
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microporous, tetraarylethylene-based polymer networks generated in a reductive polyolefination process†
E.
Preis
a,
W.
Dong
a,
G.
Brunklaus
b and
U.
Scherf
*a
aBergische Universität Wuppertal, Macromolecular Chemistry Group (buwmakro) and Institute for Polymer Technology, Gauss-Str. 20, D-42119 Wuppertal, Germany. E-mail: scherf@uni-wuppertal.de
bWestfälische Wilhelms-Universität, Institut für Physikalische Chemie, Corrensstr. 28, D-48149 Münster, Germany
First published on 19th December 2014
Abstract
Microporous Polymer Networks (MPNs) that contain aggregate-induced emission (AIE) active tetraphenylethylene (TPE) or other tetraarylethylene units have been generated in a reductive polyolefination process starting from four different tris(α,α-dichlorobenzyl)arene derivatives with dicobalt octacarbonyl or chromium(II)acetate as reductive olefination agents. Microporosity with moderately high BET surface areas up to 500 m2 g−1 could be combined with high solid state photoluminescence quantum yields up to 25.3% for polymer network P4. This unique combination should be promising for applications as optical solid state sensors, especially for MPNs with electron-deficient triazine core units.
Introduction
Microporous Polymer Networks (MPNs) comprise a rapidly growing area of materials science towards future mass applications in catalysis, gas storage/separation or as active porous materials for organic electronic devices and sensors.1–7 For such mass applications inexpensive synthesis procedures for MPN generation without use of noble metal catalysts and under mild reaction conditions are of particular appeal.8–18 Tetraphenylethylene (TPE) and related repeat units represent versatile building blocks for the rational design of MPNs due to their rigid, three-dimensional structure and interesting optical properties, especially a distinct aggregate-induced emission (AIE). Linear poly-TPEs (as representative example poly(1,4-phenylene-1,2-diphenylvinylene) DP-PPV) was first described 30 years ago by Hörhold and co-workers.19 In the following decades, a rather broad variety of TPE-based monomers, oligomers and (co)polymers have been extensively investigated by Tang and co-workers thereby documenting their broad and very promising application potential in materials science and biology.20–24 The first synthesis of MPNs that contain TPE building blocks and show a pronounced AIE effect has been reported by Jiang and co-workers.25,26 Their MPNs have been generated in a Yamamoto-type aryl-aryl coupling starting from tetrabromo-TPE with Ni(COD)2 as coupling reagent. In this study, we now introduce the synthesis of structurally related poly-TPE networks in a reductive olefination process starting from tris(α,α-dichlorobenzyl)arene monomers with dicobalt octacarbonyl27,28 or chromium(II)acetate19,29 as condensing agents. In our target networks the tetraarylethylene units (Scheme 1) are linked together via 1,3,5-substituted benzene or 2,4,6-substituted 1,3,5-triazine cores. Jiang's MPNs, in contrast, contain TPE units with the olefinic double bonds connected via biphenyl units.25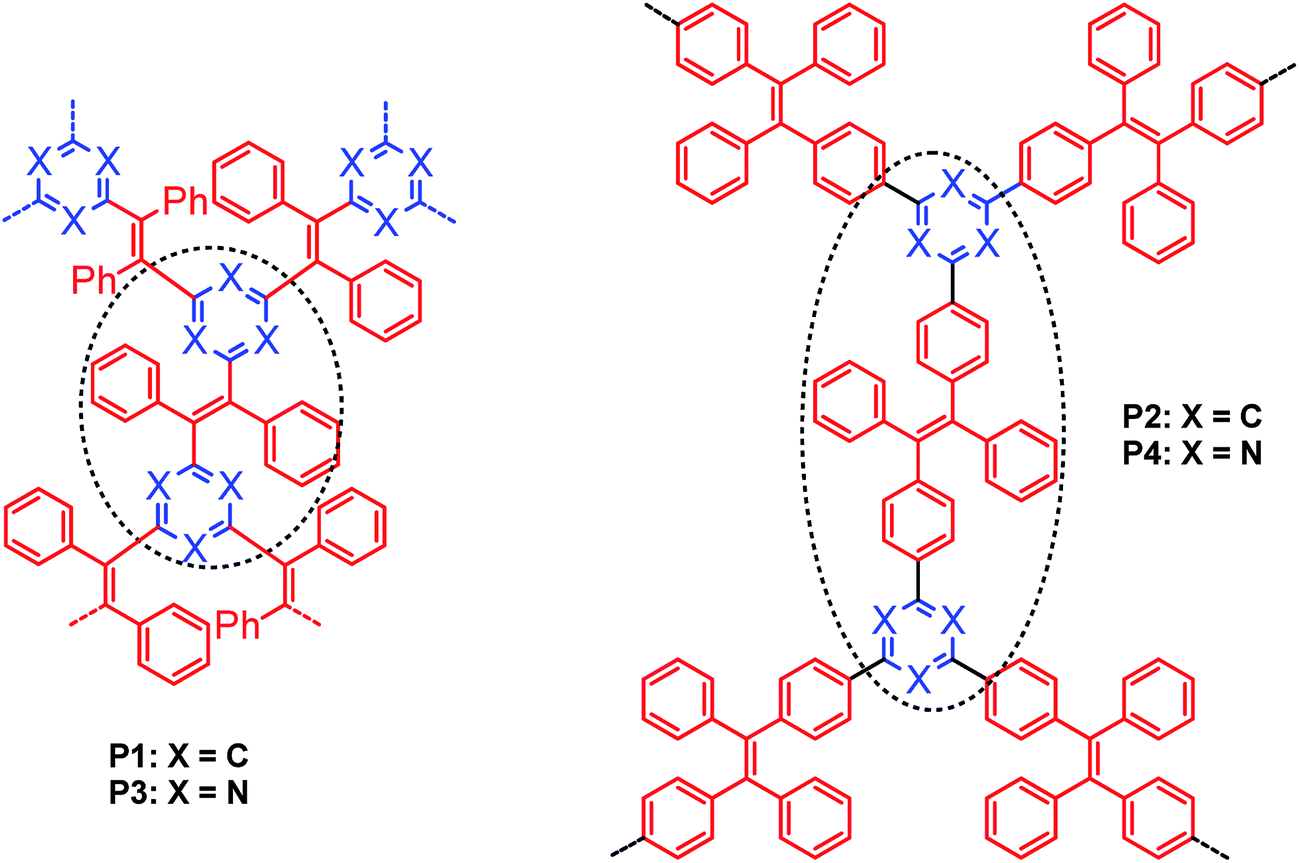 | ||
| Scheme 1 Idealized structures of the polymer networks P1–P4. | ||
The synthesis of the monomers M1–M4 is depicted in Scheme 2 (for detailed information see ESI†). The general synthesis route of the polymer networks P1–P4 is outlined in Scheme 3 for P1 as representative example. 1,3,5-Tribenzoylbenzene was prepared according to a reported procedure.30 The until now unknown, corresponding 1,3,5-tris(α,α-dichlorobenzyl)benzene MCl1 was generated by reaction with phosphorus pentachloride (PCl5) in chlorobenzene as solvent (Scheme 3). After removal of both phosphorus oxychloride (POCl3) by distillation and excess PCl5 by sublimation, respectively, the resulting oily product was used without further purification, similar to the synthesis of bifunctional analogues as 1,3-bis(α,α-dichlorobenzyl)benzene (made for polyolefination into linear coupling products).311H NMR spectroscopy indicated the presence of pure 1,3,5-tris(α,α-dichlorobenzyl)benzene MCl1 (Fig. 1). The singlet of the 1,3,5-substituted benzene core is, thereby, shifted from 8.40 ppm to 7.83 ppm.
 | ||
| Scheme 2 Synthesis of the monomers M1–M4. | ||
 | ||
| Scheme 3 Synthesis of the TPE-based MPN P1 in a reductive polyolefination. | ||
 | ||
| Fig. 1 1H NMR spectra of: (A) 1,3,5-tribenzoylbenzene M1 and (B) 1,3,5-tris(α,α-dichlorobenzyl)benzene MCl1 in C2D2Cl4. | ||
For the synthesis of monomer M2, 1,3,5-tris(4-bromophenyl)benzene was prepared following a literature procedure.32 Subsequent nucleophilic substitution with copper(I) cyanide yielded 1,3,5-tris(4-cyanophenyl)benzene (Scheme 2); the rather low yield is related to an ongoing trimerization of nitrile groups into triazine-cored byproducts.33 Addition of phenylmagnesium bromide, followed by hydrolysis with diluted hydrochloric acid afforded 1,3,5-tris[4-benzoylphenyl]benzene. The corresponding 1,3,5-tris[4-(α,α-dichlorobenzyl)phenyl]benzene MCl2 intermediate was then generated as described for 1,3,5-tris(α,α-dichlorobenzyl)-benzene, also resulting in an oily product. Furthermore, the synthesis of tris(2,4,6-benzoyl)-1,3,5-triazine M3 was accomplished following an already published approach.34–36 Its subsequent transformation into 2,4,6-tris(α,α-dichlorobenzyl)-1,3,5-triazine MCl3 was carried out as described for MCl1 and MCl2. The 1H NMR spectrum of the corresponding hexachloro-derivative MCl3 exhibits two aromatic signals, a doublet at 7.62 ppm (6H) and a multiplet at 7.36–7.30 (9H) thus demonstrating a complete transformation of carbonyl into α,α-dichloromethyl groups. The preparation of 2,4,6-tris(4-methylphenyl)-1,3,5-triazine and its subsequent oxidation to the corresponding tricarboxylic acid (Scheme 2) was achieved by applying literature methods.37,38 The reaction of the corresponding acid chloride with benzene after Friedel–Crafts resulted in monomer M4, 2,4,6-tris(4-benzoylphenyl)-1,3,5-triazine, that was finally converted into the corresponding 2,4,6-tris[4-(α,α-dichlorobenzyl)phenyl]-1,3,5-triazine MCl4 as outlined for MCl1-MCl3. In the 13C NMR spectrum of the hexachloro-substituted monomer MCl4 complete disappearance of the carbonyl signal at 196.6 ppm and formation of an aliphatic signal at 91.7 ppm (ar2CCl2) confirmed the complete conversion.
The resulting, moisture sensitive hexachloro monomers prepared from the tribenzoyl derivatives M1–M4 were used without further purification, as already mentioned. Their reductive polyolefination into the polymer networks P1–P4 was accomplished by using two different reducing/condensing agents: (i) dicobalt octacarbonyl Co2(CO)8 (ref. 27 and 28) or (ii) chromium(II)acetate Cr2ac4,19,29 respectively. The insoluble coupling products precipitated during reaction and were isolated as yellowish colored solids. The polymeric products P1, P2 and P4 show yellow photoluminescence (PL) of different intensity (P3: no PL detected, see Table 2) and were subsequently extracted with several organic solvents (methanol, ethyl acetate, 1,4-dioxane, chloroform) and finally washed with supercritical carbon dioxide sc-CO216,18 prior to further characterization.
The absence of carbonyl signals at 185–200 ppm (generated by hydrolysis of –CCl2– functions) in the solid-state 13C CPMAS spectra of the polymer networks P1–P4 (all made with Co2(CO)8 as reducing agent, for P4 see Fig. 2, for P1–P3 see Fig. S5a–c, ESI†) clearly evidence a successful reductive polycondensation. In case of polymer networks that contain triazine cores (P3 and P4), an additional signal at about 170 ppm is observed (as for the monomers) that is characteristic for the 1,3,5-triazine unit. The rather weak signal at 170 ppm for the triazine-free networks P1 and P2 may be related to carboxylic acid or ester functions that are formed in a side reaction, probably by CO insertion. In contrast to a P1 sample made with Co2(CO)8 (Fig. 3), the 13C CPMAS spectrum of another P1 sample obtained with Cr2ac4 as condensing agent exhibits two additional carbon signals at 195 ppm for leftover carbonyl groups and an aliphatic signal at 83 ppm, but, interestingly, no signal around 170 ppm. Thereby, the presence of leftover carbonyl groups suggests incomplete conversion. For this reason only samples made with Co2(CO)8 have been considered for further characterization.
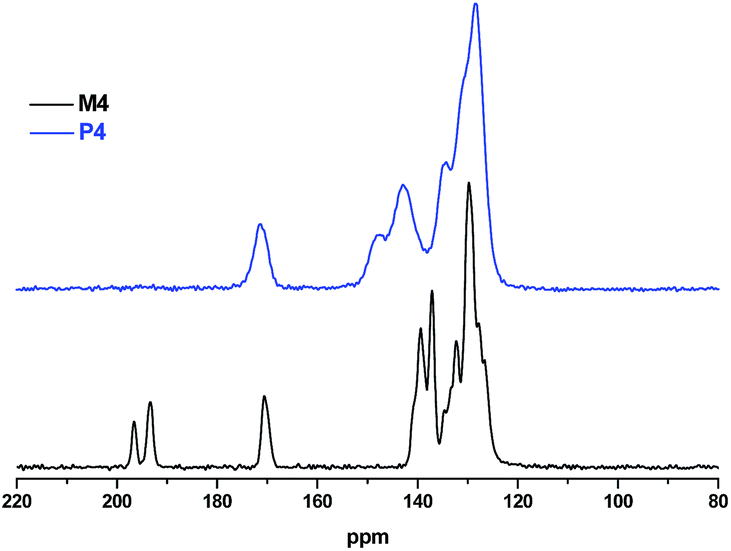 | ||
| Fig. 2 Solid state 13C NMR spectra of monomer M4 and polymer network P4. | ||
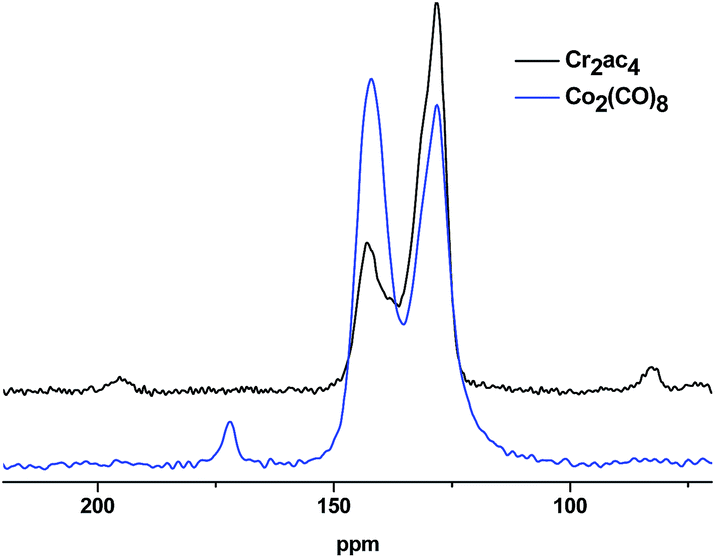 | ||
| Fig. 3 Solid state 13C NMR spectra of polymer network P1 made with Co2(CO)8 (blue) or Cr2ac4 (black) as reducing agent. | ||
The porosity of the polymer networks P1–P4 was studied by nitrogen gas adsorption and desorption at 77 K. The networks P1–P4 all show type I isotherms (see Fig. 4) with a significant amount of nitrogen adsorbed at low pressure, followed by a much increased adsorption at higher pressure (P/P0 > 0.9) attributed to gas condensation into voids between polymer particles.39
 | ||
| Fig. 4 Nitrogen adsorption (filled symbols)/desorption (open symbols) isotherms at 77 K for P1 (black squares), P2 (red circles), P3 (green stars), P4 (blue triangles); the inset shows the pressure range P/P0 from 0 to 0.7. | ||
As often observed for “soft” polymers networks, the desorption-branch did not connect to the adsorption-branch at low pressure.40 P3 additionally shows a distinct hysteresis, most probably due to capillary condensation (see Fig. 4). Moderately high BET surface areas of 462 m2 g−1 (P1), 502 m2 g−1 (P2), 200 m2 g−1 (P3), and 475 m2 g−1 (P4) could be determined for the relative pressure range of 0.01–0.15 P/P0 (see Table 1), while rather high total pore volumes of 1.05–1.87 cm3 g−1 have been found for P1–P4.
| BET surface area (m2 g−1) | Pore volume (cc) | H2 uptake (%) | CO2 uptake (%) | CH4 uptake (%) | Selectivity (CO2/CH4) (298 K) | Selectivity (N2/H2) (77 K) | |
|---|---|---|---|---|---|---|---|
| P1 | 462 | 1.76 | 0.44 | 1.46 | 0.11 | 4.7 | 24.2 |
| P2 | 502 | 1.87 | 0.57 | 2.54 | 0.26 | 3.6 | 19.3 |
| P3 | 200 | 1.05 | 0.35 | 1.58 | 0.12 | 4.6 | 18.0 |
| P4 | 475 | 1.16 | 0.55 | 2.58 | 0.26 | 3.6 | 12.6 |
| UV/Vis (λmax, nm) | PL (λmax, nm) | PLQY (%) | First decomposition step (TGA, under argon, °C) | |
|---|---|---|---|---|
| P1 | 352 | 540 | 3.5 | 272 (2.5%) |
| P2 | 363 | 540 | 6.5 | 386 (2.6%) |
| P3 | 367 | no PL detected | — | 296 (7.9%) |
| P4 | 392 | 560 | 25.3 | 408 (1.5%) |
Carbon dioxide and methane uptakes of the networks were determined at 298 K, the hydrogen uptake at 77 K up to a pressure of 1 bar. The gas uptake is significantly increased for the networks with additional phenylene “spacers” (P2 and P4) for all three gases, but most pronounced for CH4 as the largest guest molecule, thus indicating an increase of the pore diameter. The calculated selectivities for CO2/CH4 at 298 K are 4.7 (P1), 3.6 (P2), 4.6 (P3) and 3.6 (P4), for N2/H2 at 77 K are 24.2 (P1), 19.3 (P2), 18.0 P3 and 12.6 (P4) (for details see the ESI†).
Powder X-ray diffraction (PXRD) analysis of polymer network P4 (Fig. S26, ESI†) only displays three broad peaks of low intensity in the wide-angle region, thus suggesting a dominantly amorphous network structure. The peaks correspond to distances of 6.45, 4.5 and 2.1 Å, respectively, most probably reflecting periodic structural motifs of P4 (as TPE–TPE and triazine–triazine distances).
The solid state optical spectra of the polymer networks are depicted in Fig. 6. From the onset of the absorption features (Fig. S4, ESI†) HOMO–LUMO energy gaps of 2.90 and 2.78 eV are estimated for the polymer networks P2 and P4, respectively. The photoluminescence (PL) spectra of the polymer networks P1, P2, and P4 are shown in Fig. 6b. P1 (λmax = 540 nm), P2 (λmax = 540 nm) and P4 (λmax = 560 nm) display a yellow photoluminescence; P3 is non-emissive. P1 and P2 show quite moderate photoluminescence quantum yields (PLQY) of 3.5 and 6.5%, respectively, for an excitation at 340 nm. In contrast, P4 is characterized by a remarkably intense PL with a PLQY of 25.3% (excitation at 400 nm; see Fig. 5). For P3, the combination of a comparatively low BET surface area (200 m2 g−1) with the absence of photoluminescence points for the formation of structural defects during network formation.
 | ||
| Fig. 5 P4 powder (top) and P4 pellet (down) at day light (left) and under UV irradiation (right). | ||
 | ||
| Fig. 6 PL excitation spectra (λdet = 550 nm) (a) and PL spectra (λexc = 400 nm) (b) of P1 (black square), P2 (red circle), P3 (blue triangle), P4 (green star). | ||
The photoluminescence excitation (PLE) spectra of the polymer networks P1, P2 and P4 are depicted in Fig. 6a with PLE maxima at λmax = 367 nm (shoulder at ca. 450 nm) for P1, λmax = 394 nm for P2 and λmax = 410 nm (shoulder at ca. 450 nm) for P4 for a detection wavelength at 550 nm.
Finally, the microporous polymer network P4 with its electron-deficient triazine cores has been tested for its interaction with electron-rich analytes as aromatic amines (Fig. 7).
 | ||
| Fig. 7 PL quenching experiments with P4 pellets and p-toluidine or aniline as analytes. | ||
For PL quenching tests P4-pellets have been exposed to saturated vapors of the amine analytes at atmospheric pressure and room temperature.
For p-toluidine 41% PL quenching is observed after exposure for 1 min, and 73% PL quenching after 45 min. For aniline as analyte 39% PL quenching is observed after 1 min exposure and 82% after 49 min. It can be assumed that the remarkable quenching response results from donor–acceptor interactions between the electron-rich aromatic analytes and the electron-poor triazine-based MPN P4. These initial results demonstrate a remarkable potential for the solid state sensing of organic analytes. Further experiments will be, especially, focused on the selectivity issue and on the reversibility of the PL quenching.
Conclusions
In conclusion, novel microporous polymer networks P1–P4 have been successfully synthesized via a reductive polyolefination protocol starting from tris(α,α-dichlorobenzyl)-substituted aromatic monomers with dicobalt octacarbonyl (preferred) or chromium(II)acetate as condensing agents. The most promising polymer network P4 that is composed of 1,3,5-triazine cores as linkers between aggregation induced emission (AIE)-active tetraphenylethylene units, reveals an intense yellow photoluminescence peaking at 560 nm with a remarkably high PLQY of 25.3% in line with a moderately high BET surface area of 475 m2 g−1. This combination of microporosity and high solid state PLQY (based on the AIE effect) clearly qualifies P4 with its electron deficient 1,3,5-triazine cores as an attractive target for further investigations, as its interaction with electron rich guests. Notably, first, orienting PL quenching experiments have demonstrated a distinct response to aromatic amine vapors (for aniline and p-toluidine).Acknowledgements
The authors would like to thank P. Becker, J.-C. Gasse, and Dr. D. Lützenkirchen-Hecht for the powder XRD analysis.Notes and references
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- K. D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456–5459 CrossRef PubMed.
- R. R. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2007, 40, 1299–1304 CrossRef CAS.
- M. Rose, N. Klein, I. Senkovska, C. Schrage, P. Wollmann, W. Böhlmann, B. Böhringer, S. Fichtner and S. Kaskel, J. Mater. Chem., 2011, 21, 711–716 RSC.
- K. T. Jackson, M. G. Rabbani, T. E. Reich and H. M. El-Kaderi, Polym. Chem., 2011, 2, 2775–2777 RSC.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730–7732 RSC.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8875 CrossRef CAS PubMed.
- Y. Luo, B. Li, L. Liang and B. Tan, Chem. Commun., 2011, 47, 7704–7706 RSC.
- E. Preis, C. Widling, G. Brunklaus, J. Schmidt, A. Thomas and U. Scherf, ACS Macro Lett., 2013, 2, 380–383 CrossRef CAS.
- R. S. Sprick, A. Thomas and U. Scherf, Polym. Chem., 2010, 1, 283–285 RSC.
- E. Preis, C. Widling, U. Scherf, S. Patil, G. Brunklaus, J. Schmidt and A. Thomas, Polym. Chem., 2011, 2, 2186–2189 RSC.
- H.-H. Hörhold, M. Helbig, D. Raabe, J. Opfermann, U. Scherf, R. Stockmann and D. Weiß, Z. Chem., 1987, 27, 126–137 CrossRef.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef CAS PubMed.
- J. Zhou, Z. Chang, Y. Jiang, B. He, M. Du, P. Lu, Y. Hong, H. S. Kwok, A. Qin, H. Qiu, Z. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491–2493 RSC.
- J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef.
- W. Z. Yuan, P. Lu, S. Chen, J. W. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- A. Patra and U. Scherf, Chem.–Eur. J., 2012, 18, 10074–10080 CrossRef CAS PubMed.
- H. Reisch, U. Wiesler, U. Scherf and N. Tuytuylkov, Macromolecules, 1996, 29, 8204–8210 CrossRef CAS.
- E. Preis and U. Scherf, Macromol. Rapid Commun., 2006, 27, 1105–1109 CrossRef CAS.
- H.-H. Hörhold, J. Gottschaldt and J. Opfermann, J. Prakt. Chem., 1977, 319, 611–621 CrossRef.
- C. Y. K. Chan, Z. Zhao, J. W. Y. Lam, J. Liu, S. Chen, P. Lu, F. Mahtab, X. Chen, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 378–389 CrossRef CAS.
- H.-H. Hörhold and D. Raabe, Acta Polym., 1979, 30, 86–92 CrossRef.
- S. Kotha, D. Kashinath, K. Lahiri and R. B. Sunoj, Eur. J. Org. Chem., 2004, 4003–4013 CrossRef CAS.
- G. H. Miller, US Pat., 3 775 380, 1973.
- E. Ott, Ber. Dtsch. Chem. Ges. B, 1914, 52, 656–665 CrossRef.
- C. Grundmann and E. Kober, J. Org. Chem., 1956, 21, 1392–1394 CrossRef CAS.
- C. L. Schmidt and M. Jansen, Eur. J. Inorg. Chem., 2012, 34, 5649–5657 CrossRef.
- S.-H. Li, H.-P. Huang, S.-Y. Yu and X.-P. Li, Chin. J. Chem., 2006, 24, 1225–1229 CrossRef CAS.
- R. J. Milligan, C. B. Delano and T. J. Aponyi, J. Macromol. Sci., Chem., 1976, 10, 1467–1483 CrossRef.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 CAS.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc02664k |
| This journal is © The Royal Society of Chemistry 2015 |