiting nanoscale morphology with high hydroxide conductivity. The ionic conductivity was found to be higher than random copolymers with similar ion and water content, as well as its homopolymer analog.31 Elabd et al. also demonstrated a two-order increase in ionic conductivity for diblock copolymers containing the same comonomers as random copolymers to reveal the importance of a well-defined morphology for improved ion-transport.28 Gao et al. reported successful synthesis of PEG-based sulfonated polyurethanes containing sulfonates in either the soft or hard segment. The influence of charge placement on thermomechanical properties, hydrogen bonding interactions, and morphologies of PEG-based sulfonated polyurethanes was demonstrated.32
Control over ABA triblock composition allows for tailored charge content, tunable mechanical properties, and well-defined morphology.24,25,33,34 A fundamental study on the dependence of ionic conductivity on composition, morphology, and monomer selection in ionomeric triblock copolymers is essential to facilitate the intelligent design of ion-conducting polymers. Recent studies stressed that materials having low glass transition temperatures (Tg) are required to obtain high ionic conductivity, reflecting the coupling of ion-motion to polymer segmental dynamics.28,35 However, these conductive materials are of limited use as devices due to lack of mechanical strength. Block copolymers self-assemble to form physically crosslinked, mechanically tough, nanostructured polymers, and thus have attracted significant attention in the design of polymer electrolytes for electromechanical devices. Mahanthappa et al. prepared poly(imidazolium) block copolymers, and the effects of macroscopic connectivity and morphological on conductivity was demonstrated in these materials.33
In this manuscript, we demonstrate the synthesis of novel, well-defined A–BC–A triblock copolymers containing soft “BC” central blocks of DEGMEMA and imidazolium sites with polystyrene external blocks. Earlier work has demonstrated that increasing the dielectric constant of the ion-containing phase with proper monomer selection without raising Tg optimizes ionic conductivity. Diluting the ion concentration of the ion-rich phase with neutral comonomers that have high dielectric constant reduces Tg, increases ion dissociation, and potentially maximizes ionic conductivity.36,37 Lowering Tg of an ionic polymer also increases segmental mobility and thus ionic conductivity. Tendency of ethylene-oxide units to coordinate cations, forming stable crown ether-like, multi-nuclear coordination complexes, promotes solvation and dissociation of ionic aggregates.38 Thus, understanding the synergy between Tg and electrostatic solvation of ethylene-oxide containing units is crucial to observe the influence on ionic conductivity.36,38,39 Dynamic mechanical analysis (DMA) probed the thermomechanical properties of the triblock copolymers, and SAXS with complementary AFM examined the microphase-separation of the A–BC–A triblock copolymers. Dielectric relaxation spectroscopy (DRS) determined the ion-transport properties of ionomeric A–BC–A triblock copolymers and random copolymer controls of various compositions. These triblock copolymers with suitable morphology and thermomechanical properties were cast into films with ionic liquid, and fabricated into electromechanical actuators. The addition of an ionic liquid (IL) diluent will alter the polymer morphology,30 and a detailed understanding of this effect will enable precise control over the triblock copolymer morphology, ionic conductivity, and mechanical properties. For the first time to our knowledge, the devices fabricated using novel triblock copolymers performed comparably to earlier Nafion® membranes. This was attributed to optimum modulus required to obtain mechanical stability as well as improved ionic conductivity for microphase-separated morphology of triblock copolymers.
Experimental section
Materials
N,N′-Dicyclohexylcarbodiimide (DCC, 99%), 4-(dimethylamino)pyridine (DMAP, >99.0%), 1,6-hexanediamine (98%), 1,6-hexanediol (99%) were purchased from Sigma-Aldrich and used as received. 4-Cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic acid (CEP) and VBIm were synthesized according to the previous literature.2 1-Ethyl-3-methylimidazolium trifluoromethane sulfonate (EMIm-TfO, IoLiTec Inc., 99%), iodomethane (Sigma, >99.0%) and lithium bis(trifluoromethane sulfonyl)imide (Tf2N) (LiTf2N, Aldrich, 99%) were used as received. Di(ethylene glycol) methyl ether methacrylate (DEGMEMA, 95%) and styrene (Sigma, 99%) were purchased from Sigma-Aldrich and passed through an alumina packed column prior to use. 4,4′-Azobis(4-cyanovaleric acid) (V-501, Aldrich, 98%) and 2,2′-azobisisobutyronitrile (AIBN) were recrystallized from ethanol. Anhydrous dichloromethane (DCM) was obtained from a solvent purification system (Pure Solv, Innovative Technology) and all other solvents were obtained from spectrum and used as received.
Synthesis of ionomeric A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty]
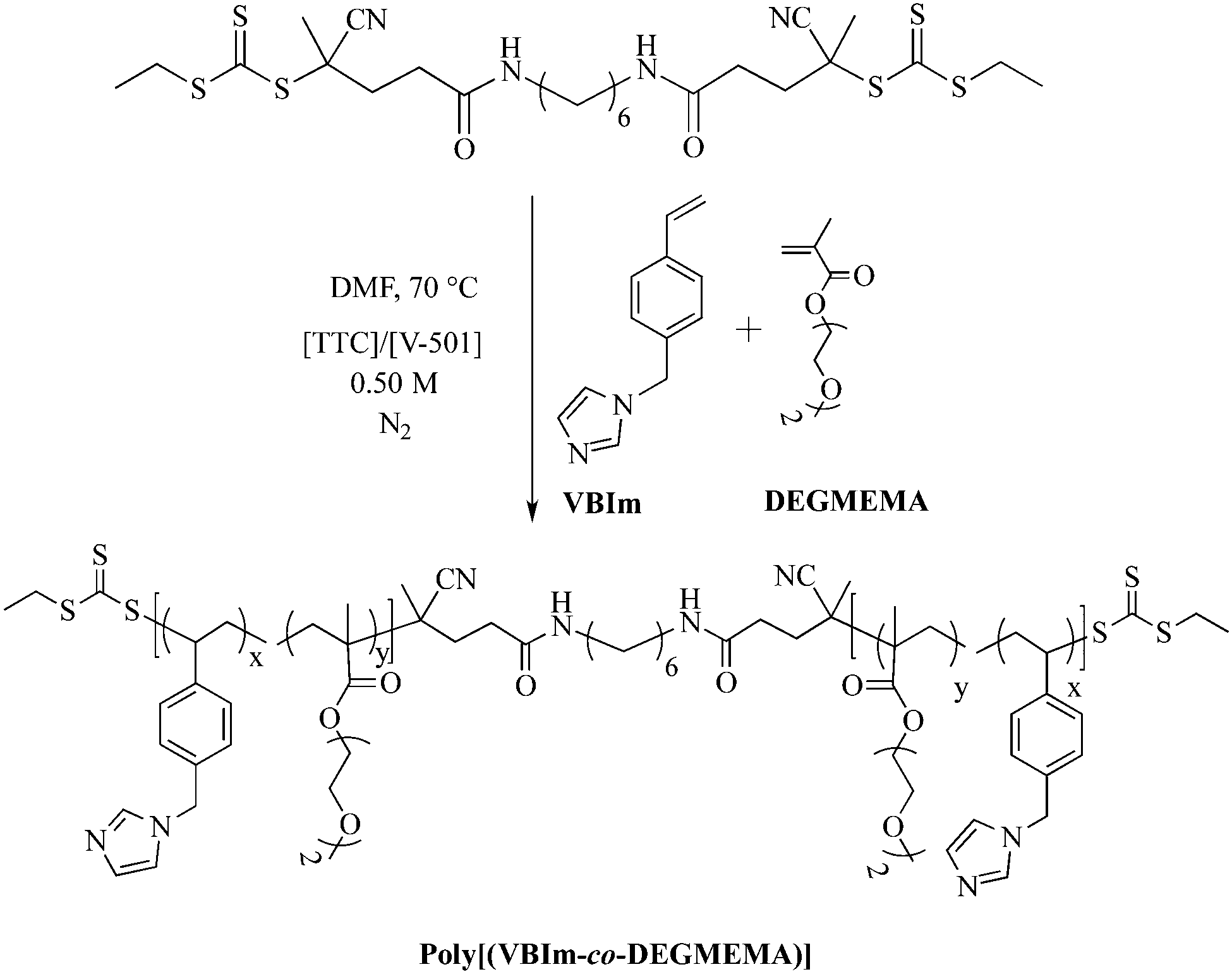
In a representative copolymerization (Scheme 1), VBIm and DEGMEMA (6.0 g), dCEP-NH2 (42.6 mg), V-501 (17.2 mg, 53.2 μmol), and DMF (42.5 mL) were added to a 100 mL, round-bottomed flask equipped with a stir bar. The reaction was sparged with argon for 30 min and placed in an oil bath thermostated at 70 °C for 4 h. Different compositions of VBIm and DEGMEMA were targeted to obtain poly(VBIm-co-DEGMEMA) copolymers. The resulting polymers were dialyzed (MWCO = 3500 g mol−1) for 3 d against methanol while changing the solvent every 24 h. Aqueous SEC was used to determine the absolute molecular weights of random copolymers of VBIm and DEGMEMA (PDI < 1.1) (SEC chromatographs are shown in ESI†).
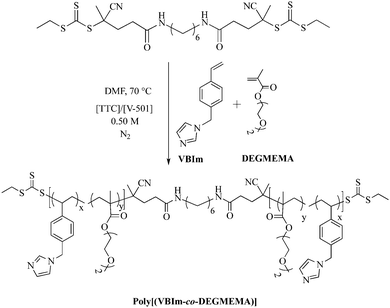 |
| | Scheme 1 Synthesis of central ‘BC’ block comprising VBIm and DEGMEMA using RAFT polymerization. V-501 (4-cyanovaleric acid) is an azo-initiator and TTC is a difunctional trithio CTA. | |
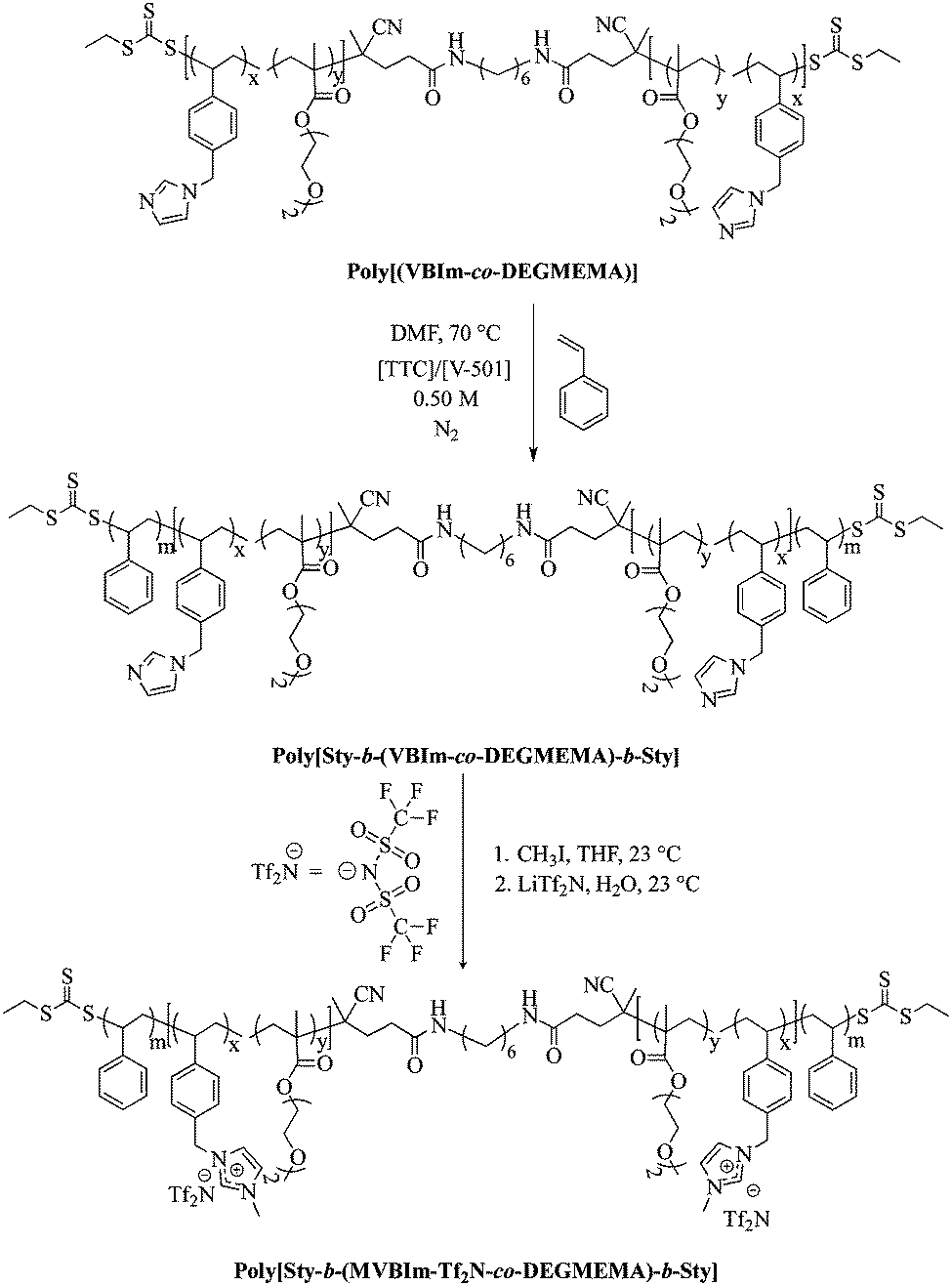
The purified macroCTA (400 mg) was redissolved in DMF (23 mL) in a 50 mL round-bottomed flask equipped with a stir bar. Styrene (0.5 g) and V-501 (3.12 mg) were added to the flask and the reaction was sparged with argon for 30 min at room temperature. The reaction was immersed in an oil bath at 70 °C for 6 h to obtain the desired amount of styrene in the external blocks. Poly[Sty-b-(VBIm-co-DEGMEMA)-b-Sty] and methyl iodide were dissolved in THF. The solution was purged with argon, and reacted at 23 °C for 24 h. The product was precipitated into ethyl acetate. Poly[Sty-b-(MVBIm-I-co-DEGMEMA)-b-Sty] was isolated through filtration and dried under reduced pressure (0.5 mm Hg) at 40 °C for 18 h. Poly[Sty-b-(MVBIm-I-co-DEGMEMA)-b]-Sty (3.0 g) and LiTf2N (67.03 g) were dissolved in separate solutions of water (50 mL each). The solutions were mixed together, immediately forming a white precipitate, and stirred at 23 °C for 24 h. Poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty] was dialyzed against methanol for 3 d to ensure removal of ionic impurities and dried at 60 °C for 24 h. The synthetic strategy is presented in Scheme 2.
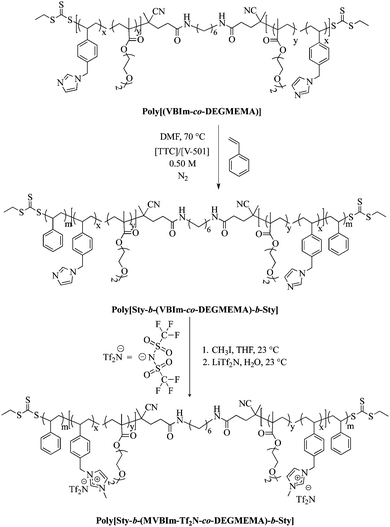 |
| | Scheme 2 Chain extension and quaternization of poly(VBIm-co-DEGMEMA) to synthesize ionomeric A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty]. | |
Synthesis of random copolymers poly(Sty-co-MVBIm-Tf2N-co-DEGMEMA)
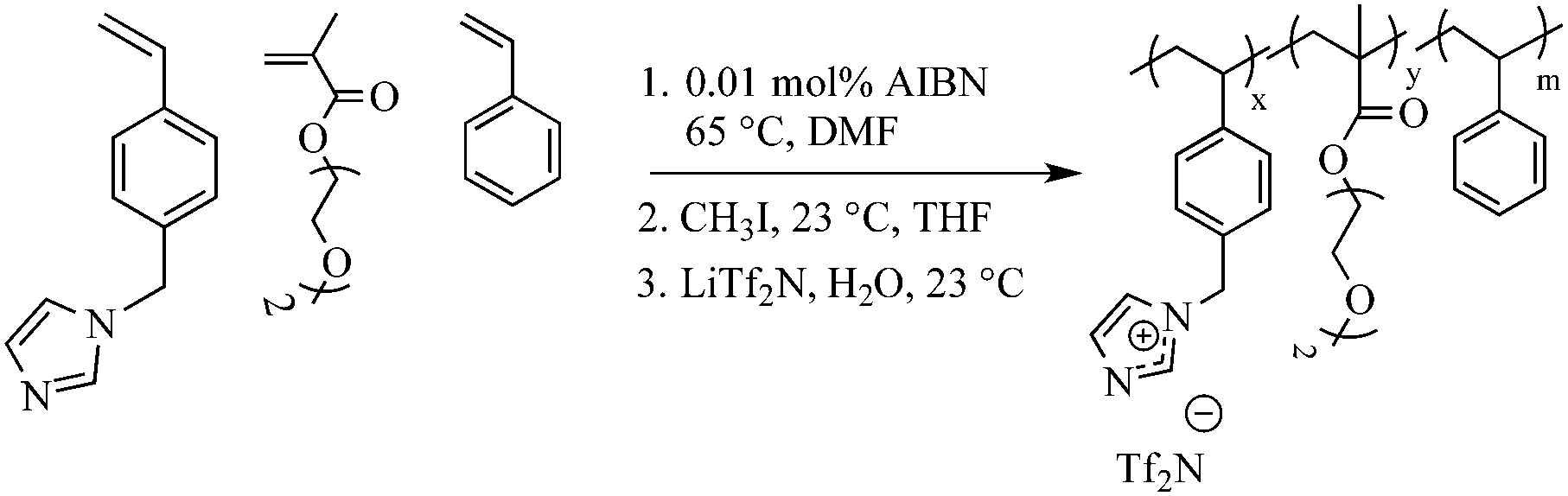
To a round-bottomed flask all monomers, styrene, VBIm and DEGMEMA were added. To dissolve the monomers, 85 mL of DMF was added. To this reaction mixture, AIBN (0.01 mol%) was added and dissolved for 5 min. The flask was sealed and sparged with argon for 30 min to remove oxygen. The polymerization was then conducted at 65 °C for 24 h. The resulting polymer was quaternized using methyl iodide and anion exchange with LiTf2N using the previously mentioned procedure. The resulting polymer solution was dialyzed against methanol for 2 d to remove monomer and DMF, which was confirmed using 1H NMR spectroscopy. Scheme 3 summarizes the overall synthetic strategy.
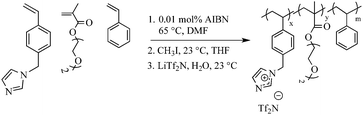 |
| | Scheme 3 Synthesis and quaternization of random copolymers of VBIm, DEGMEMA and styrene using free radical copolymerization. | |
Analytical methods
1H NMR spectroscopy (Varian Inova, 400 MHz) determined CTA, monomer, and polymer composition. Thermogravimetric analysis (TGA) of the triblock copolymers was performed on a TA Instruments thermogravimetric analyzer (TGA Q50) with a heating rate of 10 °C min−1 to 600 °C under a nitrogen atmosphere after holding the sample at 100 °C for 60 min. A TA Instruments Q5000 sorption analyzer (TGA-SA) was used to measure water uptake of triblock copolymer and random copolymer samples (relative humidity (RH) 0–95%, 5% increments) at 25 °C. Thermal transitions were determined on a TA Instruments Q1000 differential scanning calorimeter (DSC, heating rate 10 °C min−1) utilizing a heat–cool–heat cycle. Tg was determined from the second heat cycle of the DSC thermogram. Dynamic mechanical analysis (DMA) (TA Instruments Q800, 3 °C min−1 heating rate, −50 °C to 150 °C) was performed on the triblock copolymer films in tension mode at 1 Hz, oscillatory amplitude of 10 μm, and a static force of 0.01 N. Polymer molecular weight was measured using aqueous size exclusion chromatography (SEC) through two Waters ultrahydrogel linear columns with one Waters ultrahydrogel 250 column in a solvent consisting of 54/23/23 water/methanol/acetic acid (v/v/v%) with 0.1 M sodium acetate. The instrumentation consisted of a Waters 1515 isocratic HPLC pump operating at a flow rate of 0.8 mL min−1, a Waters 717plus Autosampler, a Wyatt miniDAWN multiangle light scattering detector operating at a wavelength of 690 nm. Waters 2414 differential refractive index detector was used to determine the dn/dc for absolute molecular weight determination. Dynamic light scattering (DLS) (Malvern Zetasizer NanoZS) indicated the absence of polymer aggregation in the aqueous mobile phase prior to injection onto the SEC columns. A Wyatt Optilab refractive index detector operating at 658 nm and 35 °C determined the offline specific refractive index increment values. Wyatt ASTRA SEC/LS software was used to calculate absolute polymer molecular weights and PDIs.
All triblock copolymer were dissolved in a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 CHCl3:CH3OH solvent mixture and cast from a 30 wt% solution. Films were slowly dried at ambient conditions for 2 d under reduced pressure. The films were annealed at temperatures at 120 °C for 3 d. The films were then stored in a dry box until analyzed.
:20 CHCl3:CH3OH solvent mixture and cast from a 30 wt% solution. Films were slowly dried at ambient conditions for 2 d under reduced pressure. The films were annealed at temperatures at 120 °C for 3 d. The films were then stored in a dry box until analyzed.
Atomic force microscopy (AFM)
A Veeco MultiMode scanning microscope in tapping mode provided atomic force microscopy (AFM) images of the A–BC–A triblock copolymers. Samples were imaged with a set-point ratio of 0.6 at 3 μm × 3 μm and 1 μm × 1 μm magnifications using a nanosensor silicon tip with a spring constant of 42 N m−1.
Small-angle X-ray scattering (SAXS)
A Rigaku S-Max 3000 3 pinhole small-angle X-ray scattering (SAXS) instrument outfitted with a rotating anode generated Cu Kα irradiation at a wavelength of 1.54 Å to perform bulk morphology analysis on triblock copolymer films. The sample-to-detector distance was 1.5 m and was calibrated with a silver behenate standard. Two-dimensional data sets were collected using a fully integrated 2D multiwire area detector with an hour exposure time, which was corrected for detector noise, background scattering, and sample absorption. All data processing and analysis was performed with the SAXSGUI software package to obtain intensity vs. scattering vector q plots, where q = 4πsin(θ)/λ and 2θ is the scattering angle.
Dielectric relaxation spectroscopy (DRS)
Samples for dielectric relaxation spectroscopy (DRS) measurements were solvent-cast films from mixtures of 80:20 CHCl3:CH3OH and vacuum annealed for 5 d at 120 °C. The samples were placed on a freshly polished brass electrode and dried in a vacuum oven at 353 K for 48 h, and a second brass electrode was finally placed on top of the sample. A Novocontrol GmbH Concept 40 broadband dielectric spectrometer was used to measure the dielectric permittivity. Frequency sweeps were performed isothermally from 10 MHz to 0.01 Hz in the temperature range from 233 to 453 K under dry nitrogen. The samples were initially held at a temperature above 393 K for at least 30 min to minimize water in the samples and avoid a change in water content during the experiment. The measurements were performed during subsequent cooling under a flow of dry N2. The sample was heated to the starting temperature, revealing perfectly reproducible data during the second heating to the highest temperature.
Electroactive device fabrication
For electromechanical bending actuator fabrication, two conductive network composite (CNC) layers were coated on both sides of the poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty] membrane. A layer-by-layer self-assembly technique was used to fabricate CNC layers consisting of negatively-charged gold nanoparticles (Au NPs, 3 nm diameter, 20 ppm, Purest Colloids) alternating with positively charged inert long chain poly(allylamine hydrochloride) (PAH, 10 mM, Sigma-Aldrich) to form a highly porous and conductive structure. The detailed process is described in a previous publication.40 Briefly, an automatic dipping system (StratoSequence VI Robot, nanoStrata Inc.) was used to alternatively dip a framed polymer membrane into respective solutions of PAH and Au NPs with three DI water rinses steps in between. After coating with 30 CNC bi-layers, the membrane was soaked with 1-ethyl-3-methylimidazolium trifluoromethanesulfonate (EMIm TfO) ionic liquid (>98%, Sigma-Aldrich) at 90 °C for 30 min to weight uptake of 30% as electrolyte. The transportation and accumulation of the EMIm+ cations and TfO− under the applied external electric field are responsible for the bending behavior of the actuator. Finally, the membrane was hot-pressed (90 °C, 700 psi) with 50 nm thick gold foil on both sides to provide outer electrodes for connection to the power supply and cut into 1 mm × 1 cm strips for testing. The electric stimulation was 4 V DC input, and the bending behavior was recorded using an HD CCD camera for further analysis. As a control, a second actuator was fabricated using a commercially available Nafion® membrane (N111-IP, EW 1100, 25 μm thick, Ion Power, Inc.) with an identical CNC layer and EMIm-TfO IL as electrolyte (34.2 wt%) and tested under identical condition.2,41–43
Results and discussion
Initial polymerizations involved the synthesis of poly(DEGMEMA-co-VBIm) copolymers mediated with a difunctional CTA, dCEP-NH2 in DMF to generate the low-Tg central block as shown in Scheme 1. The copolymerization of VBIm and DEGMEMA using RAFT ([DEGMEMA]/[TTC]/[V-501] ratio = 400/2/1) displayed linear, pseudo-first-order polymerization kinetics. These polymerizations proceeded at conversions 75–80% with narrow PDIs and demonstrated good agreement between experimental and theoretical molecular weights. A series of low-Tg central blocks was prepared with varying molar ratio of VBIm and DEGMEMA. All the copolymers exhibited narrow PDIs (ESI†) as shown in Table 1.
Table 1 Molecular weights and PDI of poly(VBIm-co-DEGMEMA) at various compositions of VBIm and DEGMEMA
| Polymer |
VBIma (mol%) |
M
nb (g mol−1) |
PDI |
|
1H NMR spectroscopy: 400 MHz, DMSO-d6, 25 °C.
SEC: 35 °C, 1 mL min−1, MALLS detector, 54/23/23 (v/v/v%) H2O/CH3OH/AcOH, 0.1 M NaOAc.
|
| Poly(VBIm80-co-DEGMEMA20) |
80 |
37000 |
1.02 |
| Poly(VBIm72-co-DEGMEMA28) |
72 |
36100 |
1.04 |
| Poly(VBIm52-co-DEGMEMA48) |
52 |
35200 |
1.06 |
| Poly(VBIm35-co-DEGMEMA65) |
35 |
28600 |
1.06 |
The soft central block incorporating the low Tg monomer DEGMEMA and VBIm was chain extended with styrene to obtain A–BC–A triblock copolymers, where the external polystyrene blocks provide mechanical stability and the central block poly(DEGMEMA-co-VBIm) introduces charge in the soft phase to increase ion-transport. Chain extension using styrene was accomplished using RAFT polymerization and PDI values remained relatively narrow after chain extension with increasing molecular weights as shown in Table 2.
Table 2 Thermal transitions and molecular weights of A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty]
| Polymer |
M
na (g mol−1) |
Styreneb (wt%) |
PDI |
T
g1c (°C) |
T
g2c (°C) |
T
d,5%d (°C) |
|
SEC: 35 °C, 1 mL min−1, MALLS detector, 54/23/23 (v/v/v%) H2O/CH3OH/AcOH, 0.1 M NaOAc; dn/dc = 0.2310 mL g−1.
1H NMR spectroscopy: 400 MHz, DMSO-d6, 25 °C.
DSC: 10 °C min−1, N2, second heat.
TGA: 10 °C min−1, N2.
|
| Poly(VBIm) |
ND |
— |
— |
107 |
— |
354 |
| Poly(VBIm72-co-DEGMEMA28) |
36100 |
— |
1.04 |
64 |
— |
390 |
| Poly(VBIm52-co-DEGMEMA48) |
35200 |
— |
1.06 |
43 |
— |
341 |
| Poly(VBIm35-co-DEGMEMA65) |
28600 |
— |
1.06 |
31 |
— |
327 |
| Poly[Sty-b-(VBIm72-co-DEGMEMA28)-b-Sty] |
16k-b-36k-b-16k |
47 |
1.12 |
57 |
109 |
345 |
| Poly[Sty-b-(VBIm52-co-DEGMEMA48)-b-Sty] |
18k-b-35k-b-18k |
50 |
1.23 |
40 |
96 |
415 |
| Poly[Sty-b-(VBIm35-co-DEGMEMA65)-b-Sty] |
18k-b-28k-b-18k |
56 |
1.26 |
28 |
112 |
428 |
| Poly[Sty-b-(MVBIm-Tf2N72-co-DEGMEMA28)-b-Sty] B1 |
16k-b-56k-b-16k |
37 |
— |
35 |
110 |
332 |
| Poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] B2 |
18k-b-55k-b-18k |
40 |
— |
26 |
108 |
350 |
| Poly[Sty-b-(MVBIm-Tf2N35-co-DEGMEMA65)-b-Sty] B3 |
18k-b-40k-b-18k |
47 |
— |
10 |
111 |
375 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-18 mol% ion content R1 |
ND |
48 |
— |
19 |
— |
368 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-26 mol% ion content R2 |
ND |
45 |
— |
20 |
— |
376 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-36 mol% ion content R3 |
ND |
43 |
— |
23 |
— |
365 |
The thermal properties depended on the composition, presence of charge, and counterion selection. As observed in the earlier literature, Tg of the copolymers increased after quaternization with methyl iodide and decreased after anion exchange to Tf2N− due to a large counterion size.23,28Table 2 summarizes the thermal transitions and molecular weights of central blocks as well as A–BC–A triblock copolymers. Different charge concentration enabled investigations of ionic content on thermal properties. Although the number-average molecular weight (Mn) of the ionomeric triblock copolymer remained similar with increasing ion content from 35 to 72 mol%, the Tg of the central block increased from 10 to 35 °C. Incorporation of ionic groups within the soft block restricts polymer segmental mobility and consequently, increases Tg.44 The charge placement in the soft block at various concentrations did not affect the high-Tg transition, suggesting an absence of appreciable phase mixing in the triblock copolymers. For the triblock copolymers, we observed two Tgs indicating the presence of a phase-separated morphology. The lower transition was attributed to the soft ionic block and the transition at higher temperature was assigned to the polystyrene block. The random copolymer analog did not exhibit a second Tg, suggesting the absence of phase-separation in the random copolymers. TGA showed that thermal stability decreased after quaternization due to the presence of the more basic I− anion and increased after anion exchange to the less basic couteranion Tf2N−, consistent with earlier literature.28Table 2 includes the thermal transitions and molecular weight data for random copolymers of styrene, DEGMEMA and VBIm. The random copolymer serves as a comparative control for the A–BC–A triblock copolymers.
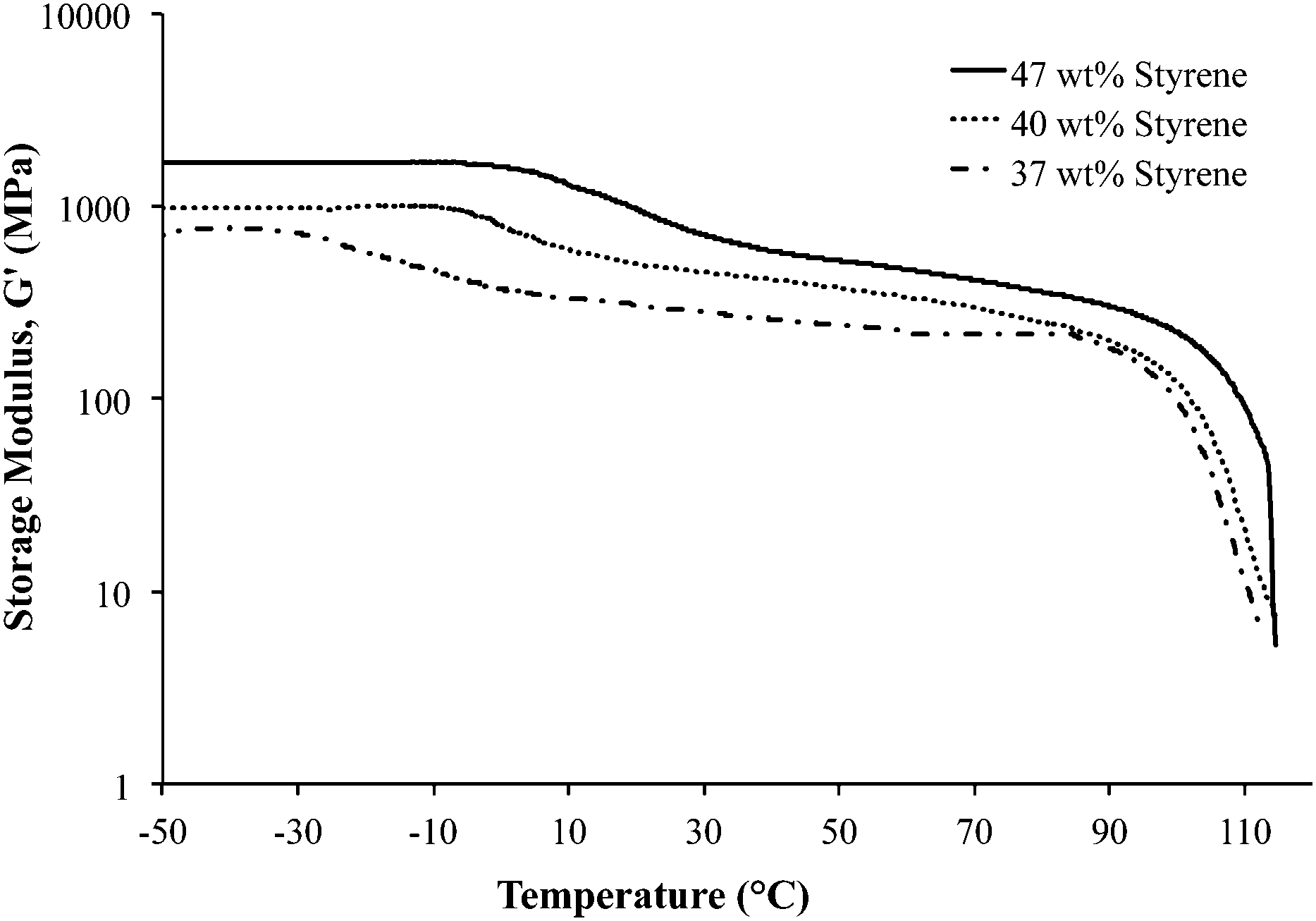
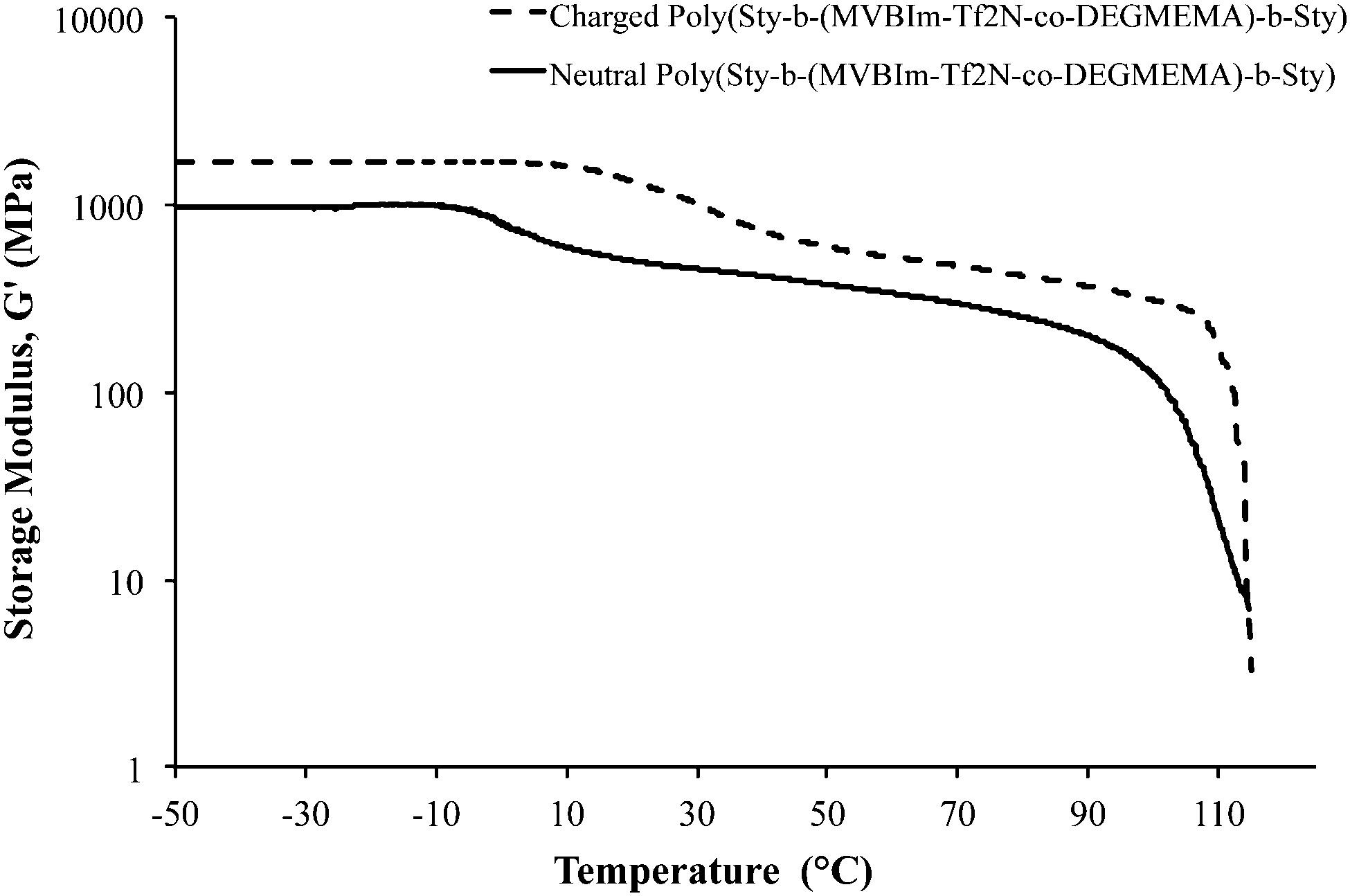
Dynamic mechanical analysis (DMA) indicated microphase-separation in the triblock copolymers at various styrene contents as shown in Fig. 1 and 2. The storage modulus (G′) vs. temperature for the ionomeric triblock copolymers is shown in Fig. 1. The triblock copolymer exhibited two distinctive transitions, i.e. Tg of the soft central block, poly(DEGMEMA-co-MVBIm-Tf2N), while the second terminal flow transition at ∼100 °C is consistent with the Tg of polystyrene hard phase. Fig. 1 depicts the DMA results for 37, 40, and 47 wt% polystyrene. The styrenic comonomer, VBIm, is presumably more compatible with polystyrene, thus an increase in the styrenic comonomer was expected to promote phase mixing. The triblock copolymer films exhibited significantly improved mechanical strength compared to either poly(VBIm-co-DEGMEMA) or poly(DEGMEMA) homopolymers as neither polymers formed a ductile film for mechanical property characterization.45Fig. 2 also confirms the mechanical stability of the neutral as well as ionic triblock copolymer films. The transition of central block shifts to higher temperature for ionomeric A–BC–A triblock copolymers.
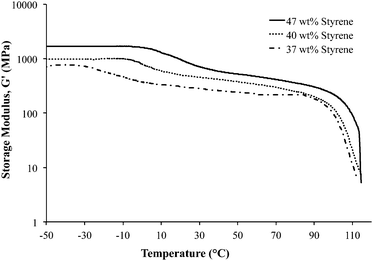 |
| | Fig. 1 Dynamic mechanical analysis (DMA) of ionic A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty] with varying compositions. | |
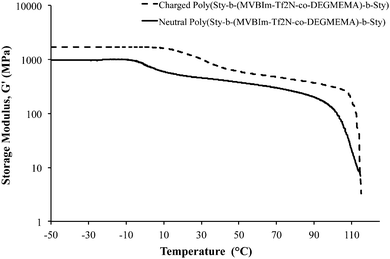 |
| | Fig. 2 Dynamic mechanical analysis (DMA) of neutral and ionomeric A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty]. | |
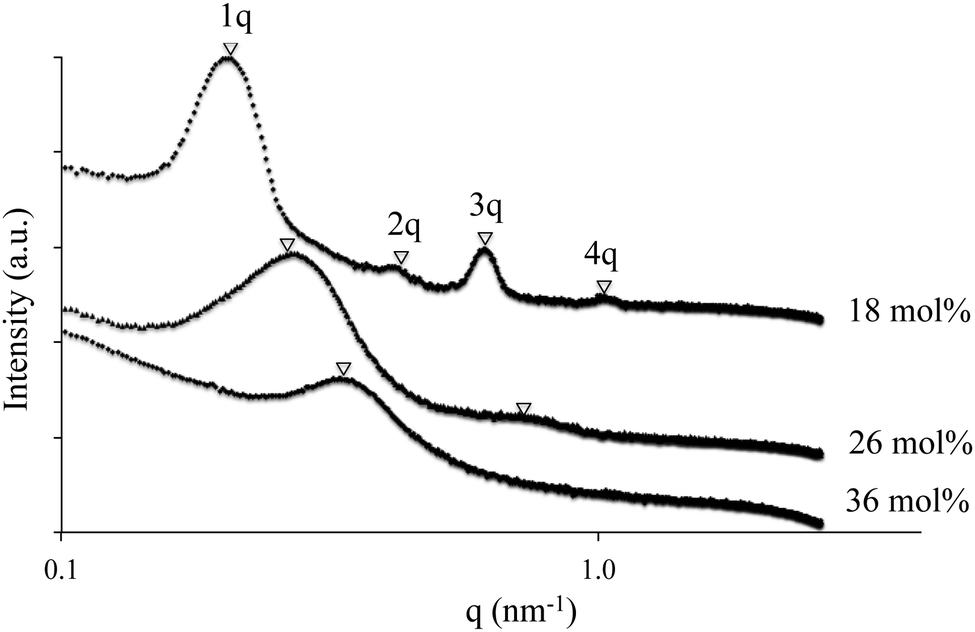
Earlier studies in the literature employed AFM to elucidate the multiphase morphology of triblock copolymers.46–49 Morphological investigations using AFM uncovered a microphase-separated surface morphology. AFM (Fig. 3) indicates the presence of lamellar morphology in a triblock copolymer with 18 mol% ion content, and morphological features changed with varying ion content. With an increase in composition of VBIm in the central block, phase mixing was observed presumably due to increased styrenic comonomer compatibility with polystyrene in the external block. The morphology became less ordered and the domain size was smaller, which is apparent in both AFM and also in SAXS data. The literature extensively describes the dynamics and structural parameters that dictate block copolymer microphase-separation.45,50 However, incorporation ions that exclusively reside in one microphase of a block copolymer changes the customary rules in such a way that maintains continuity of the ionic domains.
 |
| | Fig. 3 Atomic force microscopy (AFM) of ionomeric A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty] going from (a) 18 mol% (b) 26 mol% (c) 36 mol% ion content. | |
Past study on block copolymer morphologies where one block is an ionomer have often ignored the interaction between the ions and the presence of ion pairs or larger ionic aggregates; however, this highly asymmetric charge cohesion effect (ions only aggregate in one block) can lead phase diagrams completely different from neutral block copolymers. The recently developed hybrid self-consistent field theory-liquid state theory (SCFT-LS) permits study on ‘charge cohesion’ effect by Sing et al. that was previously neglected due to theoretical difficulty. Even though diblock copolymers with equal volume blocks were expected to form lamellar microphases when they are neutral; diblock copolymers with one ionomeric block and equal volume blocks can form nanostructures from inverse cylinders to inverse spheres (with the neutral block forming spheres and cylinders). The phase diagram of anionic diblock copolymer tilts toward the left such that nanostructure formation is enhanced at low-fA systems (A block is the ionomeric block) when ion aggregation in A block has greatest contrast compared to the mixed state; and nanostructure formation is suppressed at high-fA due to strong ion–ion interactions energetically favoring states with high charge densities.
SAXS experiments complemented AFM morphological findings and determined the presence of bulk microphase-separation (Fig. 4). X-ray scattering data plotted as intensity, I(q), vs. the momentum transfer vector, q, confirmed microphase-separation in the A–BC–A triblock copolymers. For all three triblock copolymers, scattering maxima were observed, indicating the presence of microphase-separation in the A–BC–A triblock copolymer films. The triblock copolymers with 18 mol% ion content exhibited peak maxima at q*, 2q*, 3q* and 4q*, indicating a well-ordered, lamellar morphology. The triblock copolymer with 26 mol% ion content, second peak maxima was still observed, which diminishes at 36 mol% ion content, indicating phase mixing.
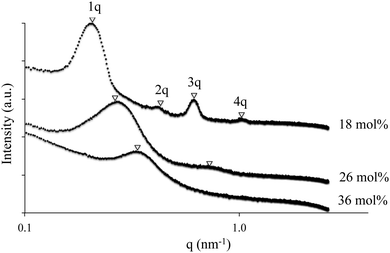 |
| | Fig. 4 Small angle X-ray scattering (SAXS) of ionomeric A–BC–A triblock copolymer poly[Sty-b-(MVBIm-Tf2N-co-DEGMEMA)-b-Sty] with various ion contents 18, 26 and 36 mol%. | |
With an increase in VBIm composition in the central block, we observed a less ordered system, since the styrenic comonomer was presumably more compatible to polystyrene external blocks, inducing phase mixing. The SAXS results agreed well with AFM images and DMA analysis of the triblock copolymer. Furthermore, there was no peak observed for ionic aggregation at q of 1.0–2.0 nm, indicating the A–BC–A triblock copolymer system behaving as single ion conductors with superior ion-transport properties as opposed to a “channel-network morphology” in Nafion®.
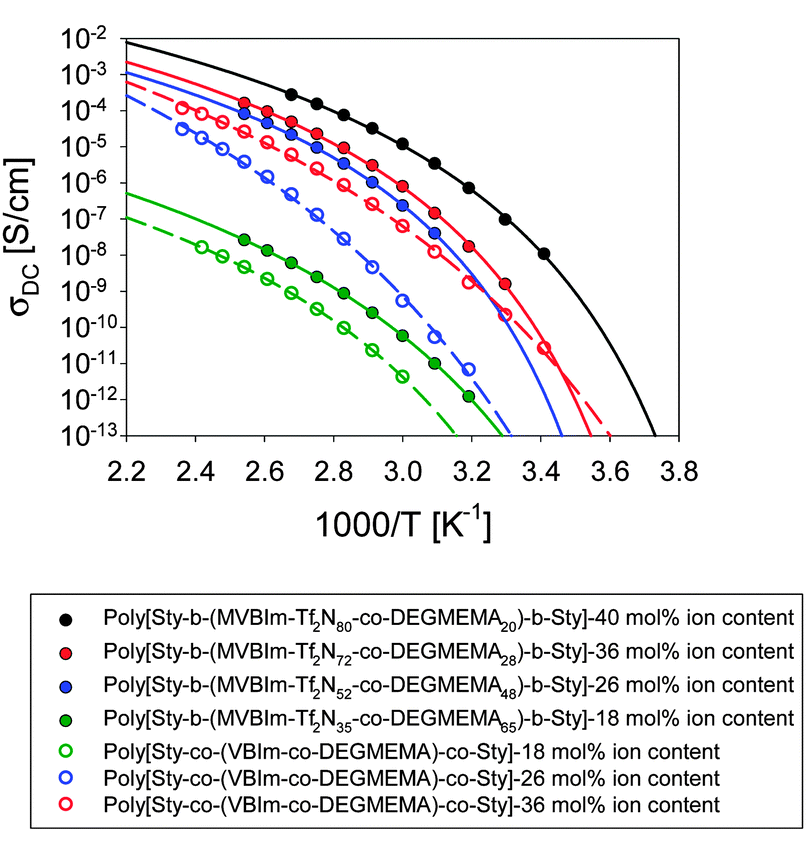
Ionomers typically exhibit the ability to absorb moisture, which is quantified using TGA-sorption analysis. These triblock copolymers showed less than 1% weight gain due to water at 25 °C and 10% relative humidity. Ionic conductivity also strongly depends on frequency and temperature for single-ion conductors.51 The DC conductivity, defined as the in-phase part of the conductivity where it is independent of frequency in a ∼3-decade frequency range, is plotted in Fig. 5. For both triblock and random copolymer ionomers, ionic conductivity increased with increasing ion content and did not correlate with the Tg of the ion-containing block; the triblock copolymer poly[Sty-b-(VBIm72-co-DEGMEMA28)-b-Sty] with the highest ion content exhibited the highest ionic conductivity. Further, addition of ethylene oxide units diluted the ion content and also changed ion aggregation behavior and morphology, resulting in decreased ionic conductivity. Moreover, all triblock copolymers showed higher ionic conductivity than random copolymers at identical monomer composition, which suggested the microphase-separated morphologies of triblock copolymers provide a continuous path to facilitate ion conduction.
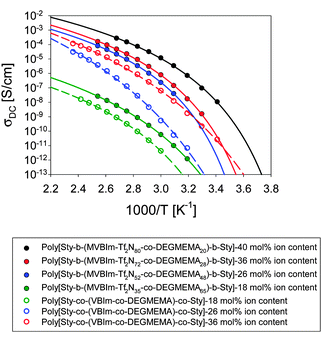 |
| | Fig. 5 Temperature dependence of ionic conductivity which increases with increase in ion content and found to be more than random copolymers (open symbols). | |
The ionic conductivity (σ) is simply expressed by
where
p,
e, and
μ are total number density of conducting ions, elementary electric charge, and conducting ion mobility, respectively. It is crucial to determine whether the increase in ionic conductivity has a larger contribution from the number density of simultaneously conducting ions or their mobility in order to understand counter anion conduction. This is assessed with electrode polarization at very low frequencies in dielectric relaxation spectroscopy (DRS).
51
In DRS, a sinusoidal ac field is applied to a thin-film sample sandwiched between two blocking electrodes. The electrode polarization (EP) occurs at frequencies low enough such that the transporting ions have sufficient time to polarize at the electrodes during each cycle. A physical model of EP,52–55 which has been applied to a number of single-ion conductors recently,14,15,51,56–59 determined the number density of simultaneous conductors and their mobility. The time scale for full polarization at the electrode is
| | 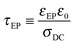 | (2) |
where
εEP is the effective permittivity after the electrode polarization is complete,
ε0 is the permittivity of vacuum, and
σDC is the DC conductivity. The time scale of conduction or when the ion motion becomes diffusive is
| | 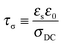 | (3) |
where
εs is the static dielectric constant. EP is regarded as a simple Debye relaxation in the Macdonald and Coelho model
52–55 with the loss tangent peak expressed as
| | 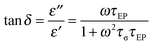 | (4) |
where the peak maximum frequency relates to the geometric mean of the two fitting parameters
τEP and
τσ, demonstrated in
Fig. 6.
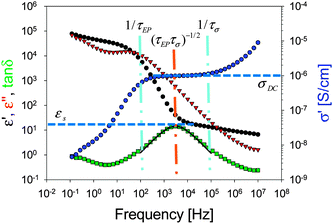 |
| | Fig. 6 Dielectric spectra and fitting of the loss tangent peak with eqn (4) for triblock copolymer poly[Sty-b-(VBIm52-co-DEGMEMA48)-b-Sty] at 70 °C. | |
Since one type of ion is attached to the polymer chain and assumed to be immobilized, the counterion's mobility μ and number density of simultaneous conducting ions p are then determined from the fitting parameters,
| | 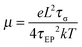 | (5) |
| | 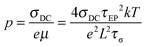 | (6) |
where
L is the sample thickness between electrodes,
k is Boltzmann's constant, and
T is the absolute temperature.
The temperature dependence of the number density of simultaneously conducting ions calculated from eqn (6) is plotted in Fig. 7 and is well-described by the Arrhenius equation,
| | 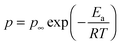 | (7) |
where
p∞ is the conducting ion concentration as
T approaches ∞ and
Ea is the activation energy for conducting ions listed in
Table 3. The activation energy is related to the Coulomb energy of a cation–anion pair, and this electrostatic attraction is the main driving force for pair and aggregate formation, mediated by an effective dielectric constant of the ion-conducting block. The Arrhenius temperature dependence of the conducting ion concentration suggests the conducting ions would be fully dissociated at infinite temperature, thus the intercept of the Arrhenius fit in
Fig. 7 is fixed to
p0, the total ion concentration calculated from a group contribution method
60,61 with stoichiometry determined from the chemical structure. The number density of simultaneously conducting ions of all random copolymers is higher than triblock copolymers, suggesting ions in random copolymers are better solvated by ether oxygen atoms. The ethylene oxide side chains in triblock copolymers are likely stretched to form the microphase-separated morphologies shown in SAXS, and the ether oxygen atoms on the stretched side chains has less ability to solvate the attached cations than the ether oxygen atoms on random copolymers because of the restricted mobility.
62
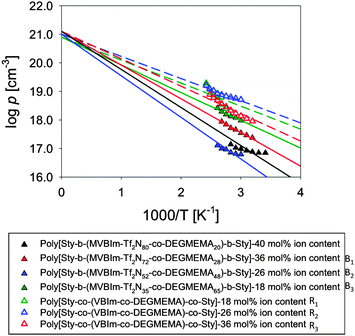 |
| | Fig. 7 Temperature dependence of conducting ion concentration for A–BC–A triblock copolymers and random copolymers (open symbols) determined using the EP model. | |
Table 3 Fitting parameters (eqn (7)) for the temperature dependence of the number density of simultaneously conducting ions with intercept p∞ fixed to the total ion content p0
| Sample |
logp∞ (cm−3) |
E
a (kJ mol−1) |
| Poly[Sty-b-(MVBIm-Tf2N80-co-DEGMEMA20)-b-Sty] |
21.1 |
25.8 |
| Poly[Sty-b-(MVBIm-Tf2N72-co-DEGMEMA28)-b-Sty] |
21.1 |
22.8 |
| Poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] |
21.0 |
28.1 |
| Poly[Sty-b-(MVBIm-Tf2N35-co-DEGMEMA65)-b-Sty] |
20.9 |
18.8 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-18 mol% ion content |
20.9 |
15.6 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-26 mol% ion content |
21.0 |
15.0 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-36 mol% ion content |
21.1 |
18.6 |
Since ionic conductivity is obtained over a much wider temperature range than the EP model allows, the activation energies obtained from the Arrhenius fit to the number density of simultaneously conducting ions where the intercept is fixed are used to fit the temperature dependence of ionic conductivity with
| | 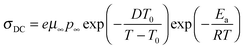 | (8) |
where
μ,
T0, and
D, listed in
Table 4, is the highest temperature limit of mobility, Vogel temperature, and the strength parameter
D that is reciprocally related to fragility, respectively. The uncertainty analysis of
Ea for
Table 3 to be 10% and for
Table 4 the uncertainty for
D is 9%.
Table 4 Fitting parameter (eqn (8)) for the temperature dependence of ionic conductivity
| Sample |
eμ
∞
p
∞ (S cm−1) |
D
|
T
0 (K) |
| Poly[Sty-b-(MVBIm-Tf2N80-co-DEGMEMA20)-b-Sty] |
16 |
4.6 |
230 |
| Poly[Sty-b-(MVBIm-Tf2N72-co-DEGMEMA28)-b-Sty] |
6.6 |
4.8 |
240 |
| Poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] |
4.2 |
4.1 |
250 |
| Poly[Sty-b-(MVBIm-Tf2N35-co-DEGMEMA65)-b-Sty] |
5.2 × 10−3 |
6.9 |
230 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-18 mol% ion content |
9.7 × 10−4 |
6.5 |
240 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-26 mol% ion content |
2.8 × 10+2 |
14 |
212 |
| Poly[Sty-co-VBIm-co-DEGMEMA]-36 mol% ion content |
5.2 × 10+1 |
12 |
201 |
The temperature dependence of the counterion mobility calculated from eqn (5) is plotted in Fig. 8. The VFT temperature dependence observed in Fig. 8 for all ionomers in this study indicates the coupling of ion transport to segmental motion of the polymer. All triblock copolymers had significantly higher ion mobility than the corresponding random copolymers at the same ion content, and this confirms our claim that the microphase-separated morphologies formed in triblock copolymers provide continuous conducting pathways that increases counterion mobility, while ions in random copolymers are hindered by polystyrene segments randomly distributed in the polymer matrix.
 |
| | Fig. 8 Temperature-dependence of ion mobility for A–BC–A triblock copolymers and random copolymers (open symbols) determined using the EP model. | |
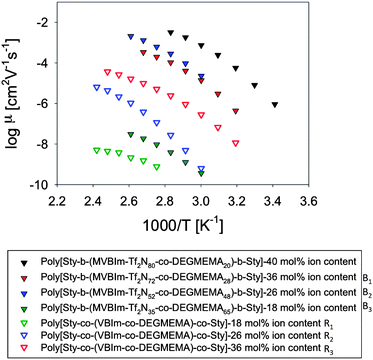
The static dielectric constant εs shown in Fig. 9, was calculated from τσ in eqn (3) and defined as the low-frequency plateau of the dielectric constant ε′(ω) before EP begins. The temperature dependence of static dielectric constant of all copolymers did not obey the Onsager prediction where thermal randomization of dipoles results in εs inversely proportional to temperature and is only applicable to segments with polar groups in the liquid state where their motion is unrestricted by neighbors (far above Tg). The very low εs ≅ 10 for the random and triblock copolymers with ∼18 mol% and their extremely low ionic conductivities suggest that placing the ionic group so close to the polymer backbone may restrict its motion.
 |
| | Fig. 9 Temperature-dependence of static dielectric constant for A–BC–A triblock copolymers and random copolymers (open symbols). | |
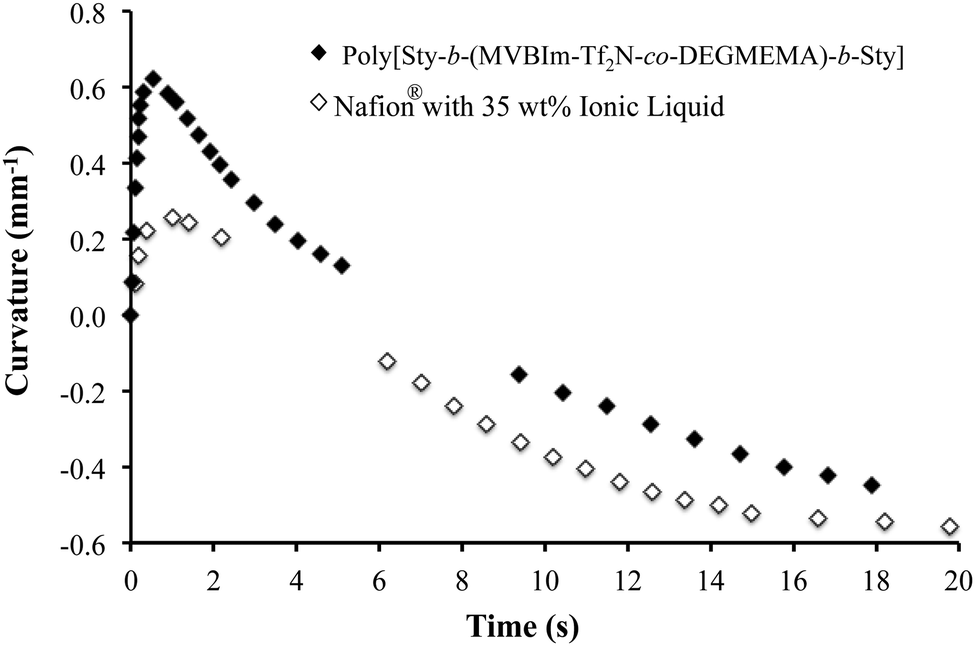
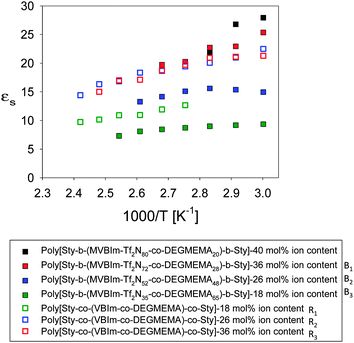
At ambient conditions (20 °C and ∼43% RH), the actuator prepared from a poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] membrane showed bi-directional bending under a 4 V DC input. Distinct from the widely reported relaxation of actuators, this bi-directional behavior is due to difference in size and transport of the two ions of the IL in the actuator, as reported previously.42 Images of the bending performance are shown in supporting figure (ESI†), where the actuator bends first toward the anode after the DC input was applied. The actuator then reverses in the opposite direction and achieves large curvature towards the cathode. The faster cations move through the membrane and rapidly accumulate on the cathode side of the CNC to swell the cathode side of the actuator and cause initial bending in the anode direction. The slower anions complete their transport and accumulate on the anode side of the CNC. The following increased swelling on the anode side of the actuator thus causes the final bending in the cathode direction. Although similar behavior was found in the Nafion® actuators in our previous study,63 as shown in Fig. 10, actuators made with poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] membrane show greater curvature and faster response in the anode direction compared to the cathode direction. This implies a more efficient transport channel for electrolytes provided by the poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] membrane and thus results in faster and larger accumulation of the cations in the cathode side of the CNC. Considering that the poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] actuator contains fewer ions (29.9% weight uptake, as compared to 34.2% of Nafion® actuator), these triblock copolymers are promising candidates for replacing Nafion® in electroactive device applications. The triblock copolymers exhibited electrochemical stability between +4 V and −4 V, as observed with cyclic voltammetry.
 |
| | Fig. 10 Curvature observed for electromechanical actuators made from poly[Sty-b-(MVBIm-Tf2N52-co-DEGMEMA48)-b-Sty] having 26 mol% ion content at ambient conditions (20 °C and ∼43% RH) under a 4 V applied voltage after soaking the film in ionic liquid [EMIm][TfO], uptaking 30 wt%. The open symbols are for similar actuators that use Nafion with an uptake of 35 wt%. | |
Conclusions
RAFT polymerization enabled the synthesis of well-defined ABA triblock copolymers using a difunctional CTA containing a low Tg monomer DEGMEMA in the central block to increase segmental motion and improve ion transport. Controlling the composition of the polystyrene external block and ion content enabled the tuning of thermomechanical properties. SAXS and AFM confirmed the microphase separation and showed that the morphology can be tuned with change in ion content in the triblock copolymer system. DRS demonstrated the importance of triblock copolymer architecture as compared to random copolymers. Ionic conductivity of the copolymers was dependent on morphology, composition and ion-content. The data suggested that these triblock copolymers provide more efficient ion transport channels for electrolytes resulting in faster and greater accumulation of the cations on the anode side of the CNC. The development of these new A–BC–A triblock copolymer systems enables the design of future electroactive devices by changing morphology, ion-content and composition to tune the macromolecular properties.
Acknowledgements
This material is based on work supported by the U.S. Army Research Laboratory and the U.S. Army Research Office under contract/grant number W911NF-07-1-0452 Ionic Liquids in Electro-Active Devices Multidisciplinary University Research Initiative (ILEAD MURI). The authors acknowledge the Laboratory for Research on the Structure of Matter at Penn (MRSEC NSF DMR11-20901) for instrument support.
References
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45 CrossRef CAS PubMed.
- M. D. Green, D. Wang, S. T. Hemp, J.-H. Choi, K. I. Winey, J. R. Heflin and T. E. Long, Polymer, 2012, 3677–3686 CrossRef CAS PubMed.
- T. Wu, D. Wang, M. Zhang, J. R. Heflin, R. B. Moore and T. E. Long, ACS Appl. Mater. Interfaces, 2012, 4, 6552 CAS.
- D. Wang, T. Wu, X. Wan, X. Wang and S. Liu, Langmuir, 2007, 23, 11866 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed.
- T. Chung, W. Janvikul and H. Lu, J. Am. Chem. Soc., 1996, 118, 705 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312 CrossRef CAS PubMed.
- A. J. Duncan, D. J. Leo and T. E. Long, Macromolecules, 2008, 41, 7765 CrossRef CAS.
- G. J. Tudryn, W. Liu, S.-W. Wang and R. H. Colby, Macromolecules, 2011, 44, 3572 CrossRef CAS.
- S.-W. Wang, W. Liu and R. H. Colby, Chem. Mater., 2011, 23, 1862 CrossRef CAS.
- B. J. Akle and D. J. Leo, Smart Mater. Struct., 2007, 16, 1348 CrossRef CAS.
- B. J. Akle, M. D. Bennett, D. J. Leo, K. B. Wiles and J. E. McGrath, J. Mater. Sci., 2007, 42, 7031 CrossRef CAS.
- J. Lin, Y. Liu and Q. Zhang, Polymer, 2011, 52, 540 CrossRef CAS PubMed.
-
A. J. Duncan, D. J. Leo, T. E. Long, B. J. Akle, J. K. Park, R. B. Moore, in The 16th International Symposium on: Smart Structures and Materials & Nondestructive Evaluation and Health Monitoring, International Society for Optics and Photonics, 2009, p. 728711.
- A. Eisenberg, B. Hird and R. Moore, Macromolecules, 1990, 23, 4098 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- M. D. Green, M. H. Allen Jr, J. M. Dennis, D. Salas-de la Cruz, R. Gao, K. I. Winey and T. E. Long, Eur. Polym. J., 2011, 47, 486 CrossRef CAS PubMed.
- M. D. Green, D. Salas-de la Cruz, Y. Ye, J. M. Layman, Y. A. Elabd, K. I. Winey and T. E. Long, Macromol. Chem. Phys., 2011, 212, 2522 CrossRef CAS.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49, 291 CrossRef CAS.
- M. D. Green, J.-H. Choi, K. I. Winey and T. E. Long, Macromolecules, 2012, 45, 4749 CrossRef CAS.
- M. H. Allen, S. T. Hemp, M. Zhang, M. Zhang, A. E. Smith, R. B. Moore and T. E. Long, Polym. Chem., 2013, 4, 2333 RSC.
- C. Jangu, J. H. H. Wang, D. Wang, S. Sharick, J. R. Heflin, K. I. Winey, R. H. Colby and T. E. Long, Macromol. Chem. Phys., 2014, 1319–1331 CrossRef CAS.
- Y. Ye and Y. A. Elabd, Polymer, 2011, 52, 1309 CrossRef CAS PubMed.
- Y. Ye, J.-H. Choi, K. I. Winey and Y. A. Elabd, Macromolecules, 2012, 45, 7027 CrossRef CAS.
- L. Gwee, J.-H. Choi, K. I. Winey and Y. A. Elabd, Polymer, 2010, 51, 5516 CrossRef CAS PubMed.
- Y. Ye, S. Sharick, E. M. Davis, K. I. Winey and Y. A. Elabd, ACS Macro Lett., 2013, 2, 575 CrossRef CAS.
- R. Gao, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Polymer, 2012, 53, 1203 CrossRef CAS PubMed.
- R. L. Weber, Y. Ye, A. L. Schmitt, S. M. Banik, Y. A. Elabd and M. K. Mahanthappa, Macromolecules, 2011, 44, 5727 CrossRef CAS.
- R. L. Weber, Y. Ye, S. M. Banik, Y. A. Elabd, M. A. Hickner and M. K. Mahanthappa, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1287 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Electrochim. Acta, 2001, 46, 1723 CrossRef CAS.
- W. Wang, G. J. Tudryn, R. H. Colby and K. I. Winey, J. Am. Chem. Soc., 2011, 133, 10826 CrossRef CAS PubMed.
- J.-H. H. Wang and R. H. Colby, Soft Matter, 2013, 9, 10275 RSC.
- W. H. Meyer, Adv. Mater., 1998, 10, 439 CrossRef CAS.
- L. Zhang, B. L. Chaloux, T. Saito, M. A. Hickner and J. L. Lutkenhaus, Macromolecules, 2011, 44, 9723 CrossRef CAS.
- R. Montazami, S. Liu, Y. Liu, D. Wang, Q. M. Zhang and J. R. Heflin, J. Appl. Phys., 2011, 109, 104301 CrossRef PubMed.
- R. Montazami, S. Liu, Y. Liu, D. Wang, Q. Zhang and J. R. Heflin, J. Appl. Phys., 2011, 109, 104301 CrossRef PubMed.
- Y. Liu, S. Liu, J. Lin, D. Wang, V. Jain, R. Montazami, J. R. Heflin, J. Li, L. Madsen and Q. Zhang, Appl. Phys. Lett., 2010, 96, 223503 CrossRef PubMed.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473 RSC.
- A. Eisenberg, Macromolecules, 1971, 4, 125 CrossRef CAS.
- L. Wu, E. W. Cochran, T. P. Lodge and F. S. Bates, Macromolecules, 2004, 37, 3360 CrossRef CAS.
- D. B. Klinedinst, E. Yilgör, I. Yilgör, F. L. Beyer and G. L. Wilkes, Polymer, 2005, 46, 10191 CrossRef CAS PubMed.
- J. P. Sheth, G. L. Wilkes, A. R. Fornof, T. E. Long and I. Yilgor, Macromolecules, 2005, 38, 5681 CrossRef CAS.
- R. S. McLean and B. B. Sauer, Macromolecules, 1997, 30, 8314 CrossRef CAS.
- J. P. Sheth, S. Unal, E. Yilgor, I. Yilgor, F. L. Beyer, T. E. Long and G. L. Wilkes, Polymer, 2005, 46, 10180 CrossRef CAS PubMed.
- M. D. Gehlsen, K. Almdal and F. S. Bates, Macromolecules, 1992, 25, 939 CrossRef CAS.
- M. Lee, U. H. Choi, R. H. Colby and H. W. Gibson, Chem. Mater., 2010, 22, 5814 CrossRef CAS.
- J. R. Macdonald, Phys. Rev., 1953, 92, 4 CrossRef CAS.
- R. Coelho, J. Non-Cryst. Solids, 1991, 131, 1136 CrossRef.
- R. Coelho, Rev. Phys. Appl., 1983, 18, 137 CrossRef.
- J. R. Macdonald, Solid State Ionics, 2005, 176, 1961 CrossRef CAS PubMed.
- R. J. Klein, S. Zhang, S. Dou, B. H. Jones, R. H. Colby and J. Runt, J. Chem. Phys., 2006, 124, 144903 CrossRef PubMed.
- D. Fragiadakis, S. Dou, R. H. Colby and J. Runt, J. Chem. Phys., 2009, 130, 064907 CrossRef PubMed.
- D. Fragiadakis, S. Dou, R. H. Colby and J. Runt, Macromolecules, 2008, 41, 5723 CrossRef CAS.
- U. H. Choi, M. Lee, S. Wang, W. Liu, K. I. Winey, H. W. Gibson and R. H. Colby, Macromolecules, 2012, 45, 3974 CrossRef CAS.
-
D. W. Van Krevelen and K. Te Nijenhuis, Properties of polymers: their correlation with chemical structure; their numerical estimation and prediction from additive group contributions, Elsevier Science, 2009 Search PubMed.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. Schubert and I. Krossing, Angew. Chem., 2007, 119, 5480 CrossRef.
- E. D. Gomez, A. Panday, E. H. Feng, V. Chen, G. M. Stone, A. M. Minor, C. Kisielowski, K. H. Downing, O. Borodin, G. D. Smith and N. P. Balsara, Nano Lett., 2009, 9, 1212 CrossRef CAS PubMed.
- Y. Liu, S. Liu, J. Lin, D. Wang, V. Jain, R. Montazami, J. R. Heflin, J. Li, L. Madsen and Q. M. Zhang, Appl. Phys. Lett., 2010, 96, 223503 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Size exclusion chromatography, TGA-sorption analysis, and cyclic voltammetry data. See DOI: 10.1039/c5tc00169b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a