
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyano substituted benzothiadiazole: a novel acceptor inducing n-type behaviour in conjugated polymers†
Abby
Casey
a,
Yang
Han
a,
Zhuping
Fei
*a,
Andrew J. P.
White
b,
Thomas D.
Anthopoulos
c and
Martin
Heeney
*a
aDept. Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: z.fei@imperial.ac.uk; m.heeney@imperial.ac.uk
bChemical Crystallography Laboratory, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
cDept. Physics and Centre for Plastic Electronics, Imperial College London, SW7 2AZ, UK
First published on 21st October 2014
Abstract
We report the synthesis of the novel acceptor, 4,7-di(thiophen-2-yl)-5,6-dicyano-2,1,3-benzothiadiazole (DTDCNBT) and compare its properties to those of the previously reported 4,7-di(thiophen-2-yl)-5,6-difluoro-2,1,3-benzothiadiazole (DTDFBT). Co-polymers of both monomers with the donor monomers indacenodithiophene (IDT) and dithienogermole (DTG) were prepared and investigated. The DTDCNBT unit was found to be a much stronger electron acceptor than DTDFBT. The electron affinity of the cyanated polymers was increased by up to ∼0.4 eV, resulting in red-shifted absorptions and reduced optical band gaps. In field effect transistors it was found that replacing the fluorine substituents of the polymers with cyano groups changed the charge transport from unipolar p-type to unipolar n-type.
1. Introduction
Research into organic field-effect transistors (OFETs) has accelerated over the past decade due to their potential to provide low cost, light weight and flexible circuitry.1–3 Low cost, large area electronics could be realised by using soluble semiconducting materials in high through-put roll-to-roll processing techniques.4–6 Organic semiconductors (OSC) can be readily chemically modified allowing properties such as the materials crystallinity, energy levels and optical band gap to be tuned.7–10 Modifying these properties influences charge transport, charge injection etc. which are critical to device performance. Understanding the relationship between chemical structure, material properties and device performance allows materials to be designed in order to manipulate their properties and optimise device characteristics.n-Type and ambipolar OSCs are hugely important for applications such as complementary logic circuits and organic solar cells. Although dramatic advancements have been made in the performance of p-type OSCs,11–13 n-type materials have generally lagged in terms of mobility, stability and the variety of structures available.3,14 One general issue with n-type materials is the sensitivity of electron transport to charge trapping in the film, particularly in the presence of oxygen and/or water. In order to promote stability and charge injection from common electrodes, electron-transporting materials with low lying LUMO levels are required, with a level below approximately −4 eV having been shown to promote ambient stability.15–17 Therefore the development of new strongly electron accepting co-monomers is important as a method to tune the polymer LUMO level.
4,7-Di(thiophen-2-yl)-2,1,3-benzothiadiazole (DTBT) is very commonly used as the electron withdrawing unit in donor–acceptor based small molecules and polymers for both OPV and OFET applications.3,18–20 Different derivatives of the 2,1,3-benzothiadiazole unit, such as naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole (NBT),21,22 benzobisthiadiazole (BBT)23–26 and thiadiazoloquinoxaline (TQ)27–31 have been synthesised in order to increase the electron deficiency. Polymers containing TQ units have shown ambipolar charge transport with electron mobilities up to 0.04 cm2 V−1 s−1.27 BBT containing polymers also show high performance ambipolar characteristics with electron mobilities up to 1.2 cm2 V−1 s−1 when co-polymerised with diketopyrrolopyrrole (DPP).23 Another method to increase the electron deficiency of DTBT is to replace the flanking thiophene rings with thiazole units (DTABT). Polymers containing DTABT were found to have a lower LUMO level and an electron mobility two orders of magnitude higher than equivalent polymers containing DTBT.32 One of the most widely explored strategies to increase the electron affinity and ionisation potential of DTBT containing polymers has been to fluorinate the BT acceptor. Although this has been shown to improve the performance of photovoltaic donor polymers,33,34 it has not been shown to promote electron transport in polymeric systems35,36 and in some instances reduces p-type mobility compared to the non-fluorinated analogues.37 The absence of electron transport is most likely due to the modest increase in electron affinity upon fluorination.
Herein, we present a simple synthetic approach to obtain the novel acceptor moiety 4,7-di(thiophen-2-yl)-5,6-dicyano-2,1,3-benzothiadiazole (DTDCNBT) through the nucleophilic aromatic substitution (SNAr) of the fluorine substituents in 4,7-di(thiophen-2-yl)-5,6-difluoro-2,1,3-benzothiadiazole (DTDFBT) with cyanide. In order to investigate the potential of this new acceptor in polymeric systems, we chose to co-polymerise it with two recently reported donor monomers, namely a tetraalkylated indacenodithiophene (IDT)37–39 and a long chain branched dialkylated dithienogermole (DTG)40–42 derivative. Both monomers were chosen partly due to the high alkyl chain density, which we believed would be important in promoting solubility due to the absence of alkyl side chains on the DTDCNBT unit. In order to further investigate the influence of the cyano group, we compare and contrast the properties of the cyanated polymers with those of the polymers containing the analogous fluorinated benzothiadiazole monomer (DTDFBT). We demonstrate the inclusion of the DTDCNBT results in an increase in polymeric electron affinity by ∼0.4 eV and that substituting the fluorine substituents with cyano groups results in a switch from p-type to n-type transport in transistor devices.
2. Results and discussion
2.1. Synthesis of monomers and polymers
The synthesis of 5,6-dicyano-2,1,3-benzothiadiazole has previously been reported by the Rosenmund von Braun cyanation of 5,6-dibromo-2,1,3-benzothiadiazole in moderate yield.43 However the subsequent electrophilic bromination of such an electron poor heterocycle appeared problematic, especially given the harsh conditions required to brominate the analogous 5,6-difluoro-2,1,3-benzothiadiazole44 (refluxing in fuming sulfuric acid) and the reported tendency for 5,6-dicyano-2,1,3-benzothiadiazole to hydrolyse under acidic conditions.45 Therefore encouraged by our recent finding that DTDFBT could undergo mild nucleophilic aromatic substitution with alkyl thiols,46 we investigated the direct substitution of fluorine with cyanide on the preformed DTDFBT monomer (Scheme 1). After some experimentation, we found that treatment of DTDFBT (1) with potassium cyanide in the presence of 18-crown-6 in refluxing THF afforded the dicyano derivative DTDCNBT (2) in a good yield of 76%. Excess KCN was used to increase the rate of reaction and ensure the complete substitution of the fluorine substituents with cyano groups.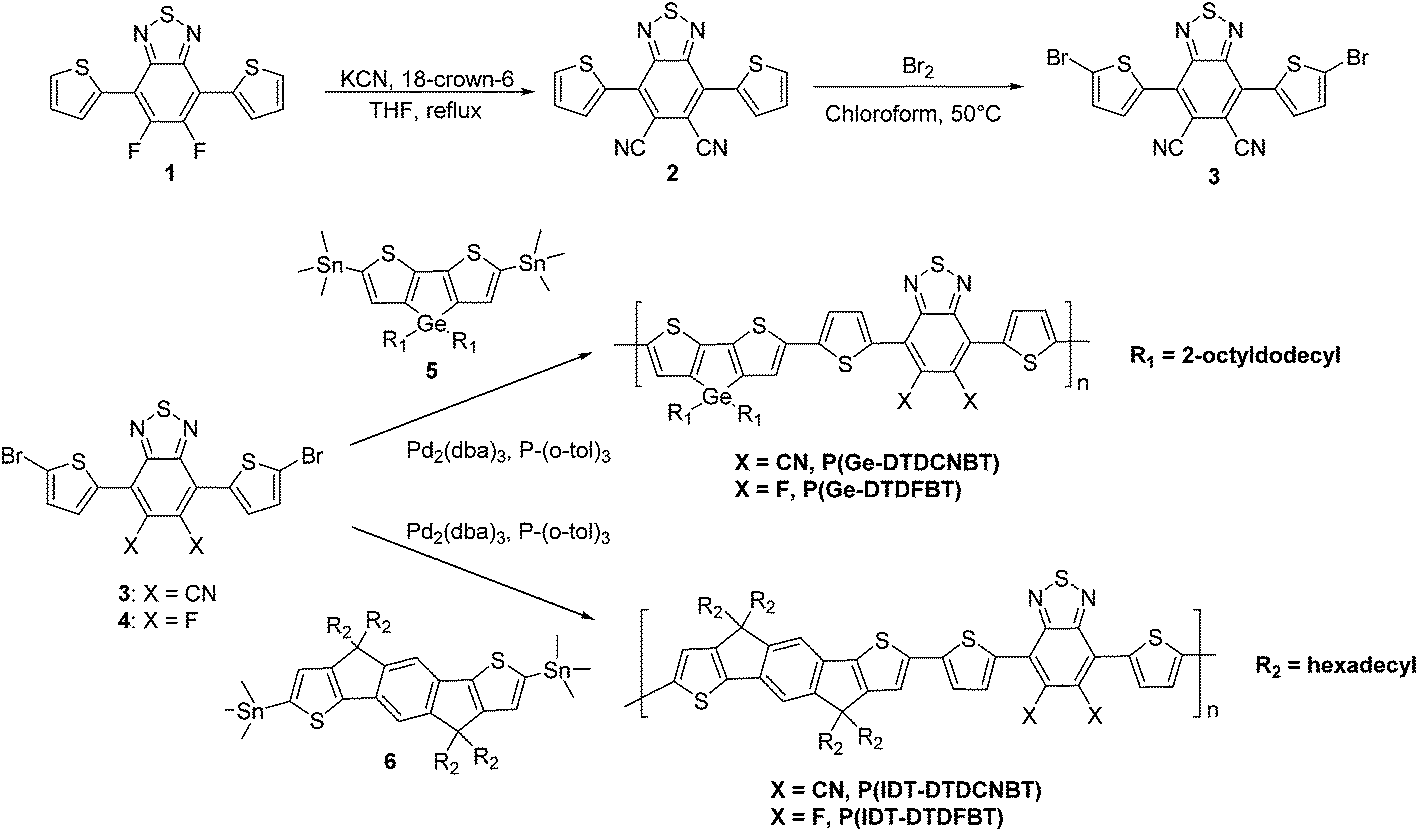 | ||
| Scheme 1 Synthetic procedure of monomer 3 and polymers P(Ge-DTDCNBT), P(Ge-DTDFBT), P(IDT-DTDCNBT) and P(IDT-DTDFBT). | ||
The subsequent bromination of product 2 required much harsher conditions than the fluorinated analogue (1). Whilst adding two equivalents of NBS at RT was sufficient to dibrominate molecule (1), an excess of molecular bromine with heating (50 °C) over 2 days was required to brominate molecule (2) in 66% yield. The reduced reactivity of the 5 and 5′ positions of the cyanated precursor (2) in comparison to the fluorinated precursor (1) is the result of the stronger electron withdrawing nature of the cyanated BT unit, reducing electron density at these positions.
The donor comonomer 4,4′-bis(2-octyldodecyl)-5,5′-bis(trimethyltin)-dithieno[3,2-b:2′,3′-d]germole (5) was prepared as previously reported.42 2,7-Bis(trimethylstannyl)-4,9-dihydro-4,4,9,9-tetrahexadecyl-s-indaceno[1,2-b:5,6-b′]-dithiophene (6), was prepared by the addition of 4 equivalents of LDA to a solution containing 4,9-dihydro-4,4,9,9-tetrahexadecyl-s-indaceno[1,2-b:5,6-b′]-dithiophene38 and 4 equivalents of trimethyltin chloride at 0 °C. The large excess of LDA and trimethyltin chloride was required to promote good yields of the distannylated monomer, since separation from the mono-stannylated species was difficult due to the poor stability of the stannyl species upon attempted chromatography over silica. The acceptor monomer DTDCNBT (3) was co-polymerised with 4,4′-bis(2-octyldodecyl)-5,5′-bis(trimethyltin)-dithieno[3,2-b:2′,3′-d]germole42 (5) and 2,7-bis(trimethylstannyl)-4,9-dihydro-4,4,9,9-tetrahexadecyl-s-indaceno[1,2-b:5,6-b′]-dithiophene (6) via Stille coupling under microwave heating to produce the low band gap polymers P(Ge-DTDCNBT) and P(IDT-DTDCNBT). Microwave-assisted Stille coupling was used as it has previously been shown to improve polymer properties by increasing molecular weight and lowering dispersity in comparison to those synthesised using conventional heating.47 The fluorinated analogues, P(Ge-DTDFBT) and P(IDT-DTDFBT) were synthesised in an analogous manner by the polymerisation of DTDFBT monomer 4. All four polymers were purified by Soxhlet extraction (methanol, acetone, hexane, chloroform) to remove low molecular weight oligomers and catalyst residues. P(IDT-DTDFBT) had a crude molecular weight of 27.7 kDa (Đ 1.8). In order to obtain a higher molecular weight fraction for comparison with P(IDT-DTDCNBT) the polymer was washed with a hexane–chloroform mix (72/28), which forms a constant boiling azeotrope, before it was extracted into chloroform. The resultant chloroform fraction had a molecular weight of 51.9 kDa, suitable for comparison with P(IDT-DTDCNBT). The crude polymers were then washed with sodium diethyldithiocarbamate dihydrate solution to remove possible palladium residues.48 The molecular weights of all polymers were determined by gel permeation chromatography in chlorobenzene at 80 °C, and are shown in Table 1. The molecular weight of P(Ge-DTDCNBT) was somewhat lower than P(Ge-DTDFBT), although we believe both are sufficiently high to be in the plateau region in terms of molecular properties, allowing a reasonable comparison of properties.
| Material | M n | M w | Đ | λ abs,max, nm (sol)b | λ abs,max, nm (film) | E g(opt) eVd |
|---|---|---|---|---|---|---|
| a Molecular weights measured using gel permeation chromatography (against polystyrene standards) in chlorobenzene at 80 °C. b Determined from solution UV-vis absorption spectroscopy in chlorobenzene. c Shoulder peak in absorption. d Determined from the absorption onset of the polymers in thin film. | ||||||
| P(Ge-DTDCNBT) | 20.5 | 44.4 | 2.17 | 754 | 833 | 1.27 |
| P(Ge-DTDFBT) | 40.0 | 92.0 | 2.30 | 651 (698)c | 647 (703)c | 1.61 |
| P(IDT-DTDCNBT) | 47.3 | 81.2 | 1.72 | 716 | 769 | 1.40 |
| P(IDT-DTDFBT) | 51.9 | 74.2 | 1.43 | 615 | 650 | 1.75 |
2.2. Optical properties
The absorption spectra of P(Ge-DTDFBT) and P(Ge-DTDCNBT) in chlorobenzene solution and thin film are shown in Fig. 1. In solution P(Ge-DTDFBT) had an absorbance maximum at 651 nm whilst the absorbance of P(Ge-DTDCNBT) peaked at 754 nm, resulting in a red-shift of 103 nm as the fluorine groups are replaced with cyano groups. P(Ge-DTDFBT) displays a clear shoulder at 698 nm in chlorobenzene solution, which becomes more defined in thin film (see Fig. 1b). This suggests the fluorinated polymer is starting to aggregate in solution and aggregates further in thin film. In contrast a very broad, featureless absorption spectra is observed for P(Ge-DTDCNBT) suggesting replacing the fluorine groups for cyano groups reduces polymer aggregation. This observation is corroborated by an investigation into the conformation of the monomers and polymers (see Section 2.4). DFT calculations predicted a planar conformation for both fluorinated polymers whilst the cyanated polymers showed significant conformational disorder between the BT and thiophene units due to steric clash with the larger cyano groups. This is likely to reduce aggregation between polymer chains. In thin film, P(Ge-DTDFBT) had an absorbance maximum very similar to solution at 647 nm, whilst the absorbance of P(Ge-DTDCNBT) peaked at 833 nm, giving a large red-shift of 186 nm between the two polymers. This large red-shift suggests the cyanated polymer is able to planarise in the solid state. The optical band gaps of P(Ge-DTDFBT) and P(Ge-DTDCNBT), measured from the absorption onset of the polymers in thin film, were 1.61 and 1.27 eV respectively. Replacing the fluorine groups on the BT unit for cyano groups therefore lead to a lowering in the optical band gap of 0.34 eV, due to the increased strength of the cyano-BT acceptor unit.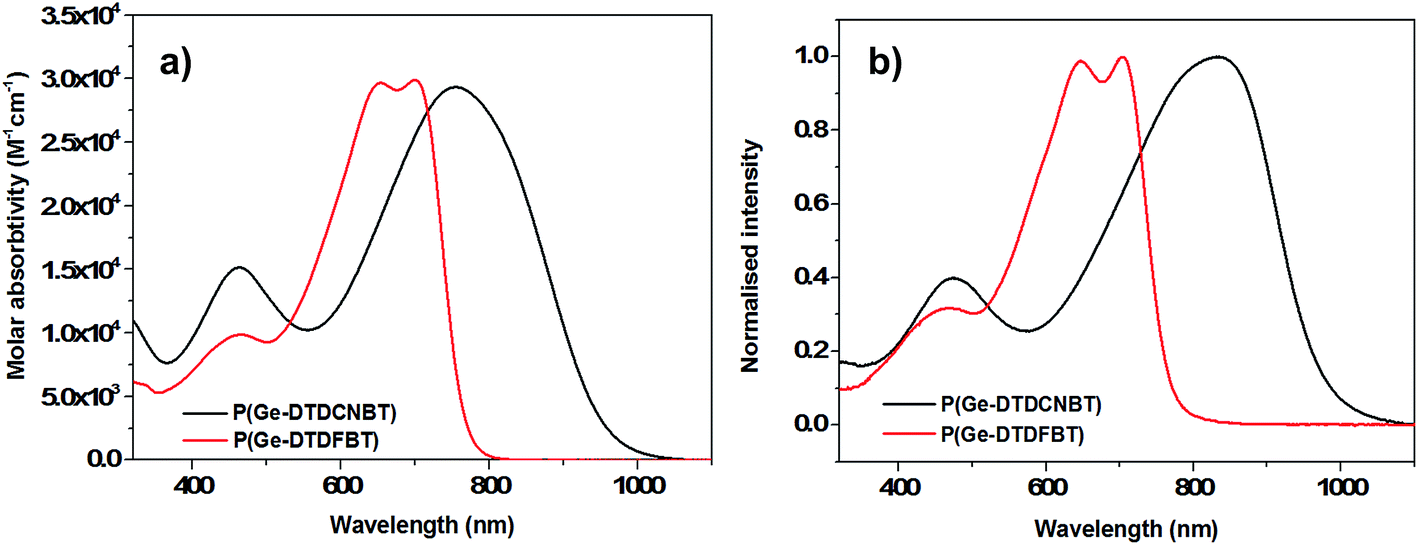 | ||
| Fig. 1 (a) Optical absorption spectra of P(Ge-DTDCNBT) and P(Ge-DTDFBT) in chlorobenzene solution (16.6 mg dm−3) and (b) in spun-cast thin film. | ||
The absorbance spectra of P(IDT-DTDFBT) and P(IDT-DTDCNBT) in chlorobenzene solution (see Fig. 2) have a maximum absorbance at 615 nm and 716 nm respectively, giving a similar red shift of 101 nm as the fluorine substituents are replaced with cyano groups. Both polymers red-shifted upon film formation with absorption maxima at 650 nm and 769 nm for P(IDT-DTDFBT) and P(IDT-DTDCNBT) respectively (see Fig. 2). The optical band gaps of P(IDT-DTDCNBT) and P(IDT-DTDFBT) were 1.40 and 1.75 eV respectively. The optical band gap of the cyanated IDT polymer is reduced by 0.35 eV in comparison to the fluorinated IDT polymer – a very similar reduction to that observed between the cyanated and fluorinated dithienogermole polymers. In both cases we note that the strength of the optical absorptions in solution are similar for both the fluorinated and cyanated polymers (see Fig. 1a and 2a).
 | ||
| Fig. 2 (a) Optical absorption spectra of P(IDT-DTDCNBT) and P(IDT-DTDFBT) in chlorobenzene solution (16.6 mg dm−3) and (b) in spun-cast thin film. | ||
2.3. Polymer electrochemistry
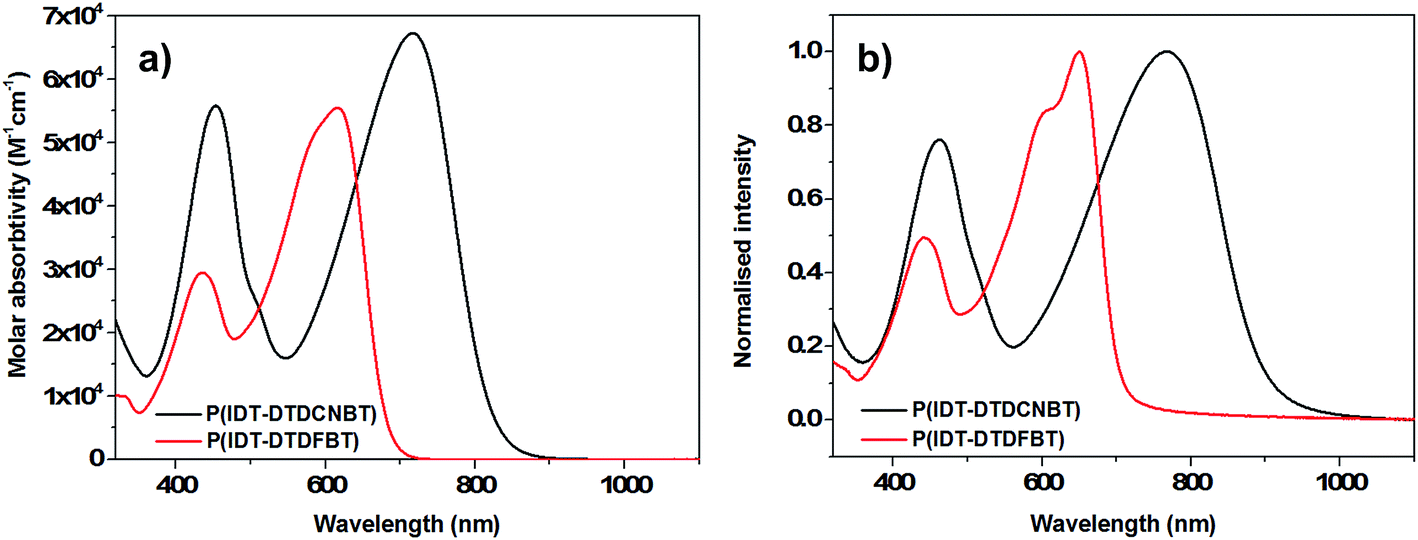
The oxidation and reduction potentials of the polymers were obtained using cyclic voltammetry (CV) measurements of the spun-cast polymer films on fluorine doped tin oxide (FTO) substrates in anhydrous, degassed solutions of acetonitrile with tetrabutylammonium hexafluorophosphate (0.1 M) electrolyte. The cyclic voltammograms of the four polymers are shown in Fig. 3 (potentials were measured against an Ag/Ag+ reference electrode). The HOMO levels were estimated by the onset of the first oxidation potential (Eox,onset – see Table 2), assuming the ferrocene/ferrocenium reference redox system is 4.8 eV below the vacuum level.49 The ionisation potentials of the polymers in thin film were also measured using photoelectron spectroscopy in air (PESA)50 (see Table 2). Although PESA measurements estimated higher ionisation potentials for all the polymers compared to CV,51 very similar trends in the movement of the HOMO level were observed with both techniques. Both CV and PESA measurements showed that replacing the fluorine substituents in P(Ge-DTDFBT) with cyano groups leads to an increase in the ionisation potential of approximately 0.1 eV. Both techniques also showed a smaller increase in the ionisation potential of 0.02–0.03 eV as the fluorine substituents in P(IDT-DTDFBT) were replaced with cyano groups.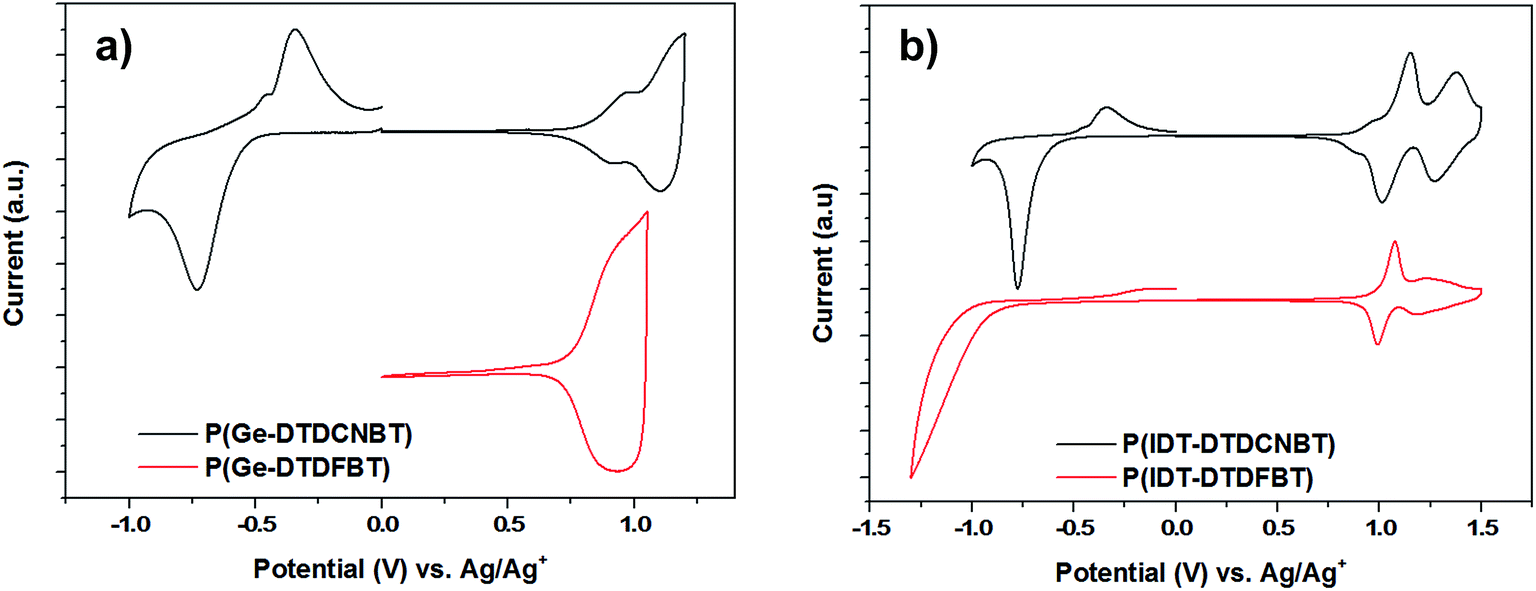 | ||
| Fig. 3 Cyclic voltammograms of thin films of (a) P(Ge-DTDCNBT) and P(DTDFBT) (b) P(IDT-DTDCNBT) and P(IDT-DTDFBT) in acetonitrile–[n-Bu4N]PF6 solutions (0.1 M) at a scan rate of 100 mV s−1. | ||
| Material | HOMO, eV (Eox,onset, V)a | LUMO, eV (Ered,onset, V)a | E g(elec) | I.P., eV (PESA)b | LUMOc, eV I.P. + Eg(opt) |
|---|---|---|---|---|---|
| a Determined by thin film cyclic voltammetry. b Determined by photoelectron spectroscopy in air (PESA) on thin films, error ± 0.05 eV. c Estimated by adding Eg(opt) to the ionization potentials determined by PESA. | |||||
| P(Ge-DTDCNBT) | −5.14 (0.82) | −3.77 (−0.55) | 1.37 | −5.30 | −4.03 |
| P(Ge-DTDFBT) | −5.07 (0.75) | — | — | −5.20 | −3.59 |
| P(IDT-DTDCNBT) | −5.34 (1.02) | −3.67 (−0.65) | 1.67 | −5.44 | −4.04 |
| P(IDT-DTDFBT) | −5.31 (0.99) | −3.39 (−0.93) | 1.92 | −5.42 | −3.67 |
The LUMO of P(Ge-DTDFBT) could not be directly measured by CV, however the reduction potential of all other polymers was observable. The LUMO levels were also estimated in all cases by adding the optical band gap of the polymer to the ionisation potential determined by PESA (see Table 2). Although this does not account for the exciton binding energy it does allow for a relative comparison between polymers. In both cases it is clear that the replacement of the fluorine substituents with cyano groups resulted in a substantial increase in the electron affinity of the polymer. In the case of the IDT polymers, this was around 0.3 eV by electrochemical measurements and slightly larger at 0.37 eV using the optical band gap estimation. For the cyanated dithienogermole polymer the estimated increase in electron affinity was 0.44 eV. Of further note was the fact that the cyclic voltammograms of both of the cyanated polymers showed clearly reversible reduction peaks in contrast to the fluorinated IDT polymer (see Fig. 3).
2.4. Backbone conformation
To compare the preferred conformation of the molecules DTDFBT (1) and DTDCNBT (2), potential energy scans were calculated by systematically changing one of the thiophene-difluorobenzothiadiazole (T-DFBT) or thiophene-dicyanobenzothiadiazole (T-DCNBT) dihedral bond angles and allowing the rest of molecule of relax to its energy minimum using density functional theory (DFT) with a B3LYP level of theory and a basis set of 6-31G(d). The potential energy curves are shown in Fig. 4. In agreement with literature calculations37 a near planar trans conformation (thiophene sulfurs pointing in an opposite direction to the BT sulfur) is predicted as the lowest energy conformation for the DTDFBT monomer, whilst a cis conformation (thiophene sulfurs pointing in same direction as BT sulfur) approximately 30° from planarity is the lowest energy conformation for the DTDCNBT monomer (see Fig. 4a). This agrees with single crystal analysis of the DTDCNBT monomer which shows that the main conformation of DTDCNBT is cis (see Fig. 4b). It is interesting to note that the energy difference between trans and cis conformations is higher for DTDCNBT (∼5 kJ mol−1) than for DTDFBT (∼2 kJ mol−1) however the barrier to rotation is much lower at ∼13 kJ mol−1 compared to ∼20 kJ mol−1 for DTDFBT. Potentially due to this relatively low barrier to rotation (∼13 kJ mol−1) the orientation of the thiophene rings is somewhat disordered in the DTDCNBT single crystal. For each thiophene two orientations were identified of ca. 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 and 86:14% occupancy for the S11 and S18-based rings respectively, the major occupancy orientations of each thiophene ring are shown in Fig. 4b. The crystal structure of DTDCNBT shows the molecule to have adopted a conformation with approximate C2 symmetry about an axis that passes through S1 and bisects the C5–C6 bond, with the dihedral angles between the DCNBT unit and the S11- and S18-based thiophene rings (ca. 35 and 24° respectively) putting the two sulfurs on opposite sides of the plane of the DCNBT unit.
:12 and 86:14% occupancy for the S11 and S18-based rings respectively, the major occupancy orientations of each thiophene ring are shown in Fig. 4b. The crystal structure of DTDCNBT shows the molecule to have adopted a conformation with approximate C2 symmetry about an axis that passes through S1 and bisects the C5–C6 bond, with the dihedral angles between the DCNBT unit and the S11- and S18-based thiophene rings (ca. 35 and 24° respectively) putting the two sulfurs on opposite sides of the plane of the DCNBT unit.
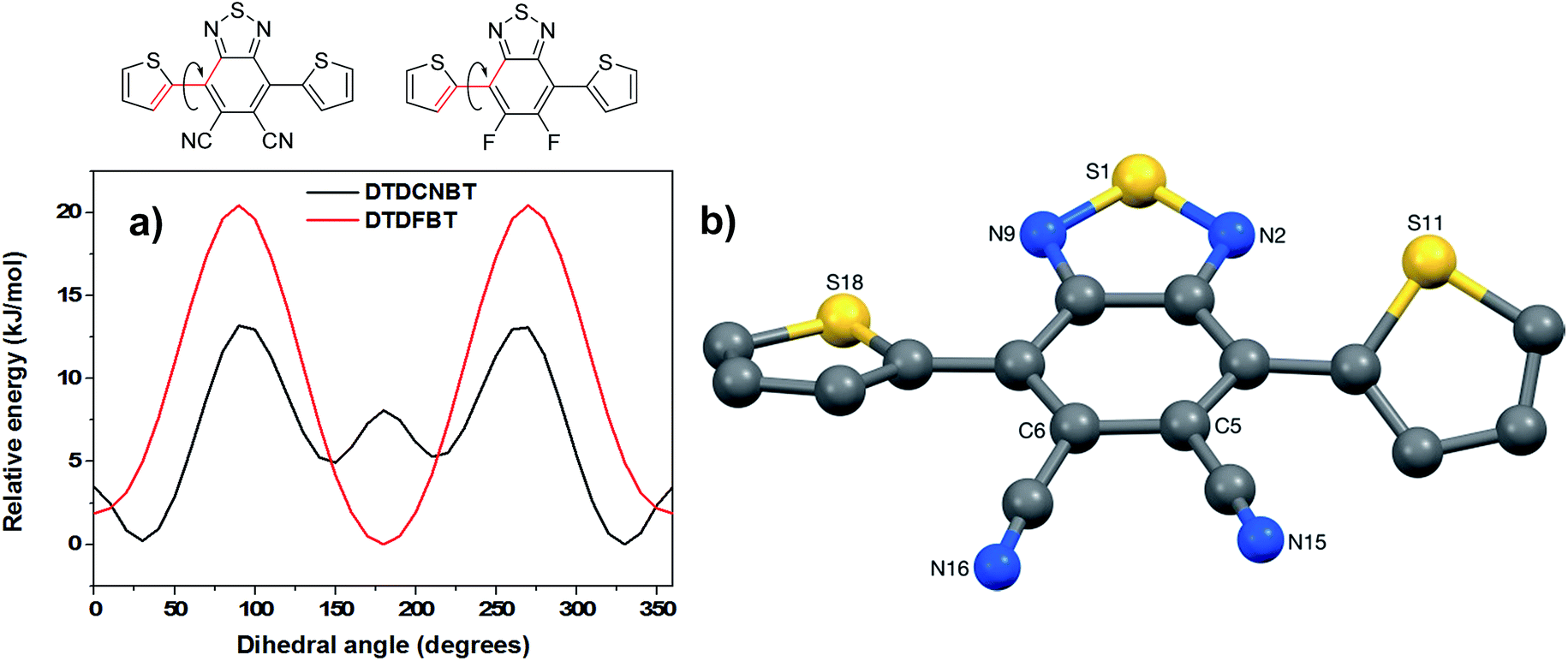 | ||
| Fig. 4 (a) Potential energy scan of the T-BT dihedral angle in a DTDCNBT unit (black) and a DTDFBT unit (red) (b) crystal structure of DTDCNBT. | ||
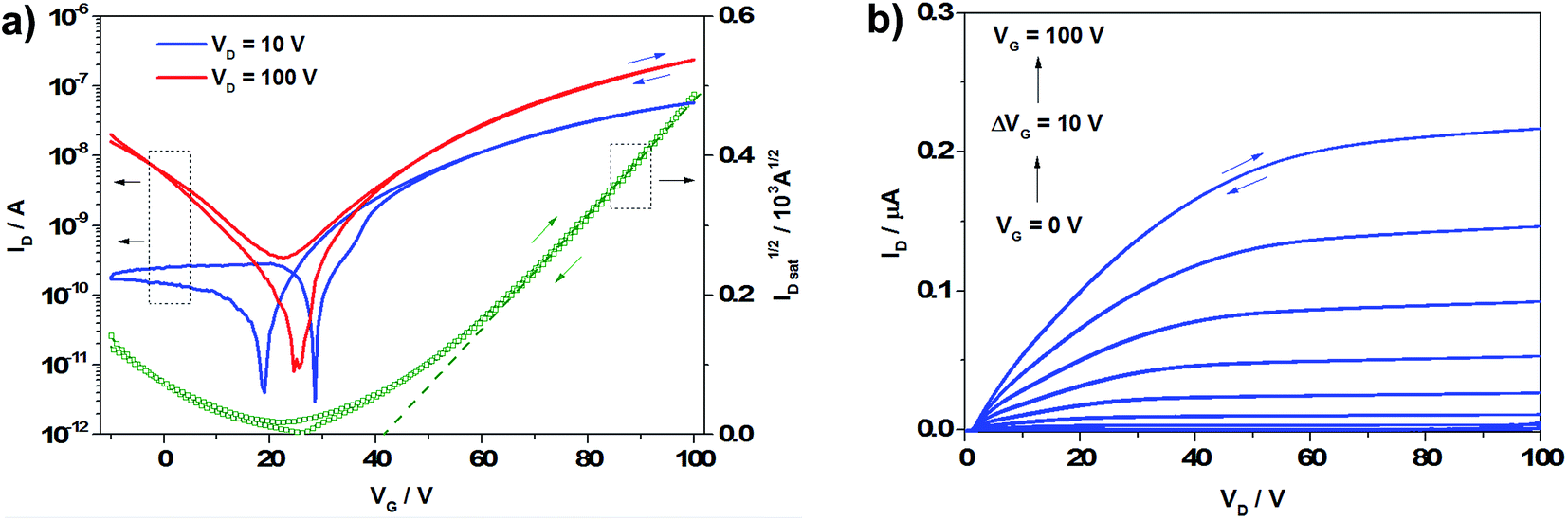 | ||
| Fig. 5 Transfer (left) and output (right) characteristics of P(DTDCNBT), under positive voltages in the TG/BC device configuration. | ||
The optimised geometry of DTDCNBT was calculated using DFT with B3LYP level of theory and a basis set of 6-31G(d). Calculations predicted both T-DCNBT dihedral angles to be ca. 30° i.e. close to those predicted in the potential energy scan (see Fig. S2† for optimised geometries). However, these calculations are performed on a single molecule in the gas phase which likely explains the slight discrepancy from the dihedral angles measured in the crystal structure.
The HOMO and LUMO molecular orbital energies, electron density plots and optimised geometries of all polymers were also calculated using DFT (B3LYP level of theory and a basis set of 6-31G(d)) and are shown in Fig. S3 and S4.† As predicted with the monomers, the fluorinated polymers had planar conformations whilst introduction of the larger cyano groups resulted in an increased dihedral angle between the thiophene and BT units reducing the planarity of the backbone. Theoretical HOMO and LUMO energy levels, as well as the polymer band gaps were also predicted using DFT (see Table S1†). Experimentally measured HOMO and LUMO levels were found to be deeper than those predicted using DFT, however DFT calculations predicted similar trends to those observed experimentally. For both the DTG and IDT based polymers replacing the fluorine substituents with cyano groups resulted in a large deepening of the calculated LUMO, and a smaller deepening of the HOMO, and reduced band gaps.
The thermal behaviour of the polymers was also investigated using DSC (differential scanning calorimetry), cycling between 30 and 300 °C. All of the thermograms were featureless with no obvious thermal transisitons, except P(IDT-DTDFBT) which showed a small, broad endotherm on heating at ∼200 °C and a small exotherm upon cooling, with an onset of 116 °C and peak at ∼105 °C (see Fig. S13†).
2.5. OFET devices
The charge carrier mobilities of the polymers were measured in either bottom gate, top contact (BG/TC) or top gate, bottom contact (TG/BC) devices. In BG/TC devices the Si/SiO2 substrates were treated with octadecyltrichlorosilane (OTS) in order to minimise the concentration of charge traps due to the presence of hydroxyl groups. In TG/BC devices made with P(Ge-DTDFBT) the Au source–drain electrodes were treated with pentafluorobenzene thiol (PFBT) to aid hole injection, however untreated Au was used for devices made with P(Ge-DTDCNBT). CYTOP was used as the gate dielectric in both cases. For both IDT polymers, the films were annealed at 200 °C near the thermal transition observed in DSC, and for the DTG polymers an annealing temperature of 160 °C was used. The charge carrier mobilities (calculated in saturation) of the polymers measured from these device configurations are summarised in Table 3.| Polymer | Device configuration | Electrode treatment, dielectric type | Field-effect carrier mobility (cm2 V−1 s−1) | |
|---|---|---|---|---|
| Holes | Electrons | |||
| P(Ge-DTDCNBT) | TG/BC | Au, CYTOP | Not observed | 2.8 × 10−3 |
| P(Ge-DTDFBT) | TG/BC | Au–PFBT, CYTOP | 6.2 × 10−2 | Not observed |
| P(IDT-DTDCNBT) | BG/TC | Au/Al, Si/SiO2–OTS | Not observed | 4.9 × 10−4 |
| P(IDT-DTDFBT) | BG/TC | Au/Al, Si/SiO2–OTS | 6.1 × 10−2 | Not observed |
TG/BC transistors based on P(Ge-DTDFBT) were found to function as p-channel devices yielding a peak hole mobility of 6.2 × 10−2 cm2 V−1 s−1. Replacing the fluorine substituents in P(Ge-DTDFBT) with cyano groups in P(Ge-DTDCNBT) resulted in a change in the dominant carrier from holes (p-type) to electrons (n-type), with an electron mobility of 2.8 × 10−3 cm2 V−1 s−1 in the TG/BC device configuration (Fig. 5). In these devices Au source–drain electrodes were left untreated and P(Ge-DTDCNBT) showed no hole transport when tested under negative gate voltages (see Fig. S5†). Treatment of Au electrodes with PFBT had little effect on the electron transport, however it induced weak ambipolar transport, with hole mobilities an order of magnitude lower at ∼10−4 cm2 V−1 s−1. We ascribe the emergence of electron conduction in P(Ge-DTDCNBT) to the substantial lowering of the LUMO in comparison to P(Ge-DTDFBT), which facilitates electron injection. The decrease in hole mobility may relate to the increase in ionisation potential for P(Ge-DTDCNBT) which increases the barrier to hole injection.
BG/TC transistors made with the fluorinated IDT polymer P(IDT-DTDFBT) were found to operate as p-channel devices with a peak hole mobility of 6.1 × 10−2 cm2 V−1 s−1. Similarly to the DTG based polymers, the introduction of the cyano groups to the polymer backbone induced a change in the type of dominant charge carrier from holes (p-type) to electrons (n-type). For optimised BG/TC transistors made with P(IDT-DTDCNBT) an electron mobility value of 4.9 × 10−4 cm2 V−1 s−1 was calculated. Both cyanated polymers showed a reduction in charge mobility in comparison to their fluorinated analogues, P(Ge-DTDCNBT) had a charge mobility approximately an order of magnitude lower whilst P(IDT-DTDCNBT) had a charge mobility approximately two orders of magnitude lower. This reduction in the charge mobility may be due to reduced planarity of the backbone upon cyanation. This might be more significant in the IDT based polymers than the DTG case because in the later, the long and branched 2-octyldodecyl chains may already cause some backbone disorder by interacting with the adjacent thiophene monomers. We have observed similar effects in bridged dithienogermolodithiophene based polymers with 2-octyldodecyl side chains.52 For full device characterisation, including threshold voltages and on/off ratios see Table S2.†
In order to examine the difference in mobilities between the polymers further, the morphology of the spin coated films were investigated using atomic force microscopy (AFM). Films of P(Ge-DTDCNBT) and P(Ge-DTDFBT) were spun onto glass under the same conditions as for the TG/BC devices. The topography and phase images of P(Ge-DTDCNBT) and P(Ge-DTDFBT) are shown in Fig. 6. The films of the both polymers look fairly similar with a uniform polymer grain size. Both films had a very low surface roughness with a route mean square (RMS) roughness of 0.72 nm and 0.92 nm for P(Ge-DTDCNBT) and P(Ge-DTDFBT) respectively. Both polymers have a similar morphology in thin film, which may be one of the reasons they have a closer performance in OFET devices than the IDT-based polymers.
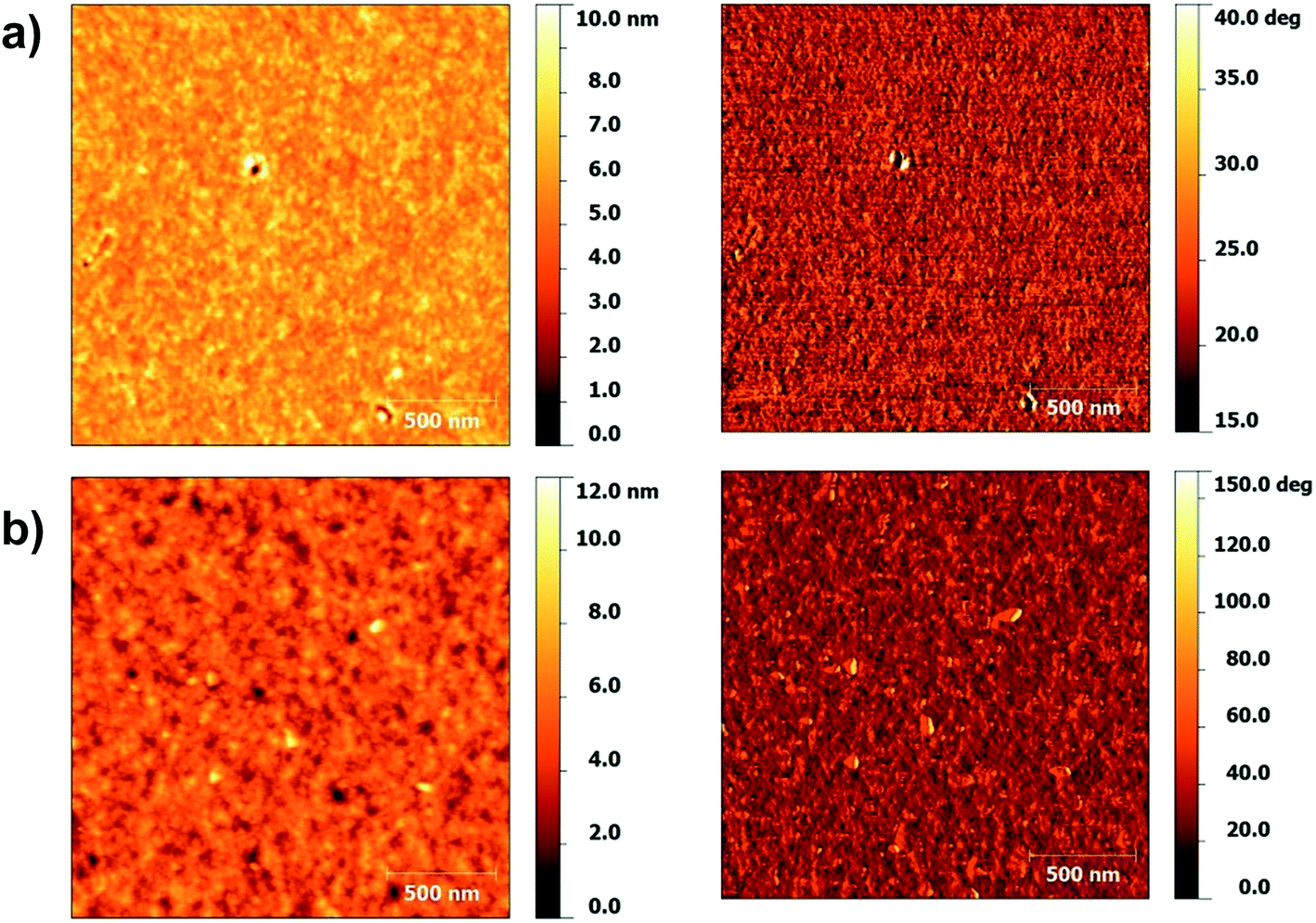 | ||
| Fig. 6 AFM topography (left) and phase (right) of (a) P(Ge-DTDCNBT) and (b) P(Ge-DTDFBT). | ||
Fig. 7 shows the topography and phase images of P(IDT-DTDCNBT) and P(IDT-DTDFBT), films were spun onto OTS treated Si/SiO2 substrates as BG/TC device mobilities are compared. In this case the surfaces look quite different, large terraces seem to be present in the film of P(IDT-DTDCNBT) giving a less continuous film than P(IDT-DTDFBT). These observations are reflected in the surface roughness of the films. The surface of P(IDT-DTDFBT) is fairly smooth with an RMS of 0.93 nm whilst the surface of P(IDT-DTDCNBT) is approximately three times as rough with an RMS of 2.94 nm. The less continuous morphology of the P(IDT-DTDCNBT) film and rougher surface may help to explain the greater reduction in device mobility of P(IDT-DTDCNBT) in comparison to P(IDT-DTDFBT). However, it should be noted that it is the morphology of the bottom of the film, in contact with the dielectric that is more relevant for charge transport in the BG/TC device architecture.
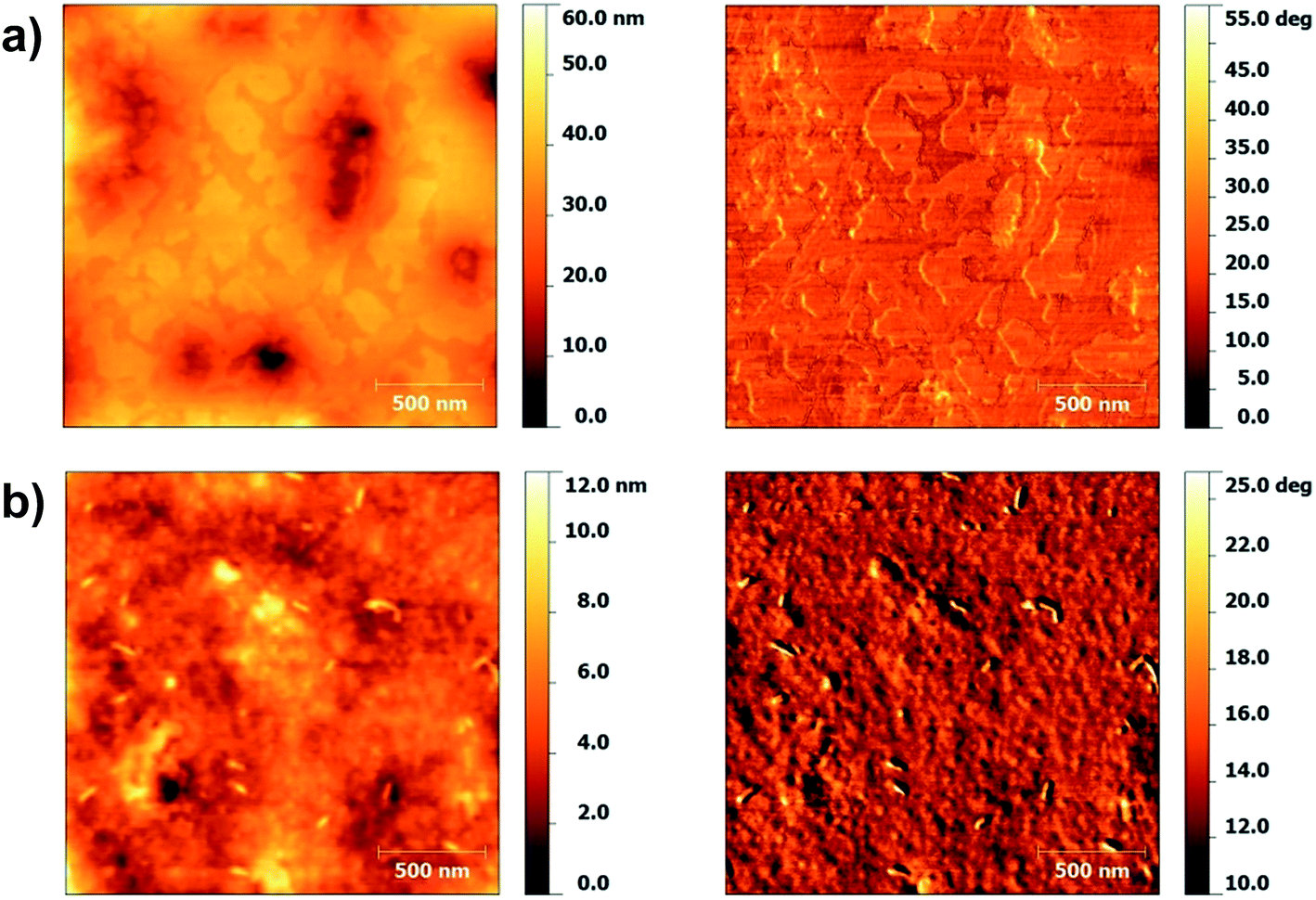 | ||
| Fig. 7 AFM topography (left) and phase (right) of (a) P(IDT-DTDCNBT) and (b) P(IDT-DTDFBT). | ||
3. Conclusion
Here we report the synthesis of a new electron accepting monomer, 4,7-di(thiophen-2-yl)-5,6-dicyano-2,1,3-benzothiadiazole (DTDCNBT) in which cyano groups are incorporated onto the 2,1,3-benzothiadiazole. The cyano groups can be readily introduced from a fluorinated precursor 4,7-di(thiophen-2-yl)-5,6-difluro-2,1,3-benzothiadiazole DTDFBT through substitution of the fluorine substituents under SNAr conditions. Co-polymers of both DTDCNBT and DTDFBT with electron rich dithienogermole (DTG) and indacenodithiophene (IDT) co-monomers were prepared and investigated. Replacing the fluorine substituents with cyano groups in both the DTG and IDT based polymers was found to significantly red-shift the absorbance of the polymers in both solution and thin-film. Such large red-shifts (up to 186 nm) were a surprising result given the fairly subtle structure change. In both cases the cyanated polymers showed a small reduction in HOMO energy (<0.1 eV) and a large reduction in LUMO energy (∼0.4 eV) in comparison to the fluorinated analogues, explaining the large reduction in band gap.Investigating the charge mobility of the polymers in OFET devices, it was found that replacing the fluorine substituents of the polymers with cyano groups changed the predominant charge transport from p-type to n-type. This work demonstrates a facile approach to switching the type of charge transport through a relatively subtle change in chemical structure. The large stabilisation of the LUMO levels, increased electron mobility and reversible reduction processes may also make these cyanated polymers interesting candidates for acceptor materials in all polymer solar cells.
4. Experimental
4.1. General
All solvents and chemicals, were purchased from Sigma-Aldrich and used without further purification. 4,7-Bis(thiophen-2-yl)-5,6-difluoro-2,1,3-benzothiadiazole46 and monomer 542 and were synthesised using literature procedures. P(Ge-DTDFBT) were prepared as reported.53 Microwave reactions were performed in a Biotage initiator V 2.3. in constant temperature mode. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker AV-400 (400 MHz) spectrometers in chloroform-d solutions. Number-average (Mn) and weight-average (Mw) molecular weights were determined with an Agilent Technologies 1200 series GPC detected using the refractive index signal in chlorobenzene at 80 °C using two PL mixed B columns in series, and calibrated against narrow polydispersity polystyrene standards. UV-visible absorption spectra were measured using a Shimadzu UV-1800 UV-vis Spectrophotometer. 16.6 mg dm−3 polymer solutions in chlorobenzene were used for solution spectra whilst 5 mg mL−1 polymer solutions in chlorobenzene were used to spin coat thin films at 1000 rpm for 60 seconds. Solutions and films were not de-oxygenated for these measurements. Preparative GPC used a customer build Shimadzu recSEC system comprising a DGU-20A3 degasser, an LC-20A pump, a CTO-20A column oven, an Agilent PLgel 10 μm MIXED-D column and a SPD-20A UV detector. Ionisation potentials were measured by Photo-Electron Spectroscopy in Air (PESA) on a Riken Keiki AC-2 PESA spectrometer. Polymer thin films were prepared by spin-coating from 5 mg mL−1 polymer solutions in chlorobenzene onto glass substrates. The PESA samples were run with a light intensity of 5 nW and data processed with a power number of 0.5. Density functional theory using a B3LYP54 functional and a basis set of 6-31G(d) was used to calculate the ground state geometries and electron density plots of monomers and trimer molecules. All calculations were carried out using Gaussview 5.0.55 Cyclic voltammograms were recorded using a Metrohm Autolab PGStat101 Potentiostat/Galvanostat. The experimental set-up consisted of an Ag/Ag+ reference electrode, a platinum wire counter electrode and an FTO working electrode, all measurements were carried out under argon at room temperature (∼25 °C). Measurements of the polymers were done on spun-cast films in anhydrous, degassed solutions of acetonitrile with tetrabutylammonium hexafluorophosphate (0.1 M) electrolyte. Thin films were spin coated onto FTO on glass substrate from a 5 mg mL−1 solution. A ferrocene internal standard was used to calibrate the results. Atomic force microscopy images were obtained with a Picoscan PicoSPM LE scanning probe in tapping mode under ambient conditions. Thin films of polymers were fabricated identically to the OFET devices except that the evaporation of the contact electrodes in the BG/TC configuration was not performed and films were spun directly onto glass for the TG/BC configuration. Preparative Gel Permeation Chromatography was performed in hexane at 40 °C. The system consists of a DGU-20A3 degasser, an LC-20A pump, a CTO-20A column oven, an Agilent PLgel 10 μm MIXED-D column and a SPD-20A UV detector.
4.2. OFET device fabrication
Transistor characterization was carried out under nitrogen using a Keithley 4200 parameter analyzer. All films were prepared and characterized under inert atmosphere. Bottom gate/top contact devices were fabricated on heavily doped n+-Si (100) wafers with 400 nm-thick thermally grown SiO2. The Si/SiO2 substrates were treated with trichloro(octadecyl)silane (OTS) to form a self-assembled monolayer. Both polymers (P(IDT-DTDFBT) and P(IDT-DTDCNBT)) were dissolved in chlorobenzene (5 mg mL−1) and spun cast at 2000 rpm for 60 s from hot solution before being annealed at 200 °C for 10 minutes. Au/Al (30/30 nm) source and drain electrodes were deposited under vacuum through shadow masks, where the asymmetric bilayer electrodes enables ambipolar injection in a single device, as previously described.56 The channel width and length of the transistors are 1000 μm and 40 μm, respectively. Mobility was extracted from the slope of ID1/2vs. VG.Top gate/bottom contact devices were fabricated on glass substrates using Au (30 nm) source–drain electrodes and CYTOP dielectric. Devices made with P(Ge-DTDFBT) used Au electrodes treated with pentafluorobenzene thiol (PFBT) SAM to increase the work function. The channel width and length of the transistors are 1000 μm and 30 μm, respectively. Both polymers (P(Ge-DTDFBT) and P(Ge-DTDCNBT)) were dissolved in chlorobenzene (5 mg mL−1) and spun cast at 2000 rpm from a hot solution for 60 s before being annealed at 160 °C for 30 min. Mobility was extracted from the slope of ID1/2vs. VG.
4.3. Synthesis of monomer and polymers
:3 (v:v)) giving an orange powder (210 mg, 0.6 mmol). Yield: 76%; mp 249–250 °C. 1H NMR (400 MHz, CDCl3) δ 8.18 (dd, J = 3.8, 1.1 Hz, 2H), 7.79 (dd, J = 5.1, 1.1 Hz, 2H), 7.32 (dd, J = 5.1, 3.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 153.48, 133.37, 133.17, 132.63, 132.38, 128.03, 116.34, 111.09. Anal. calcd for C16H4Br2N4S3: C, 54.84%; H, 1.73%; N, 15.99%. Found: C, 54.83%; H, 1.77%; N, 15.91%.
:72, chloroform) under argon. The chloroform fraction was concentrated to ∼60 mL in a round bottom flask, a solution of aqueous sodium diethyldithiocarbamate dihydrate solution (∼100 mg in 60 mL) was then added. The round bottom flask was equipped with a reflux condenser and the mixture stirred vigorously at 60 °C for 1 h to allow palladium extraction. The chloroform layer was then separated and washed with water (3 × 100 mL). The solution was then concentrated to ∼8 mL, precipitated in methanol (100 mL) and filtered. This precipitation was repeated to yield the dark green polymer P(Ge-DTDCNBT) (190 mg, yield: 63%) as long fibres. Chloroform fraction: Mn: 20.52 kDa, Mw: 44.42 kDa, Mn/Mw (Đ): 2.16. 1H NMR (400 MHz, TCE) δ 8.38 (m, 3H), 7.51–7.42 (m, 3H), 1.71 (m, 4H), 1.47 (m, 4H), 1.40–1.24 (m, 62H), 0.96–0.91 (m, 12H).
4.4. P(IDT-DTDCNBT)
Monomer 6 (190 mg, 0.128 mmol), monomer 3 (65 mg, 0.128 mmol), Pd2(dba)3 (2.3 mg, 0.0026 mmol) and P(o-tol)3 (3.1 mg, 0.010 mmol) were reacted using the same procedure as P(Ge-DTDCNBT). The polymer was purified using a Soxhlet extractor (methanol, acetone, hexane, chloroform). The chloroform fraction was washed with aqueous sodium diethyldithiocarbamate dihydrate (to remove palladium) then concentrated and precipitated in methanol (100 mL) to yield P(IDT-DTDCNBT) as a dark green fibres (175 mg, 90%). GPC: chloroform fraction: Mn = 47.3 kDa, Mw = 81.2 kDa, Mw/Mn (Đ) = 1.7. 1H NMR (500 MHz, TCE-d2) δ 8.45–8.35 (m, 2H), 7.50–7.32 (m, 6H), 2.13 (m, 4H), 2.04 (m, 4H), 1.37–1.25 (m, 104H), 1.13 (m, 4H), 1.06 (m, 4H), 0.98–0.93 (m, 12H).4.5. P(IDT-DTDFBT)
Monomer 6 IDT (168 mg, 0.113 mmol), monomer 3 (56 mg, 0.113 mmol), Pd2(dba)3 (2.1 mg, 0.0022 mmol) and P(o-tol)3 (2.8 mg, 0.0090 mmol) were reacted using the same procedure as P(Ge-DTDCNBT). The polymer was purified using a Soxhlet extractor (methanol, acetone, hexane, hexane–chloroform 28:72, chloroform). Both the chloroform–hexane and chloroform fractions were washed with aqueous sodium diethyldithiocarbamate dihydrate (to remove palladium) then concentrated and precipitated in methanol (100 mL) to yield P(IDT-DTDFBT) as a dark blue fibres (135 mg, 80%). GPC: chloroform–hexane fraction (72/28) (111 mg): Mn = 25.4 kDa, Mw = 38.0 kDa, Mw/Mn (Đ) = 1.5, chloroform fraction (12 mg): Mn = 51.9 kDa, Mw = 74.2 kDa, Mw/Mn (PDI) = 1.4. 1H NMR (500 MHz, TCE-d2) δ 8.33 (m, 2H), 7.46–7.31 (m, 6H), 2.13 (m, 4H), 2.04 (m, 4H), 1.36–1.24 (m, 104H), 1.13 (m, 4H), 1.05 (m, 4H), 0.95 (m, 12H).
Acknowledgements
We thank the UK's Engineering and Physical Sciences Research Council (EPSRC) for financial support via the Doctoral Training Centre in Plastic Electronics EP/G037515/1 (AC, MH) and the EPSRC platform grant EP/G060738/1 (ZF, MH). We gratefully acknowledge Dr Scott E Watkins (CSIRO) for the PESA measurements.References
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- K. Fukuda, Y. Takeda, M. Mizukami, D. Kumaki and S. Tokito, Sci. Rep., 2014, 4, 3947 Search PubMed.
- M. Jung, J. Kim, J. Noh, N. Lim, C. Lim, G. Lee, J. Kim, H. Kang, K. Jung, A. D. Leonard, J. M. Tour and G. Cho, IEEE Trans. Electron Devices, 2010, 57, 571–580 CrossRef CAS.
- K.-J. Baeg, M. Caironi and Y.-Y. Noh, Adv. Mater., 2013, 25, 4210–4244 CrossRef CAS PubMed.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- B. J. Jung, N. J. Tremblay, M.-L. Yeh and H. E. Katz, Chem. Mater., 2011, 23, 568–582 CrossRef CAS.
- R. Y. C. Shin, P. Sonar, P. S. Siew, Z.-K. Chen and A. Sellinger, J. Org. Chem., 2009, 74, 3293–3298 CrossRef CAS PubMed.
- H.-R. Tseng, H. Phan, C. Luo, M. Wang, L. a. Perez, S. N. Patel, L. Ying, E. J. Kramer, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Adv. Mater., 2014, 2993–2998 CrossRef CAS PubMed.
- H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS PubMed.
- Y. Diao, B. C.-K. Tee, G. Giri, J. Xu, D. H. Kim, H. a. Becerril, R. M. Stoltenberg, T. H. Lee, G. Xue, S. C. B. Mannsfeld and Z. Bao, Nat. Mater., 2013, 12, 665–671 CrossRef CAS PubMed.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- H. Zhong, J. Smith, S. Rossbauer, A. J. P. White, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2012, 24, 3205–3211 CrossRef CAS PubMed.
- T. D. Anthopoulos, F. B. Kooistra, H. J. Wondergem, D. Kronholm, J. C. Hummelen and D. M. deLeeuw, Adv. Mater., 2006, 18, 1679–1684 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- P. Sonar, S. P. Singh, P. Leclère, M. Surin, R. Lazzaroni, T. T. Lin, A. Dodabalapur and A. Sellinger, J. Mater. Chem., 2009, 19, 3228–3237 RSC.
- T. C. Parker, D. G. Dan Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015 10.1039/C4MH00102H , Advance Article.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS PubMed.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS PubMed.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J.-H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186–2190 CrossRef CAS PubMed.
- T. L. Dexter Tam, T. Salim, H. Li, F. Zhou, S. G. Mhaisalkar, H. Su, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem., 2012, 22, 18528–18534 RSC.
- T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898–13901 CrossRef CAS PubMed.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190–1193 CrossRef CAS PubMed.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- A. P. Zoombelt, M. Fonrodona, M. M. Wienk, A. B. Sieval, J. C. Hummelen and R. a J. Janssen, Org. Lett., 2009, 11, 903–906 CrossRef CAS PubMed.
- H. Li, T. L. Tam, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 46–49 CrossRef CAS PubMed.
- Y. Cao, T. Lei, J. Yuan, J.-Y. Wang and J. Pei, Polym. Chem., 2013, 4, 5228–5236 RSC.
- Z. Li, J. Lu, S.-C. Tse, J. Zhou, X. Du, Y. Tao and J. Ding, J. Mater. Chem., 2011, 21, 3226–3233 RSC.
- H. Zhou, L. Yang, S. C. Price, K. J. Knight and W. You, Angew. Chem., Int. Ed., 2010, 49, 7992–7995 CrossRef CAS PubMed.
- J.-F. Jheng, Y.-Y. Lai, J.-S. Wu, Y.-H. Chao, C.-L. Wang and C.-S. Hsu, Adv. Mater., 2013, 25, 2445–2451 CrossRef CAS PubMed.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- H. Bronstein, D. S. Leem, R. Hamilton, P. Woebkenberg, S. King, W. Zhang, R. S. Ashraf, M. Heeney, T. D. Anthopoulos, J. De Mello and I. McCulloch, Macromolecules, 2011, 44, 6649–6652 CrossRef CAS.
- Z. Fei, J. S. Kim, J. Smith, E. B. Domingo, T. D. Anthopoulos, N. Stingelin, S. E. Watkins, J.-S. Kim and M. Heeney, J. Mater. Chem., 2011, 21, 16257 RSC.
- Z. Fei, Y. Kim, J. Smith, E. B. Domingo, N. Stingelin, M. a. McLachlan, K. Song, T. D. Anthopoulos and M. Heeney, Macromolecules, 2012, 45, 735–742 CrossRef CAS.
- Z. Fei, M. Shahid, N. Yaacobi-Gross, S. Rossbauer, H. Zhong, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Commun., 2012, 48, 11130–11132 RSC.
- C. Burmester and R. Faust, Synthesis, 2008, 8, 1179–1181 Search PubMed.
- L. Dou, C. Chen, K. Yoshimura, K. Ohya, W. Chang, J. Gao, Y. Liu, E. Richard and Y. Yang, Macromolecules, 2013, 46, 3384–3390 CrossRef CAS.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045–7052 CAS.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- S. Tierney, M. Heeney and I. McCulloch, Synth. Met., 2005, 148, 195–198 CrossRef CAS.
- K. T. Nielsen, K. Bechgaard and F. C. Krebs, Macromolecules, 2005, 38, 658–659 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. Mahrt, H. Bassler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- H. Kirihata and M. Uda, Rev. Sci. Instrum., 1981, 52, 68–70 CrossRef CAS.
- R. J. Davis, M. T. Lloyd, S. R. Ferreira, M. J. Bruzek, S. E. Watkins, L. Lindell, P. Sehati, M. Fahlman, J. E. Anthony and J. W. P. Hsu, J. Mater. Chem., 2011, 21, 1721–1729 RSC.
- H. Zhong, Z. Li, E. Buchaca-Domingo, S. Rossbauer, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. A, 2013, 1, 14973–14981 CAS.
- A more detailed description of the performance of P(Ge-DTDFBT) in organic solar cells, Z. Fei, E. Collado-Fregoso, S. Ashraf, J. R. Durrant, M. Heeney, manuscript in preparation.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- Z. Fei, X. Gao, J. Smith, P. Pattanasattayavong, E. Buchaca Domingo, N. Stingelin, S. E. Watkins, T. D. Anthopoulos, R. J. Kline and M. Heeney, Chem. Mater., 2013, 25, 59–68 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1020253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc02008a |
| This journal is © The Royal Society of Chemistry 2015 |