
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An isoindigo containing donor–acceptor polymer: synthesis and photovoltaic properties of all-solution-processed ITO- and vacuum-free large area roll-coated single junction and tandem solar cells†
Rasmus Guldbaek
Brandt‡
ab,
Wei
Yue‡
ab,
Thomas Rieks
Andersen
c,
Thue Trofod
Larsen-Olsen
c,
Mogens
Hinge
d,
Eva
Bundgaard
c,
Frederik C.
Krebs
c and
Donghong
Yu
*a
aDepartment of Chemistry and Bioscience, Aalborg University, Fredrik Bajers Vej 7H, DK-9220, Aalborg East, Denmark. E-mail: yu@bio.aau.dk
bSino-Danish Centre for Education and Research (SDC), Niels Jenses Vej 2, DK-8000, Aarhus, Denmark
cDepartment of Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, DK-4000, Roskilde, Denmark
dDepartment of Engineering, Aarhus University, Hangoevej 2, 8200 Aarhus N, Denmark
First published on 16th January 2015
Abstract
In this work, the design, synthesis, and characterization of a donor–acceptor polymer from dithieno[3,2-b:2′,3′-d]pyrrole and isoindigo (i-ID) are presented. The synthesized polymer has been applied in the fabrication of large area ITO-free organic photovoltaic devices, both by spin coating and roll coating; the latter was used to construct both single junction and multi-junction organic photovoltaic (OPV) architectures.
In recent years, there has been an increasing focus on the development of electron deficient heteroaromatic moieties for the synthesis of donor–acceptor (D–A) type conjugated polymers for organic photovoltaic devices (OPVs), such as phthalimide (PI),1 naphthalene diimide (NDI),2 perylenediimide (PDI),3 benzothiadiazole (BT) and benzobis(thiadiazole) (BBT),4 diketopyrrolopyrrole (DPP),5 benzodipyrrolidone (BDP),6 thieno[3,4-c]pyrrole-4,6-dione (TPD),7 and isoindigo (i-ID).8 When incorporated with donor units (alternating) in the polymer backbone, the electron richness and electron deficiency ensure a low band gap, thus allowing for more efficient absorbance of incident solar photons over a wider part of the solar spectrum.9
Electron deficient monomers are essential for the design of low band gap (LBG) conjugated polymers, as such moieties in the polymer main chain exhibit high electron affinity and strong ability to reduce the band gaps of D–A polymers compared to the corresponding homo-polymers. They can especially tune the energy level of the lowest unoccupied molecular orbital (LUMO) of the given polymer, which is the key parameter, being associated with the charge transfer process in OPVs. The optimal driving force for the charge transfer process has been commonly recognized as 0.3 eV, i.e. the LUMO energy difference between the donor (such as in D–A polymers) and the acceptor (state-of-the-art phenyl-C61-butyric acid methyl ester (PC[60]BM)).10 Among those previously mentioned acceptor moieties, i-ID is especially interesting as an efficient acceptor block; due to its strong electron-withdrawing characteristics and a large optical transition dipole, the interest was first sparked by Reynolds et al.11 who showed that an i-ID containing oligothiophene can be used as a p-type OPV material, with a power conversion efficiency (PCE) of 1.76%. Independent reports have been published since 2011 by Zhang et al.,12 Liu et al.,13 and Wang et al.14 with PCE ranging from 1 to 3%, the high PCE of 6.3% was obtained by Wang et al.15 in 2011. This was until very recently the best performing i-ID containing polymer, but it was overtaken by Geng et al.16 who reported a PCE of 8.2% in 2014, which is now the highest PCE for i-ID based LBG polymers. On the other hand, for D–A LBG conjugated polymers, the fused ring donor unit dithieno[3,2-b:2′,3′-d]pyrrole (DTP) is a widely used electron rich moiety and has been proven to have excellent electron donating properties and good molecular planarity. A similar D–A combination of bisthiophene–DTP (BT–DTP) has been reported by Wu et al., poly(bisthiophenedithieno[3,2-b:2′,3′-d]pyrrole-isoindigo) (PBTDTPI) afforded OPV devices with efficiencies in the range of around 1.86%.17 However, all the above-mentioned “hero” i-ID based LBG polymer solar cells including PBTDTPI are single junction solar cells with small active areas (not exceeding 0.1 cm2) on rigid glass substrates using ITO as the semitransparent front electrode and vacuum evaporated metal back electrode. Indium scarcity, its high market demand and the high energy input required for ITO production using vacuum techniques have become the bottlenecks in developing/commercializing OPV devices due to the resulting non-sustainability, high cost and high energy payback time.18 Therefore the replacement of indium becomes essential and the substitute should preferably be a solution-based ambient processable system. A possible combination of poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) with a metal grid remaining on flexible plastic (e.g. PET) substrates turns out to be the most readily scalable, facile alternative to ITO coated glass.18 Large area ITO- and vacuum-free OPV device can be fabricated in a fast and scalable manner by roll-to-roll (R2R) slot-die coating, which has been identified and explored using a large array of techniques by few groups, but with great success so far. However, all but a few reports have used poly(3-hexylthiophene) (P3HT) as a polymer donor.19 Although the above-introduced i-ID containing D–A LBG polymers have outperformed P3HT with regard to PV performance with PCEs up to 7.2%, there is no guarantee on transferring this high PV performance when upscaled to a R2R setting from conventional spin-coating and vacuum deposition techniques. It is due to the inherent differences in processing, many of the achievements for small area OPV devices are likely to prove difficult to reproduce using the R2R methods. In order to minimize/close this gap of knowledge, more effort must be devoted to the testing of new promising materials in R2R fabricated PSCs. As a consequence of the discrete nature of the absorbance bands of conjugated organic molecules, a necessity to absorb different regions of the solar spectrum emerges. To capture the entirety of the solar spectrum, tandem solar cells with compensated light absorption can be employed. Such a tandem structure will in principle lead to a higher performance than the single junction in spite of the expense of extra processing steps and enhanced material consumption. In this paper, a novel D–A polymer PDTPI incorporating DTP and i-ID as donor and acceptor moieties, respectively, was successfully synthesized via Pd-catalyzed Stille coupling reaction. Vacuum- and ITO-free OPV devices from PDTPI have been roll-coated as single junction and tandem solar cells with P3HT and PBDTTTz-4 in the area scale of 1 cm2, and the photophysical and electrochemical properties of PDTPI and photovoltaic properties of spin coated single junction and roll coated large area (∼1 cm2) single junction and tandem cells were fully investigated.
Experimental
Materials and synthesis
The monomers 2,6-di(trimethyltin)-N-(1-pentylhexyl)dithieno[3,2-b:2′,3′-d]-pyrrole and 6,6′-dibromodi(2-ethylhexyl)isoindigo were synthesized following the procedures reported in the literature.11,20 Poly-3-hexylthiophene (P3HT from Plextronics) had a Mn of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da. PBDTTTz-4 was synthesized according to literature procedures,37,38 phenyl-C61-butyric acid methyl ester (PC[60]BM, from Solenne) had a purity of 99%. PEDOT:PSS was purchased from Agfa (Orgacon EL-P 5010), or Heraeus (Clevious P VP Al 4083 and Clevious F-010). Electron transport layers (ETL) were coated using a stabilized ZnO nanoparticle solution in acetone. Thermally curable Ag (PV-410) was purchased from Dupont. All other chemicals were used as received, unless noted.
000 Da. PBDTTTz-4 was synthesized according to literature procedures,37,38 phenyl-C61-butyric acid methyl ester (PC[60]BM, from Solenne) had a purity of 99%. PEDOT:PSS was purchased from Agfa (Orgacon EL-P 5010), or Heraeus (Clevious P VP Al 4083 and Clevious F-010). Electron transport layers (ETL) were coated using a stabilized ZnO nanoparticle solution in acetone. Thermally curable Ag (PV-410) was purchased from Dupont. All other chemicals were used as received, unless noted.
Polymerization of PDTPI
A mixture of 2,6-di(trimethyltin)-N-(1-pentylhexyl)dithieno[3,2-b:2′,3′-d]-pyrrole (104.30 mg, 0.158 mmol), 6,6′-dibromodi(2-ethylhexyl)isoindigo (100.00 mg, 0.155 mmol), tris(dibenzylideneacetone)dipalladium (Pd2(dba)3, 2.84 mg, 3.10 μmol), and tri-o-tolylphosphine (P(o-Tol)3, 7.55 mg, 24.8 μmol) was preliminarily degassed with N2, and then toluene (16 mL) was added. The mixture was further purged with N2 for 20 min and heated to 120 °C for 48 h. Subsequently the reaction was cooled to room temperature and the solution was precipitated in methanol. The crude polymer was collected by filtration and then purified on a Soxhlet extractor using acetone and hexane in succession. The polymer was extracted with chloroform and obtained by precipitating the chloroform solution in methanol, yielding 105 mg (55%) solid dark polymer. GPC: 8860 Da; PDI 1.52. 1H NMR (600 MHz, CDCl3, δ, ppm): 9.11 (d, 2H), 6.65–7.31 (m, 6H), 4.39 (s, br, 1H), 3.75 (s, br, 4H), 0.95–2.27 (m, br, 52H).Measurements
1H NMR spectra were recorded on a Bruker AvanceIII 600 MHz spectrometer. High temperature gel permeation chromatography (HT-GPC) measurements were conducted on PL-GPC220 equipment at 150 °C with 1,2,4-trichlorobenzene as eluent and polystyrene as standards. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer STA6000 simultaneous thermal analyzer at a heating rate of 10 °C min−1 under nitrogen flow. UV-vis absorption was recorded on a Varian 50 Bio UV-vis spectrophotometer. Cyclic voltammetry (CV) was performed on a CHI660B electrochemical analyzer (CH Instruments) using a three-electrode cell set-up with 0.1 M TATBF4 in acetonitrile as the electrolyte at a scan rate of 1 V s−1. A glassy carbon working electrode with a diameter of 1 mm, a platinum-counter electrode, and an Ag/Ag+ pseudo reference electrode were applied. The potential was calibrated against the ferrocene/ferrocenium redox couple (Fc/Fc+, 4.8 eV below the vacuum level). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were estimated by the equations: HOMO = −(4.80 + Eonsetox) and LUMO = HOMO + Eopticalbg.Solar cell fabrication and characterization
Spin-coated single junction solar cell
Single junction devices were prepared on a flexible poly(ethylene terephthalate) (PET) substrate using a highly conductive PEDOT:PSS (Heraeus Clevious PH1000) for charge collection, the electron transport layer (ETL) was spin-coated using an aluminium doped ZnO nanoparticle solution in acetone (49 mg mL−1) at 1000 rpm followed by spin coating of a dichlorobenzene solution of PDTPI:PC[60]BM (1:2, 20 mg mL−1) at 600 rpm. The hole transport layer (HTL) of PEDOT:PSS (Agfa EL-P-5010:isopropanol 2:1 w/w) was spin-coated at 3000 rpm, and finally Ag was vacuum evaporated as the back electrode (Ag). The active area of the OPV cell was 0.25 cm2.
Roll-coated single junction solar cells
All roll coated devices were printed on a substrate known as the Flextrode comprising PEDOT:PSS (Heraeus Clevious PH1000) and ZnO on a barrier material with a large scale roll-to-roll flexographic printed silver grid electrode. The active layer PDTPI:PC[60]BM (1:X) and HTL PEDOT:PSS (Clevious P VP Al 4083 and Clevious F-010) were all slot-die coated as reported by Andersen et al.21,22 The back silver electrode was applied by flexographic printing of a heat curing silver paste PV410 (Dupont) as previously reported.22 All cells possess an active area of 1 cm2. All layer thicknesses can be seen in ESI Table S1.†
Roll-coated tandem solar cells
Tandem cells were coated by a method comparable to that used for roll-coated single junction solar cells as described above. LBG polymers PBDTTTz-4:PC[60]BM (1:1) and PDTPI:PC[60]BM (1:2) were slot-die coated as the first and second active layers, respectively, isolated by PEDOT:PSS F010, Al 4083 and ZnO layers in between. To minimize the risk of solvent penetration through the intermediate layer the second active layer was coated at 60 °C for faster evaporation. Prior to flexographic printing of the silver electrode, three PEDOT:PSS layers namely F010, 4083 and F010 (seen above) were employed by slot-die coating. Detailed printing processes were described by Andersen et al.21,22 All layer thicknesses can be found in ESI Table S2.†
Solar cell performance
Solar cells were measured with a Keithley 2400 source meter under a KHS 575 solar simulator with an AM1.5G 1000 W m−2 intensity. The external quantum efficiency (EQE) was determined using a QEX10 system (PV Measurements).Results and discussion
Synthesis
The structures and synthetic route to the PDTPI polymer are illustrated in Scheme 1. PDTPI was synthesized via Stille cross-coupling reaction between 2,6-di(trimethyltin)-N-(1-pentylhexyl)dithieno[3,2-b:2′,3′-d]-pyrrole and 6,6′-dibromodi(2-ethylhexyl)isoindigo that were prepared according to the literature procedure,11,20 resulting in a reasonable reaction yield of 55%. The obtained polymer had a relatively low Mn of 8860 g mol−1 resulting from the poor solubility of PDTPI in toluene. The thermal decomposition temperature (5% mass loss) was found to be 388 °C (see ESI Fig. S1†), providing a sufficient thermal stability for device fabrication and application.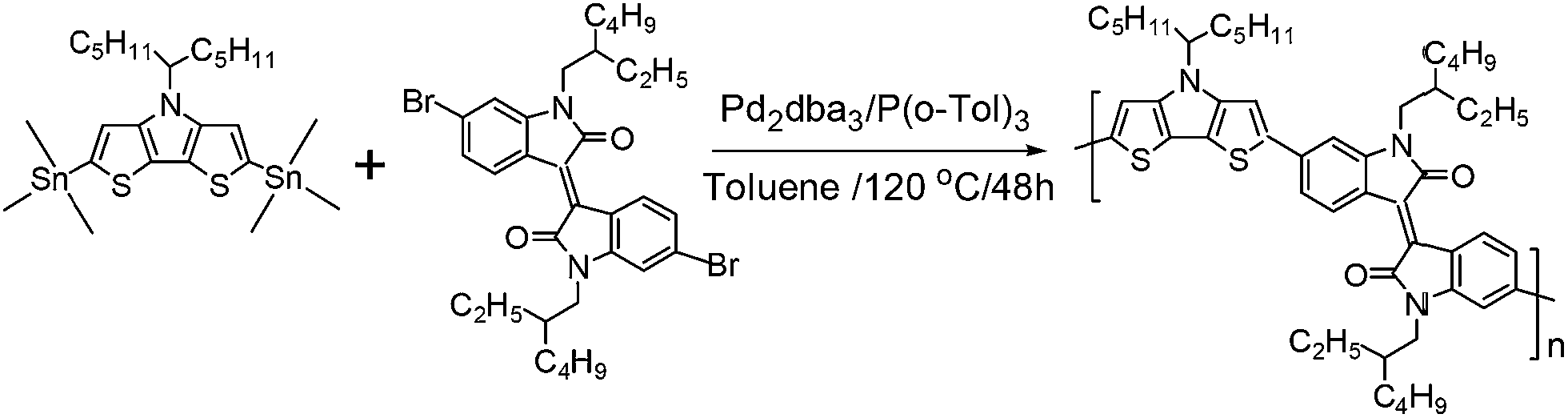 | ||
| Scheme 1 Stille cross-coupling of 2,6-di(trimethyltin)-N-(1-pentylhexyl)dithieno[3,2-b:2′,3′-d]-pyrrole and 6,6′-dibromodi(2-ethylhexyl)isoindigo affording the polymer PDTPI. | ||
Photophysical properties
Fig. 2 presents the UV-vis absorption spectrum of PDTPI acquired in both chloroform solution and thin film. Compared to the weak absorption for the π–π* transition in the high energy band range of 300–500 nm, the intense absorption in the low-energy band range of 550–900 nm evidences a strong electron-withdrawing effect of the i-ID moiety and the intramolecular charge transfer (ICT) between DTP and i-ID entities. As depicted, the maximum absorption bands of PDTPI in solution were obtained at 400 and 820 nm, while in film they were unusually blue-shifted to 390 and 790 nm. We suppose that such blue shifts in the absorption of polymer films may originate from two possible aspects: (a) the aggregation of the polymer chains formed in solution; or (b) the presence of two branched alkyl chains on both the D and A moieties, in particular on the i-ID unit, has a larger effect on the spatial arrangement of the copolymers to retard the intermolecular π–π interactions, therefore weak π–π stacking could be assumed. For the former, we have reported, in our previously published work the blue shifts of 5–19 nm from solution to film absorption on D–A LBG polymers comprising dithieno[3,2-b:2′,3′-d]pyrrole as the donor unit and phthalimide or thieno[3,4-c]pyrrole-4,6-dione as the acceptor.20 This has been further proved by a blue-shift of 5–13 nm in chlorobenzene (CB) solution absorption of the same polymers at 70 °C, compared to those measured at 25 °C also in CB. Such blue shifts in solution could be attributed to breakup of interchain aggregates in solution at the elevated temperature23,24 also as suggested by another report on poly-benzodipyrrolidone and dithieno[3,2-b:2′,3′-d]pyrrole.6 For the latter, as reported by Su et al., the existence of aliphatic segments on poly(cyclopentadithiophene-alt-isoindigo) in the form of both long linear- and short branched-side chains on either donor or acceptor units has proven the same blue-shift of low-energy band absorption from solution to films.25 Stalder et al. observed a similar phenomenon on i-ID and the dithieno[3,2-b:2′,3′-d]silole copolymer exhibited a slight blue shift of about 5 nm from its absorption in solution to that in films.26 Meanwhile, the broader ICT band in films than that in solution implies strong and more efficient charge transfer occurred in the solid state facilitating charge transport. The optical band gap (Eopticalbg), according to the equation Eopticalbg = 1240/λedge, was calculated to be 1.38 eV. | ||
| Fig. 1 Schematic structure of the slot-die coated tandem solar cell (left) and the single junction solar cell (right). | ||
 | ||
| Fig. 2 UV-vis spectra of PDTPI in chlorobenzene solution (black line) and film (red line). | ||
Molecular modelling
Density functional theory (DFT) was used to investigate the optimal geometric structure and the electronic configuration of PDTPI, this was done at the B3LYP/6-31G(d) level using Gaussian 09 and visualized with GaussView 5.0.9. To reduce the computational load, the aliphatic side-chains are reduced to methyl groups, as this has been shown in the literature not to affect the frontier orbital, while affecting the torsion angle to some extent.26 As the system investigated was a polymer, two different methods of evaluation were set up. The first being simply one repetition of a donor unit and an acceptor unit, and the other was 3 consecutive donor acceptor units, thereby simulating a rather extended conjugation of polymers. Salvatori et al.27 showed that this trimer setup gives decent results when simulating a similar system. The orbitals that are interesting in DFT modelling are the frontal orbitals i.e. the HOMO and LUMO. The geometrically optimized structures of both the single D–A and the three repeating D–A moieties are shown in Fig. 3. The dihedral angle between the single D–A model is 21.99°, and the 3 repeating D–A moieties all show similar angles ranging from 20.87 to −20.45° as seen in ESI Fig. S3.† | ||
| Fig. 3 DFT modelling at the B3LYP/6-31G(d) level by Gaussian 9 of the donor–acceptor model compound and for three consecutive D–A configurations. | ||
This continuous twist will afford a torsion on the backbone, thus explaining why there is practically no difference between the absorption of the film and the solution with less than 25 nm onset redshift.28 The modelled torsion angle corresponds to the findings of Lei et al.29 and Wang et al.30 with angles similar to the modelling results, likewise this affords a small red shift onset of the solid state absorbance. While Wang et al. argue that the small change/redshift of the absorbance onset is ascribed to tight aggregation of the polymers in solution, this is seemingly also the case in PDTPI having this continuous torsion. Wang et al. also modeled a fluorinated i-ID moiety, which affords a very small torsion angle between the donor and the acceptor moiety (∼0.01°), following the argument the solvated polymeric interaction (π-stacking) should be significantly increased thus expecting a large red shift, which is not observed, indicating that even though the torsion angles are continuous, the angles are negligible and thus have a low effect on the π-stacking in both film and in solution. Although simplifications with regards to the side-chains were made, the true tendencies influencing the torsion angle are still valid.27
From the single D–A model, it is clear that the HOMO is primarily located on the donor moiety (DTP) and the LUMO is primarily located mainly on the (i-ID) moiety, as expected. This is also supported by the clear intermolecular charge transfer seen in the absorbance spectra, as described earlier.
The predicted HOMO and LUMO levels are −4.80 eV and −2.52 eV giving a band gap of 2.28 eV. This band gap differs from the experimentally determined levels, which is much lower (as the onset of absorbance is approximately 898 nm (Fig. 2)). The 3 repeating D–A units show the same tendencies with only a slight change in the predicted HOMO level given at −4.77 eV while the LUMO level is −2.69 eV. The computed band gap of the 3 repeating D–A units is 2.08 eV which still deviates from the experimental values. The modelling is in line with the findings of Zhuang et al.31 who found that the frontier orbital of the LUMO and HOMO of a donor–acceptor polymer was primarily attributed to the acceptor and donor, respectively.
Frontier orbitals
To experimentally determine the frontier orbitals a CV experiment was performed as presented in ESI Fig. S2.† The oxidation onset was determined to be 0.98 V affording a HOMO level of −5.23 eV. The onset of the absorbance is determined to be 898 nm yielding an optical band gap of 1.38 eV. Using the optical band gap, the LUMO level is calculated to be −3.85 eV. Stalder et al.32 reported that the placement of the LUMO should be in the range of −3.8 to −4 eV, which is supported by the CV findings. The found LUMO aligns well (as mentioned above) with the energy level −4.03 eV of PC[60]BM, although it is generally considered that 0.3 eV is the optimal LUMOACCEPTOR − LUMODONOR difference to ensure the highest charge separation. Even though the found difference of our system is slightly lower (0.2 eV), this is expected to be still sufficient to ensure charge separation. The LUMO of PDTPI is slightly higher than the LUMO reported by Wu et al.17 where the LUMO of a similar polymer was determined to be −3.61 eV. The polymer reported by Wu et al. differs from PDTPI only in one extended thiophene on each side of the DTP moiety. Likewise the HOMO level of a similar polymer was found to be −4.94 eV, therefore the removal of thiophene will make PDTPI more stable under ambient conditions, while still maintaining the favorable LUMO level of i-ID. Comparing with the DFT simulations, the difference is rather large. This difference can be ascribed to two major contributions, first the fact that the relative conjugation length is rather low compared to the experimental data, which in reality is approximately 11 continuously linked D–A couples. Also the spatial dimension of the polymer is neglected in the DFT model applied, this means that the π–π stacking of the polymers is not evaluated. Despite the mismatch with the experimental values, the DFT model data are very similar to the previously reported DFT data at the B3LYP level, which are in the range of 2.8–3 eV.27,31Photovoltaic properties
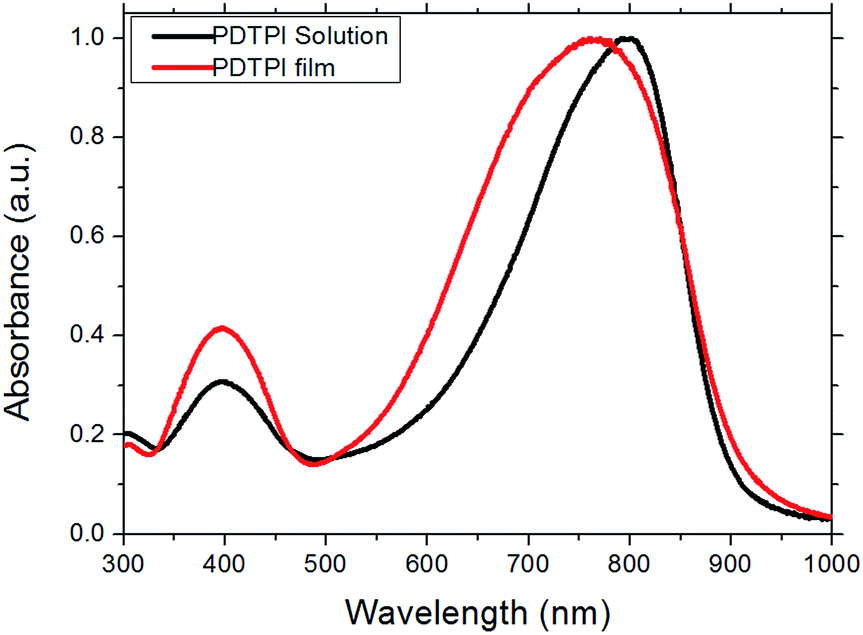
The photovoltaic properties of PDTPI were investigated by preparing flexible ITO-free organic solar cells in an inverted geometry with a bulk heterojunction active layer morphology consisting of PDTPI and PC[60]BM. Layer deposition was conducted both by spin coating and the more industrial scale suitable slot-die coating on a mini roll coater. The corresponding I–V curves and photovoltaic data, PCE, open circuit voltage (VOC), short circuit current (JSC), and fill factor (FF), can be seen in Fig. 4 and Table 1, respectively. The spin-coated devices exhibited a lower PCE (0.72%) than slot-die coated devices with the same composition. This is exclusively due to an increased JSC for the slot-die coated devices. The improvement could be seen as an abnormality, since not only is the deposition method changed from spin-coating to slot-die coating, but also the area increases from 0.25 cm2 to 1 cm2, which would usually lead to a decreased JSC. The observed increase can be explained by the utilization of a different PEDOT:PSS top-electrode where the spin-coated devices were based on Agfa EL-P5010, the roll-coated devices had the 3 layer PEDOT:PSS electrode, as seen in Fig. 1. Meanwhile there must exist different morphologies caused by different solvent evaporation rates between the roll coating and spin coating process, this would have an impact, thus resulting in different short-circuit current density. However this is not the focus of this work, further studies on morphology effects, when going from spin coated to roll coated devices, could be pursued in the near future.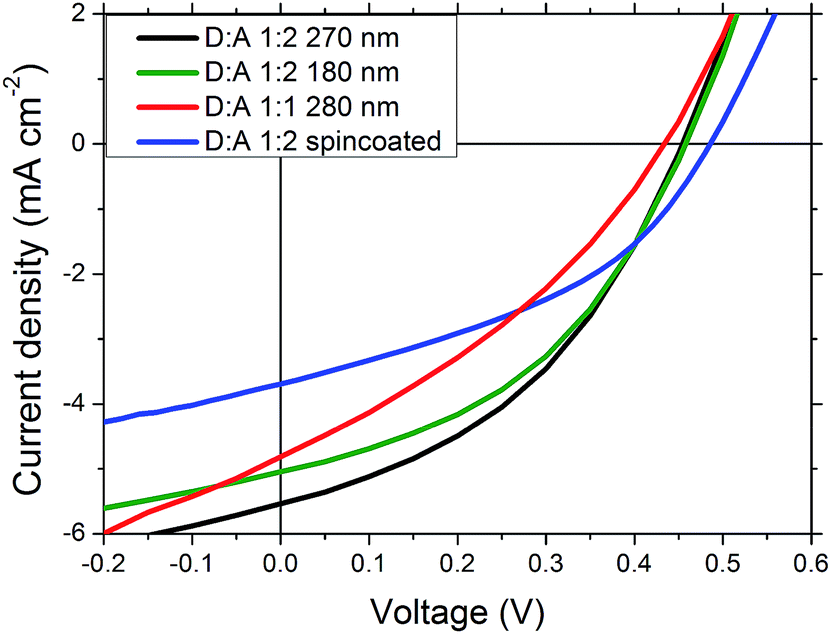 | ||
| Fig. 4
I–V curves of roll coated and spin coated single junction solar cells with different PDTPI/PC[60]BM ratios and film thicknesses. The estimated thickness of the active layer in the spin coated cell (D:A 1:2) was 160–200 nm. | ||
| D:A |
L (nm) | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| a D: PDTPI, A: PC[60]BM, L: thickness. b Spin-coated sample. c Estimated active layer thickness was 160–200 nm. | |||||
| 1:1 |
280 | 0.42 ± 0.01 | −4.6 ± 0.2 | 32.3 ± 0.6 | 0.62 ± 0.05 |
| 1:2b |
0.49 | −3.69 | 40.3 | 0.72 | |
| 1:2 |
180 | 0.45 ± 0.004 | −4.95 ± 0.18 | 40.9 ± 1.1 | 0.92 ± 0.05 |
| 1:2 |
270 | 0.46 ± 0.004 | −5.25 ± 0.28 | 41.3 ± 1.4 | 0.99 ± 0.08 |
To optimize the PDTPI:PC[60]BM composition two different compositions (1:1 and 1:2) were tested. The results in Table 1 clearly reveal that the 1:2 composition is better, which besides an increase in PCE from 0.62% to 0.99% also showed improvement in the remaining photovoltaic characteristics.
The increase in FF by approximately 25% is the most significant, which may be explained by the improved interface or more likely improved carrier transport in the active layer, which together with a possible increase in exciton dissociation accounts for the increase in JSC due to a reduced recombination. The film thickness was investigated by producing devices with 270 nm and 180 nm thick active layers. Reducing the film thickness resulted in a slight decrease in device efficiency due to a decrease in JSC most likely due to a reduced absorption whereas the remaining photovoltaic characteristics were unaffected.
Better performing devices were therefore prepared with an active layer composition between PDTPI and PC[60]BM of 1:2 and a thickness of 280 nm, achieving a PCE of 0.99%, the JSC of which was verified by EQE (seen in Fig. 5) which was found to be 5.30 mA cm−2 correlating nicely with the measured 5.25 mA cm−2 (see Table 1). These results however do not show the full potential of PDTPI since the Flextrode has a decreasing transmission from 60% to 35% in the interval of 600 nm to 900 nm (ref. 33) where PDTPI is photoactive (see Fig. 2).
 | ||
| Fig. 5 EQE of the best performing single junction device, with a PDTPI/PC[60]BM ratio of 1:2 and an active layer thickness of 280 nm. | ||
The absorption characteristics of PDTPI which has a primary absorbance band in the 600–900 nm range (Fig. 2) could make it a prime candidate for the low band gap junction in tandem solar cells. P3HT and PBDTTTz-4 were chosen as high band gap polymers due to their ideal spectral match with PDTPI as shown in Fig. 6.
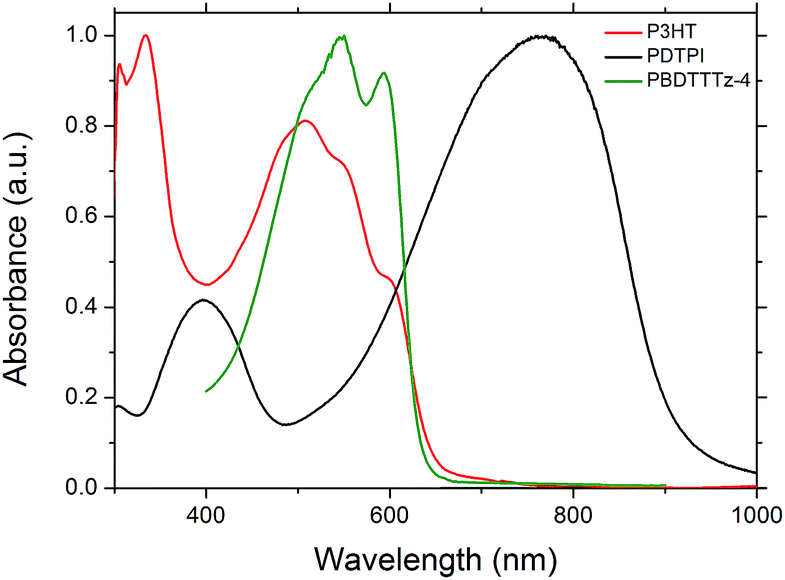 | ||
| Fig. 6 The film absorbance of PBDTTTz-4 (green line), PDTPI (black line) and P3HT (red line). | ||
Tandem solar cells were prepared using a roll coater by slot-die coating and flexographic printing via a method presented by Andersen et al.22,34 the layer structure is shown in Fig. 1. The results from the three different tandem solar cell combinations prepared can be seen in Table 2. Fig. 7 shows the I–V characteristics of the 3 best performing devices with PBDTTTz-4 and P3HT combined with PDTPI. It is clear that the VOC is lower than the theoretical maximum estimated from the sum of the single junction VOC's. This has however been reported previously in several cases when using flexible substrates22,35,36 in which the tandem cell open-circuit voltages are 10–15% lower. Despite a close to ideal spectral overlap between P3HT and PDPTI, it is clear that the tandem solar cell consisting of PDPTI and P3HT underperforms, due to both lower JSC and FF.
| Composition | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) | Best PCE [%] |
|---|---|---|---|---|---|
| 180 nm P3HT 270 nm PDTPI | 0.85 ± 0.01 | −2.49 ± 0.07 | 35.17 ± 0.05 | 0.74 ± 0.01 | 0.75 |
| 155 nm PBDTTTz-4 350 nm PDTPI | 0.95 ± 0.02 | 2.83 ± 0.98 | 33.85 ± 3.84 | 1.12 ± 0.23 | 1.40 |
| 290 nm PBDTTTz-4 350 nm PDTPI | 1.08 ± 0.09 | 4.60 ± 0.32 | 32.93 ± 0.52 | 1.63 ± 0.05 | 1.73 |
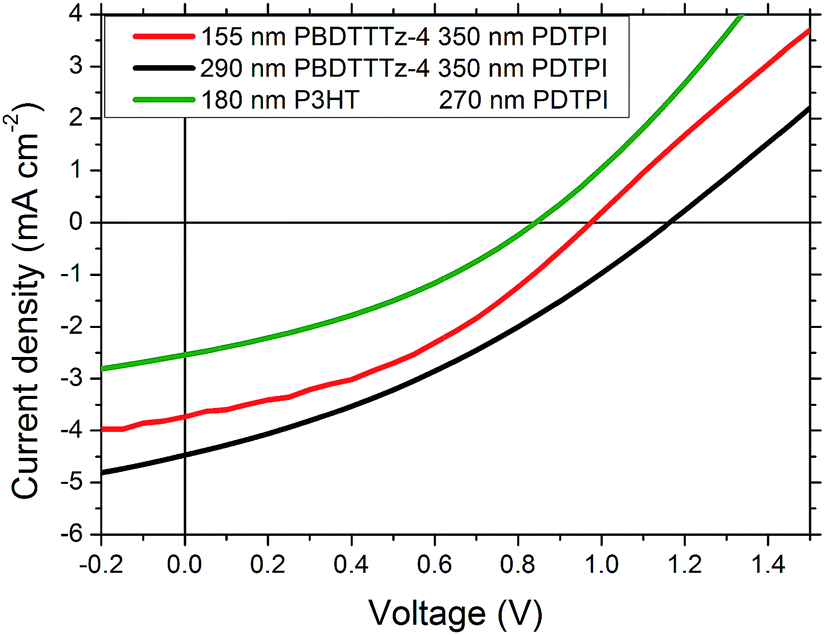 | ||
| Fig. 7 I–V characteristics of the tandem solar cells with the following combinations, 155 nm PBDTTTz-4 350 nm PDTPI (red line), 290 nm PDBTTTz-4 350 nm PDTPI (black line) and 180 nm P3HT 270 nm PDTPI (green line). | ||
The efficiencies obtained for the PBDTTTz-4 and PDTPI combination outperformed the P3HT:PDTPI combination with similar active layer thicknesses, mostly due to an increase in VOC. The effects of layer thickness of tandem devices are clear when comparing the large difference in PCE for devices prepared with front junction thicknesses of 155 nm and 290 nm.
The increase in VOC with increasing front junction thickness is likely due to the usage of the Flextrode which has been previously shown to contain rather large silver spikes;22,35 as a larger number of spikes will be embedded in the front junction relative to the back junction, increasing the front junction thickness will result in fewer penetrating silver spikes and hence the level of shunting will be reduced. The increase in JSC can be ascribed to a better current matching between the junctions.
The best performing device showed a PCE of 1.73% and consists of a 290 nm PBDTTTz-4 front junction and a 350 nm back junction of PDTPI, shown as the black line in Fig. 7.
Conclusions
A low-band gap polymer consisting of alternating di(2-ethylhexyl)isoindigo and N-(1-pentylhexyl)dithieno[3,2-b:2′,3′-d]-pyrrole moieties was synthesized. The synthesized polymer had a band gap of 1.38 eV with a HOMO level of −5.23 eV (determined by CV) and a suitably aligned LUMO level of −3.85 eV; which make it an ideal donor for the PC[60]BM. Density functional theory (DFT) computations showed that the LUMO was primarily located on the acceptor while the HOMO was primarily distributed on the donor moiety, i-ID and DTP, respectively. The DFT simulations also showed a torsion angle between the donor and acceptor moiety at around 22°, this combined with a simulation of an extended D–A structure showed a continuous torsion. This torsion could explain the relatively small onset redshift (∼25 nm) in absorbance when going from solution to film. Large area (0.25 to 1 cm2), ITO-free single junction photovoltaic devices showed efficiencies ranging from 0.72% to 0.99% for spin-coated and roll-coated devices, respectively. The low band gap and the EQE characteristics of PDTPI combined with the nice spectral overlap with PBDTTTz-4 and P3HT made it an ideal candidate for a back cell in a tandem solar cell. Large area (0.8 cm2), ITO-free roll-coated tandem junction photovoltaic devices were produced with efficiencies ranging 0.72% to 1.72%, for PDTPI combined with P3HT and PBDTTTz-4, respectively.Acknowledgements
The authors would like to acknowledge the financial support from the Danish Council for Strategic Research through the WAPART project (Water-based particulate approach to organic photovoltaics with controlled morphology, 11-116380), and the Danish National Research Foundation (DNRF) for the Danish-Chinese Center for Organic based Photovoltaic Cells with Morphological Control. Support from the Sino-Danish Centre for Education and Research (SDC) is also fully acknowledged.Notes and references
- H. Xin, X. Guo, F. S. Kim, G. Ren, M. D. Watson and S. A. Jenekhe, J. Mater. Chem., 2009, 19, 5303–5310 RSC.
- X. Guo, F. S. Kim, M. J. Seger, S. A. Jenekhe and M. D. Watson, Chem. Mater., 2012, 24, 1434–1442 CrossRef CAS.
- X. Zhan, Z. Tan, E. Zhou, Y. Li, R. Misra, A. Grant, B. Domercq, X.-H. Zhang, Z. An, X. Zhang, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 5794–5803 RSC.
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Jr and J. Dupont, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC.
- W. Yue, X. Huang, J. Yuan, W. Ma, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 10116–101119 CAS.
- B. R. Aïch, S. Beaupré, M. Leclerc and Y. Tao, Org. Electron., 2014, 15, 543–548 CrossRef PubMed.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- E. Bundgaard and F. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954–985 CrossRef CAS PubMed.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414–1420 CrossRef CAS.
- B. Liu, Y. Zou, B. Peng, B. Zhao, K. Huang, Y. He and C. Pan, Polym. Chem., 2011, 2, 1156–1162 RSC.
- E. Wang, Z. Ma, Z. Zhang, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, Chem. Commun., 2011, 47, 4908–4910 RSC.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471–476 CrossRef CAS PubMed.
- F. Wu, H. Yang, C. M. Li and J. Qin, Polym. Adv. Technol., 2013, 24, 945–950 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 1636–1641 CrossRef CAS PubMed.
- F. C. Krebs, Org. Electron., 2009, 10, 761–768 CrossRef CAS PubMed.
- W. Yue, T. T. Larsen-Olsen, X. Hu, M. Shi, H. Chen, M. Hinge, P. Fojan, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 1785–1793 CAS.
- D. Angmo, S. A. Gevorgyan, T. T. Larsen-Olsen, R. R. Søndergaard, M. Hösel, M. Jørgensen, R. Gupta, G. U. Kulkarni and F. C. Krebs, Org. Electron., 2013, 14, 984–994 CrossRef CAS PubMed.
- T. R. Andersen, H. F. Dam, B. Andreasen, M. Hösel, M. V. Madsen, S. A. Gevorgyan, R. R. Søndergaard, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2014, 120, 735–743 CrossRef CAS PubMed.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed.
- C.-C. Ho, S.-Y. Chang, T.-C. Huang, C.-A. Chen, H.-C. Liao, Y.-F. Chen and W.-F. Su, Polym. Chem., 2013, 4, 5351–5360 RSC.
- R. Stalder, C. Grand, J. Subbiah, F. So and J. R. Reynolds, Polym. Chem., 2012, 3, 89–92 RSC.
- P. Salvatori, E. Mosconi, E. Wang, M. Andersson, M. Muccini and F. De Angelis, J. Phys. Chem. C, 2013, 117, 17940–17954 CAS.
- S. Zhang, H. Fan, Y. Liu, G. Zhao, Q. Li, Y. Li and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2843–2852 CrossRef CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, Y. Li and Q. Peng, Polym. Chem., 2014, 5, 4984–4992 RSC.
- W. Zhuang, M. Bolognesi, M. Seri, P. Henriksson, D. Gedefaw, R. Kroon, M. Jarvid, A. Lundin, E. Wang, M. Muccini and M. R. Andersson, Macromolecules, 2013, 46, 8488–8499 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- M. Hösel, R. R. Søndergaard, M. Jørgensen and F. C. Krebs, Energy Technol., 2013, 1, 102–107 CrossRef.
- T. R. Andersen, H. F. Dam, M. Hösel, M. Helgesen, J. E. Carlé, T. T. Larsen-Olsen, S. a. Gevorgyan, J. W. Andreasen, J. Adams, N. Li, F. Machui, G. D. Spyropoulos, T. Ameri, N. Lemaître, M. Legros, A. Scheel, D. Gaiser, K. Kreul, S. Berny, O. R. Lozman, S. Nordman, M. Välimäki, M. Vilkman, R. R. Søndergaard, M. Jørgensen, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 2925–2933 CAS.
- T. R. Andersen, H. F. Dam, B. Burkhart, D. Angmo, M. Corazza, B. C. Thompson and F. C. Krebs, J. Mater. Chem. C, 2014, 2, 9412–9415 RSC.
- N. Li, D. Baran, G. D. Spyropoulos, H. Zhang, S. Berny, M. Turbiez, T. Ameri, F. C. Krebs and C. J. Brabec, Adv. Energy Mater., 2014, 4, 1400084 Search PubMed.
- M. Zhang, Y. Sun, X. Guo, C. Cui, Y. He and Y. Li, Macromolecules, 2011, 44, 7625–7631 CrossRef CAS.
- M. Helgesen, J. E. Carle and F. C. Krebs, Adv. Energy Mater., 2013, 3, 1664–1669 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc02877e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |