
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Establishing the most favorable metal–carbon bond strength for carbon nanotube catalysts
Fredrik
Silvearv
*ab,
Peter
Larsson
c,
Sarah. L. T.
Jones
d,
Rajeev
Ahuja
be and
J. Andreas
Larsson
abd
aApplied Physics, Division of Materials Science, Department of Engineering Sciences and Mathematics, Luleå University of Technology, 971 87 Luleå, Sweden. E-mail: fredrik.silvearv@ltu.se
bDivision of Materials Theory, Department of Physics and Astronomy, Uppsala University, Box 530, 751 21 Uppsala, Sweden
cNational Supercomputer Center, Linköping University, 581 83, Sweden
dTyndall National Institute, University College Cork, Lee Maltings, Prospect Row, Cork, Ireland
eApplied Materials Physics, Department of Materials and Engineering, Royal Institute of Technology (KTH), 100 44 Stockholm, Sweden
First published on 17th February 2015
Abstract
We have studied a wide range of transition metals to find potential carbon nanotube (CNT) catalysts for chemical vapor deposition (CVD) production. The adhesion strengths between a CNT and a metal cluster were calculated using first principle density functional theory (DFT) for all 1st, 2nd and 3rd row transition metals. We have developed the criterion that the metal–carbon adhesion strength per bond must fulfill a Goldilocks principle for catalyzing CNT growth and used it to identify, besides the well known catalysts Fe, Co and Ni, a number of other potential catalysts, namely Y, Zr, Rh, Pd, La, Ce and Pt. Our results are consistent with previous experiments performed either in a carbon arc discharge environment or by a CVD-process with regard to CNT catalyst activity.
1 Introduction
Carbon nanotubes (CNTs) have properties that make them attractive materials for a range of applications including strong materials, thermal conduction and electronic devices.1–5 For most information and communication technology (ICT) applications there is a CNT manufacturing barrier to their usage, since these require selective growth of all metallic or all semiconducting single-walled carbon nanotubes (SWNTs). The SWNTs' electronic properties are governed by their structure (the diameter and roll direction of the graphene sheet = chirality), which is specified by an index (n,m). Traditional attempts to achieve index-controlled growth of SWNTs are done through the catalytic chemical vapor deposition (CVD) method, where efforts are made to use an effective catalyst, with a narrow distribution of catalyst particle size, and carefully optimizing the chemicals and process parameters, which results in the selective distribution of SWNTs with only a few different indices.6–8 In addition, these experimental attempts use different variations of metal catalyst particle compositions in a more or less trial and error fashion because there are no systematic studies of which metals are suitable CNT catalysts and why. Recently, index-controlled growth has been achieved from both metal–particle templates,9 and from synthesized organic molecular templates.10CNT growth occurs at the interface of the metal particle and the growing tube.11–14 Firstly, CNT growth is nucleated/seeded by formation of hemisphere shaped graphitic fragments on the surface of the metal catalyst particle. When the edge of these carbon domes matches the size of the metal particle, their edges bind in-plane with the surrounding metal atoms. Thus, the edges of the graphitic structures have M–C chemical bonds, while the interior is bound to the metal through physisorption similar to graphene on metal surfaces.15,16 Addition of carbon atoms at the edges of the forming capped nanotube fragment will put strain on and eventually break the physisorption between the metal and the interior sp2 carbon atoms, and the final phase of continued tube growth follows. For a metal to be catalytically active it must form particles that are able to fulfill three key parameters: (i) decompose the carbon feed-stock gas, (ii) form graphitic caps at their surface and (iii) maintain the CNT hollow structure by stabilizing the growing end.17,18 Criterion (iii) follows a Goldilocks principle where the metal–carbon bonds should be strong enough to allow dissociation of the catalytic metal particle and the CNT unfavorable (fulfilled by Fe, Co and Ni),17 but not too strong favoring the formation of metal carbides (which occurs for Mo and W). Too weak metal–carbon bonds cannot stabilize the hollow structure (which is the case for Cu, Au and Pd).17 This Goldilocks principle has been proven by tuning the metal–carbon bond strength by alloying weakly bonding metals (Cu or Pd) with strongly bonding metals (Mo or W)19,20 to achieve CNT growth with mixtures of metals that do not individually act as catalysts. This also shows that criterion (iii) expressed through the strength of the metal–carbon bonds is one of the key parameters for CNT growth. Furthermore, alloying has interest of its own in that it could give a possibility to tune the cluster properties in such a way as to control the quality of the product, as recently shown using WCo by Yang et al.9 All three criteria (i)–(iii) are related to M–C bond strengths, e.g. the temperature for feed-stock gas decomposition is different for the different metals, which may or may not be related to how well a particular metal catalyzes CNT growth, however, criterion (i) is a necessary requirement. Some metals, like Cu, have the ability to catalyze graphene growth and under certain conditions graphitic caps form on its surface (criterion (ii) fulfilled) but this leads to the production of so called ‘giant fullerenes’ rather than CNTs.21,22 In addition, both Cu and Pd can be used as catalysts for the growth of carbon nanofibres,23,24i.e. they fulfill criteria (i) and (ii) but not (iii). Studying criterion (iii) separately from criterion (ii) also relates directly to CNT growth protocols where the formation of graphitic caps is circumvented by seeding the growth with cut segments of pre-produced CNTs or even synthesized rings.25–29 And by seeding using organic molecular templates, as shown by Sanchez-Valencia et al.10 Some work has been done to study these three criteria using different atomistic simulations:
Criterion (i) has been studied for Ni by ab initio electronic structure theory computations of the transition state energies for the decomposition of typical feed-stock molecules on various Ni–surfaces,30 which could be done for a range of metals; criterion (ii) has been studied for Ni at reaction temperatures using tight-binding based Monte Carlo (MC)31–33 and using density functional tight-binding (DFTB) based molecular dynamics (MD) simulations for Ni and Fe,34–36 which could also be done for a range of metals. Criteria (i) and (ii) share common traits with the growth of fibers that also require freeing up of building material on the catalytic metal surface and is dependent on the strength of the binding of carbon in the metal carbide. These have been related to the catalytic ability of a set of metals for CNT growth through the heterogeneous catalysis model in the seminal work of Robertson.37 But criterion (iii) is specific to tube growth, which is due to their hollow structure resulting in an unstable growing end that has to be stabilized through a metal that has the M–C bond strength in the Goldilocks zone (in addition to building material decomposition, surface diffusion, and product precipitation). In this study we are concerned with criterion (iii), which we explore through first principles DFT calculations of the metal–CNT (M–C) adhesion energy for a range of metals, using pure metal particles and perfect CNTs. With this approach we are able to isolate criterion (iii) and study it without the influence of defects, contaminants, temperature, pressure, etc., which we compare with criterion (ii) through bulk cohesive energies calculated from experimental data. Our M–C bond strength could be used as a basis for new parameters for large scale MD and MC simulations of CNT growth on a range of metals to study criteria (ii) and (iii) using dynamics. The behavior of many metals as CNT catalysts is largely unknown. We have studied the metal–carbon bond strength for the 1st, 2nd and 3rd row transition metals in order to find out if there are other metals that have similar values to the traditional CNT catalysts Fe, Co and Ni, and to provide information on how different metals can be alloyed to function as CNT catalysts.
2 Computational methods
All the first principle DFT calculations were performed using the Vienna ab initio Simulation Package,38 the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional39 and the projector augmented-wave method (PAW).40 The carbon nanotube–metal cluster complexes (CNT–metal-complexes), and cluster and tube segments were all modeled in a box of (15 × 15 × 20) Å for the small system and for the large system (20 × 25 × 20) in order to avoid as much as possible unphysical interactions between the CNT–metal-complexes in each supercell. For the k-point mesh a γ-centred (1 × 1 × 1) was used. All calculations performed were spin polarized with no symmetry constraints and the energy cutoff was set to 500 eV. The Gaussian smearing was tuned so that the difference between the free energy and the total energy is less than 1 meV per atom. The energy convergence was set to 1 × 10−5 eV. We have optimized the complex and the metal cluster taking precautions to retain the symmetric structure shown in Fig. 1 and 2 as much as possible for all clusters so that the shape of the free clusters is as similar in shape as possible to that of the CNT–metal-complexes. To be able to make a just comparison of bond strength between systems of the same size we have chosen the same symmetry for all clusters so the metal–carbon coordination is the same.41 Our small model system consists of a (5,0)-tube and a thirteen atom large metal cluster, and our large system constitutes a (10,0)-tube and a cluster of fifty-five atoms. Three separate relaxations were done to obtain the total energies, the CNT–metal-complex, the cluster and the nanotube. From these energies the metal–carbon adhesion energies per bond were calculated as a measure of M–C bond strength (eqn (1)).EM–C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) bond = (ECNT–cluster − (Ecluster + ECNT))/n bond = (ECNT–cluster − (Ecluster + ECNT))/n | (1) |
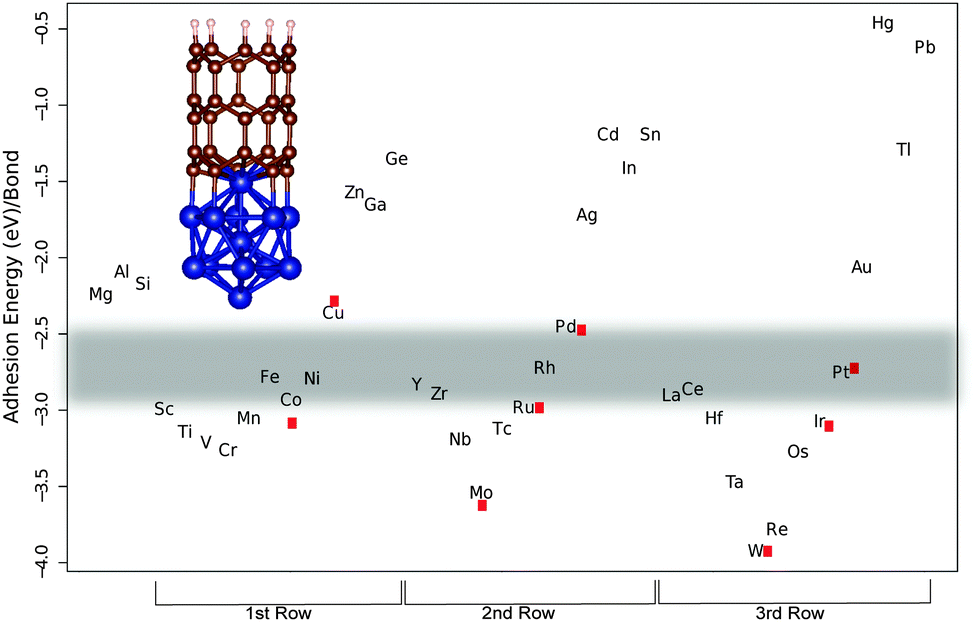 | ||
| Fig. 1 The calculated adhesion energies per bond for the first three transition metal rows. The red dots are the results received from TURBOMOLE calculations. The shaded region marks the ‘Goldilocks zone’. The inset in the figure: a picture of the (5,0)-M13 complex. | ||
 | ||
| Fig. 2 The calculated adhesion energies per bond for the first three transition metal rows. The shaded region marks the ‘Goldilocks zone’. The inset in the figure: a picture of the (10,0)-M55 complex. | ||
3 Results and discussion
We have studied three unit cell long (5,0) and (10,0) SWNT segments. On one end the dangling bonds are hydrogen terminated to simulate a capped end and the other end is attached to the M13 and M55 metal cluster, respectively (see Fig. 1 and 2). There are considerable differences in the bond strength of armchair and zigzag SWNT and graphene edges,17,18,43–46 which originate from fragment stabilization in the form of triple bond formation on armchair edges.47,48 This has been shown to be a collective effect that occurs when several neighboring bonds are broken, however, the individual M–C bonds are of equal strength even for the two extreme armchair and zigzag ends.47,48 We have thus opted to calculate the M–C bond strengths for the zigzag tubes (10,0) and (5,0) that do not undergo triple-bond fragment stabilization. In Fig. 1 and 2 we show three rows of transition metals in the order of 1st, 2nd and 3rd from left to right. The main trend that can be observed is that the bond strength decreases when going from left to right along a row. If we look at the first row that begins with Sc, there is a slight increase in bond strength that culminates with Cr (bold marks the values for the (10,0)-M55 complex) (−2.85/−3.24 eV). After that a steady decrease in bond strength follows until the end of the row. Similarly, the second row beginning with Y starts with a sharp increase in bond strength down to Mo (−3.42/−3.61 eV). Then there is a steady decrease in bond strength up to Cd (−1.34/−1.19 eV) and Sn (−1.21/−1.19 eV). The third row begins with La and again the pattern is repeated with an increase in bond strength down to W (−3.40/−3.68 eV) followed by a steep decrease in bond strength. We have used experimental data on successful CNT production to outline the borders of the ‘Goldilocks zone’. In the first row we have the well known catalyst metals Fe, Co, and Ni that have adhesion energies per M–C bond of (−2.56/−2.78 eV), (−2.63/−2.93 eV), and (−2.65/−2.79 eV), respectively. Candidates for other catalyst metals should be in the same or at least close to this energy range. Values that are particularly close are for Y (−2.44/−2.83 eV), Zr (−2.60/−2.89 eV), La (−2.67/−2.90 eV) and Ce (−2.59/−2.78 eV). Of these, Y and Ce have been used as pure catalysts in trials using the laser ablation and the arc discharge method, of which the latter produced SWNTs with both metals.49–52 However, as far as we know they have not been tried as catalysts in CVD. Y is commonly alloyed with Ni to promote SWNT49,50,53,54 or even DWNT growth by controlling the composition ratio of Y and Ni.54Deck and Vecchio have tested a range of metals beside Fe, Co and Ni, consisting of Cr, Mn, Zn, Cd, Ti, Zr, La, Cu, V, and Gd in a CVD-process55 without success. They claim from studying metal–carbon binary phase diagrams that either the carbon solubility is too low, as in the case of Cu, Zn and Cd, which is in line with our calculated bond strengths that are weak, or they form many different intermediate carbides as in the case of Cr, Mn, Ti, Zr, La and V. This is also consistent with our results in that all of them except Zr and La bind stronger to carbon than Co thus forming more stable carbides than Fe, Co and Ni, making it difficult for the diffusion required for graphitic precipitation to occur. We should also mention that it has been for a long time the common opinion that a prerequisite for nanotube growth is the formation of carbide. This seems not to be the case since pure Fe metal clusters have been found to result in nanotube growth.56 Zr and La did not act as catalysts may be because the catalytic process is thwarted by the reported chlorine contamination. In support of this hypothesis is that they have used lanthanum chloride (LaCl3) and zirconium chloride (ZrCl) with no success, while Saito et al. found La in the form of La2O3 to be highly active for SWNT growth in an carbon arc discharge environment,52 and Wu et al. found Zr in the form of a Zr plate to produce DWCNTs and triple walled nanotubes in CVD.57
Other metals that are close in energy to the catalytic window are Sc (−2.86/−2.99 eV), Cr (−3.24/−3.25 eV), Mn (2.86/−3.05 eV), Ru (−3.02/−2.98 eV), Rh (−2.74/−2.72 eV), Ir (−2.96/−3.07 eV) and Pt (−2.58/−2.75 eV). Saito et al. have tested the platinum-group metals (Ru, Rh, Pd, Os, Ir, and Pt) in an arc discharge environment.58 Only Rh, Pd and Pt showed catalytic activity for SWNT growth. Recently Pt has been used to successfully grow SWNTs in CVD.10,59–61 It seems from analyzing our calculations in the light of the above experimental data that Rh constitutes the lower end of bonding strength since both Sc and Ru that bind a little bit stronger do not promote CNT growth.52 We judge from our calculations that Pd is the upper end of bond strength for CNT catalytic activity, since Saito et al. have found Pd to be active in SWNT growth. But the length of the tubes grown from Pd particles was shorter (30–50 nm) compared to Rh (10–>200 nm) and Pt (10–100 nm). This difference in length follows the calculated bond strength trend with regard to catalyst suitability, and can be explained by slower tube growth when the bond strength is weak. Other studies report that Pd grows carbon nanowires, not nanotubes, through CVD,20 and previous computational work has shown Pd to be a borderline case for CNT catalytic activity.17 Another study by Saito and Nishikubo shows that CNT growth on Pd nanoparticles in an arc discharge environment is highly dependent on the pressure, high He pressure (600 torr) produced CNTs in abundance (approximately 40 nm long), but at low pressure (100 torr) they were more scarce. Instead they report finding more naked Pd particles (neither CNT nor graphitic layers on their surfaces) and graphite wrapped Pd particles.62 It seems for Pd to act as a CNT catalyst it needs 'help' to get the carbon to stay on the particle surface, either high pressure or a dopant like Mo.20 We speculate that maybe Cu could act as a CNT catalyst if the ambient pressure is as high or possibly even higher than in the case of Pd, since Cu also, like Pd, can be made catalytic when alloyed with Mo or W.19 As mentioned above, Sc did not have any catalytic activity for tube growth. In an earlier paper Saito et al. tested a series of rare-earth elements (Sc, Y, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu) in a carbon arc discharge environment and only Y, La and Ce showed high activity for SWNT growth.52 Some elements, Gd, Tb, Ho, Er and Lu, had an intermediate activity and two elements, Pr and Nd, had low activity. The rest did not show any activity at all. Our results of how a metal particle enables CNT growth by stabilizing the growing end according to criterion (iii) can be compared to criterion (ii) through the combination of the experimental cohesive energies of metals and carbides studied in the work of Guillermet et al.63–65 (see Fig. 3), for the carbide forming metals (no value could be got for e.g. Cu that actually fulfills criterion (ii)). Comparing these two measures shows that there are similarities but only a subset of the metals that fulfills criterion (ii) also fulfills criterion (iii), which cements the importance of studying criteria (ii) and (iii) independently, and the separation of bulk properties from properties arising from clusters.
 | ||
| Fig. 3 The bulk cohesive energies per bond at zero Kelvin and one atmosphere, calculated from experimental thermodynamic data (Guillermet et al.). The shaded region marks the ‘Goldilocks zone’ as established in Fig. 1 and 2. | ||
We also include Mg, Al, and Si in our calculations as they are commonly used as substrates in the form of oxides. Future projects could be used to calculate the elements not covered in this study and in addition, metal oxides such as TiO2, Al2O3, ZrO2 and SiO2 that have been reported to be active CNT catalysts,66–69 to see if they follow the bond strength trend, and to compute the metals in this study as carbides and/or with the carbon-covered surface to see if the bond strengths are altered. However, as Robertson37 points out when establishing the heterogeneous catalyst model of CNT growth, our arguments of bond strength will probably not differ between the pure metal and the carbide, since the nanoparticle surface will always be partly metal and partly carbide when CNT growth occurs. As mentioned above one can mix two metals one with weak and one with strong bond strength to produce nanoparticles that are in the ‘Goldilocks zone’.19,20 Here a plethora of possibilities opens up if one considers any of the metals above the ‘Goldilocks zone’ combined with any metal under the zone. E.g. one could combine Ag with Ta and judging from the distances from the zone the mix ratio should be expected to be somewhere around 1:1.5. And more examples are Cu and Nb in a ratio around 1:2 to 1:3, or Au and Os in a ratio of 1:1, or Al and Sc in a ratio of 1:5 to 1:6. In addition, we speculate that metal catalysts with similar metal–carbon bond strength to the traditional catalyst Fe, Co and Ni, but with a different atomic size/lattice constant could promote the growth of SWNTs with a specific index. E.g. if the M–M distance is significantly longer it could favor growth of zigzag tubes, since these have the longest length between M–C bonds at the interface to the catalyst. In analogy, metals with very short M–M distances could favor armchair tubes.
4 Conclusions
A wide range of transition metals have been studied with respect to the carbon–metal adhesion energy per bond using first principle DFT, a number of metals (Y, Zr, Rh, Pd, La, Ce, Pt) are found to have potential catalytic properties for CNT growth apart from the traditional catalysts Fe, Co and Ni. Comparison with experiments in the arc discharge environment has shown that these metals indeed show CNT catalytic activity. We have used these findings to establish the optimal M–C bond strength for CNT growth, which forms a ‘Goldilocks zone’. In this zone the M–C bonds are strong enough to stabilize the CNT hollow structure but not too strong favoring the formation of stable carbides. It will be interesting to see how these metals function in a CVD environment, since this is only known for some of them, when other parameters come into play such as substrate choice, impurities, temperature and pressure. For example, these non-traditional catalysts could help in the quest for CVD growth of CNTs at lower temperature and lower pressure. We hope our findings will inspire further research in testing new metals for CVD growth of CNTs, and also ideas about new metal mixtures.Acknowledgements
The authors thank National Supercomputer Center in Linköping (NCS) and High Performance Computing Center North (HPC2N) for allocation of time and resources, through the Swedish National Infrastructure for Computing (SNIC), and the Irish Centre for High-End Computing (ICHEC). We would like to acknowledge the Swedish Research Council (VR), Carl Tryggers Stiftelsen and Kempestiftelserna for financial support.References
- I. Ivanov and A. Puretzky, Appl. Phys. Lett., 2006, 89, 223110 CrossRef PubMed.
- W. Kim, R. Wang and A. Majumdar, Nano Today, 2007, 2, 40 CrossRef.
- P. Avouris, Z. Chen and V. Perebeinos, Nat. Nanotechnol., 2007, 2, 605 CrossRef CAS PubMed.
- G. F. Close, S. Yasuda, B. Paul, S. Fujita and H. S. P. Wong, Nano Lett., 2008, 8, 706 CrossRef CAS PubMed.
- J. Chaste, L. Lechner, P. Morfin, G. T. K. Feve, J. M. Berroir, D. C. Glattli, H. Happy, P. Hakonen and B. Placais, Nano Lett., 2008, 8, 525 CrossRef CAS PubMed.
- H. R. Barzegar, F. Nitze, T. Sharifi, M. Ramstedt, C. W. Tai, A. Malolepszy, L. Stobinski and T. Wågberg, J. Phys. Chem. C, 2012, 116, 12232 CAS.
- A. R. Harutyunyan, G. G. Chen, T. M. Paronyan, E. M. Pigos, O. A. Kuznetsov, K. Hewaparakrama, S. M. Kim, D. Zakharov, E. A. Stach and G. U. Sumanasekera, Science, 2009, 326, 116 CrossRef CAS PubMed.
- L. T. Qu, F. Du and L. M. Dai, Nano Lett., 2008, 8, 2682 CrossRef CAS PubMed.
- F. Yang, et al. , Nature, 2014, 510, 522 CrossRef CAS PubMed.
- J. R. Sanchez-Valencia, T. Dienel, O. Gröning, I. Shorubalko, A. Mueller, M. Jansen, K. Amsharov, P. Ruffieux and R. Fasel, Nature, 2014, 512, 61 CrossRef CAS PubMed.
- S. Helveg, C. Lopez-Cartes, J. Sehested, P. L. Hansen, B. S. Clausen, J. R. Rostrup-Nielsen, F. Abild-Pedersen and J. K. Norskov, Nature, 2004, 427, 426 CrossRef CAS PubMed.
- S. Hofmann, R. Sharma, C. Ducati, G. Du, C. Mattevi, C. Cepek, M. Cantoro, S. Pisana, A. Parvez, F. Cervantes-Sodi, A. C. Ferrari, R. Dunin-Borkowski, S. Lizzit, L. P. Goldoni and A. Robertson, J. Nanosci. Lett., 2007, 7, 602 CrossRef CAS PubMed.
- Y.-H. Shin and S. Hong, Appl. Phys. Lett., 2008, 92, 043103 CrossRef PubMed.
- C. Journet, M. Picher and V. Jourdain, Nanotechnology, 2012, 23, 142001 CrossRef PubMed.
- M. Vanin, J. J. Mortensen, A. K. Kelkkanen, J. M. Garcia-Lastra, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 081408 CrossRef.
- I. Hamada and M. Otani, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 153412 CrossRef.
- F. Ding, P. Larsson, J. A. Larsson, R. Ahuja, H. Duan, A. Rosén and K. Bolton, Nano Lett., 2008, 8, 463 CrossRef CAS PubMed.
- P. Larsson, J. A. Larsson, R. Ahuja, F. Ding, B. Yakobson, H. Duan, A. Rosén and K. Bolton, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 115419 CrossRef.
- Z. Li, J. A. Larsson, P. Larsson, R. Ahuja, J. M. Tobin, J. M. O'Byrne, A. Morris, G. Attard and J. D. Holmes, J. Phys. Chem. C, 2008, 112, 12201 CAS.
- J. O'Byrne, J. M. Tobin, J. A. Larsson, P. Larsson, R. Ahuja, Z. Li and J. D. Holmes, J. Phys. Chem. C, 2010, 114, 8115 Search PubMed.
- T. Cabioch, J. P. Riviere and J. Delafond, J. Mater. Sci., 1995, 30, 4787 CrossRef CAS.
- F. L. Normand, L. Constant, G. Ehret and C. Speisser, J. Mater. Res., 1999, 14, 560 CrossRef.
- B. Farmer, D. Holmes, L. Vandeperre, R. Stearn and W. Clegg, MRS fall meetingon nanomaterials for structural applications, PA Boston, 2002, p. 81 Search PubMed.
- X. Tao, X. Zhang, J. Cheng, Y. Wang, F. Liu and Z. Luo, Chem. Phys. Lett., 2005, 409, 89 CrossRef CAS PubMed.
- Y. Wang, M. J. Kim, H. Shan, C. Kittrell, H. Fan, L. M. Ericson, W.-F. Hwang, S. Arepalli, R. H. Hauge and R. E. Smalley, Nano Lett., 2005, 5, 997 CrossRef CAS PubMed.
- R. E. Smalley, Y. Li, V. C. Moore, B. K. Price, R. Colorado, H. K. Schmidt, R. H. Hauge, A. R. Barron, J. T. Iwasaki, J. Robertson and H. Kawarada, Nano Lett., 2008, 8, 886 CrossRef PubMed.
- M. Tour, J. Am. Chem. Soc., 2006, 128, 15824 CrossRef PubMed.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572 CrossRef CAS PubMed.
- J. Liu, C. Wang, X. Tu, B. Liu, L. Chen, M. Zheng and C. Zhou, Nat. Commun., 2012, 3, 1199 CrossRef PubMed.
- I. Lebedeva, A. Knizhnik, A. Gavrikov, A. Baranov, B. Potapkin, S. Aceto, P.-A. Bui, C. Eastman, U. Grossner, D. Smith and T. Sommerer, Carbon, 2011, 49, 2508 CrossRef CAS PubMed.
- M. Diarra, H. Amara, C. Bichara and F. Ducastelle, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 245446 CrossRef.
- M. Diarra, A. Zappelli, H. Amara, F. Ducastelle and C. Bichara, Phys. Rev. Lett., 2012, 109, 185501 CrossRef CAS.
- M. Diarra, H. Amara, F. Ducastelle and C. Bichara, Phys. Status Solidi B, 2012, 249, 2629 CrossRef CAS.
- A. J. Page, H. Yamane, Y. Ohta, S. Irle and K. Morokuma, J. Am. Chem. Soc., 2010, 132, 15699 CrossRef CAS PubMed.
- A. J. Page, S. Minami, Y. Ohta, S. Irle and K. Morokuma, Carbon, 2010, 48, 3014 CrossRef CAS PubMed.
- Y. Ohta, Y. Okamoto, A. J. Page, S. Irle and K. Morokuma, ACS Nano, 2009, 3, 3413 CrossRef CAS PubMed.
- J. Robertson, J. Mater. Chem., 2012, 22, 19858 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS.
- M. J. Piotrowski, P. Piquini and J. L. F. D. Silva, Phys. Rev. Lett., 2010, 81, 155446 Search PubMed.
- R. Ahlrichs, M. Bär, H. Häser, H. Horn and C. Kölmel, Chem. Phys., 1989, 162, 165 CAS.
- Y. Liu, A. Dobrinsky and B. I. Yakobson, Phys. Rev. Lett., 2010, 105, 235502 CrossRef.
- H. Dumlich and S. Reich, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 085421 CrossRef.
- A. Börjesson and K. Bolton, J. Phys. Chem. C, 2011, 115, 24454 Search PubMed.
- F. Ding, A. R. Harutyunyan and B. I. Yakobson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2506 CrossRef CAS PubMed.
- J. D. Baran, W. Kołodziejczyk, P. Larsson, R. Ahuja and J. A. Larsson, Theor. Chem. Acc., 2012, 131, 1270 CrossRef PubMed.
- Y. Li, R. Ahuja and J. A. Larsson, J. Chem. Phys., 2014, 140, 091102 CrossRef PubMed.
- E. Muñoz, W. K. Maser, A. M. Benito, M. T. Martínez, G. F. de laFuente, A. Righi, J. L. E. A. Sauvajol and Y. Maniette, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 145 CrossRef.
- M. Yudasaka, N. Sensui, M. Takizawa, S. Bandow, T. Ichihashi and S. Iijima, Chem. Phys. Lett., 1999, 312, 155 CrossRef CAS.
- D. Zhou, S. Seraphin and S. Wang, Appl. Phys. Lett., 1994, 65, 1593 CrossRef CAS PubMed.
- Y. Saito, K. Kawabata and M. Okuda, J. Phys. Chem., 1995, 99, 16076 CrossRef CAS.
- J. Gavillet, A. Loiseau, F. Ducastelle, S. Thair, P. Bernier, O. Stéphan, J. Thibault and J.-C. Charlier, Carbon, 2002, 40, 1649 CrossRef CAS.
- T. Sugai, T. Okazaki, H. Yoshida and H. Shinohara, New J. Phys., 2004, 6, 21 CrossRef.
- C. P. Deck and K. Vecchio, Carbon, 2006, 44, 267 CrossRef CAS PubMed.
- C. T. Wirth, B. C. Bayer, A. D. Gamalski, S. Esconjauregui, R. S. Weatherup, C. Ducati, C. Baehtz, J. Robertson and S. Hofmann, Chem. Mater., 2012, 24, 4633 CrossRef CAS.
- H. Wu, C. Huang, M. Youh, C. Tseng, H. Chen, Y. Li and A. Sakoda, Carbon, 2010, 48, 1897 CrossRef CAS PubMed.
- Y. Saito, K. Nishikubo, K. Kawabata and T. Matsumoto, J. Appl. Phys., 1996, 80, 3062 CrossRef CAS PubMed.
- Y. Mizutani, N. Fukuoka, S. Naritsuka, T. Maruyama and S. Iijima, Diamond Relat. Mater., 2012, 26, 78 CrossRef CAS PubMed.
- T. Iokawa, T. Tsutsui, S. Naritsuka and T. J. Maruyama, J. Appl. Phys., 2012, 51, 1 Search PubMed.
- N. Fukuoka, Y. Mizutani, S. Naritsuka, T. Maruyama and S. J. Iijima, J. Appl. Phys., 2012, 51, 1 Search PubMed.
- Y. Saito and K. Nishikubo, J. Phys. Chem. Solids, 1996, 57, 243 CrossRef CAS.
- J. Haglund, G. Grimvall, T. Jarlborg and A. F. Guillermet, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 14400 CrossRef.
- A. F. Guillermet, J. Haglund and G. Grimvall, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 11557 CrossRef.
- A. F. Guillermet, J. Haglund and G. Grimvall, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 11673 CrossRef.
- Q. Cai, Y. Hu, Y. Liu and S. Huang, Appl. Surf. Sci., 2012, 258, 8019 CrossRef CAS PubMed.
- H. Liu, D. Takagi, H. Ohno, S. Chiashi, T. Chokan and Y. Homma, Appl. Phys. Express, 2008, 1, 014001 CrossRef.
- S. A. Steiner III, T. F. Baumann, B. C. Bayer, R. Blume, M. A. Worsley, W. J. Moberly-Chan, E. L. Shaw, R. Schlögl, A. J. Hart, S. Hofmann and B. L. Wardle, J. Am. Chem. Soc., 2009, 131, 12144 CrossRef PubMed.
- A. Bachmatiuk, F. Börrnert, F. Schäffel, M. Zaka, G. S. Martynkowa, D. Placha, R. Schönfelder, P. M. Costa, N. Ioannides, J. H. Warner, R. Klingeler, B. Büchner and M. H. Rümmeli, Carbon, 2010, 48, 3175 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |