
Synergic effect of unsaturated inner bridges and polymorphism for tuning the optoelectronic properties of 2,3-thieno(bis)imide based materials†
Massimo
Zambianchi
a,
Laura
Favaretto
a,
Margherita
Durso
a,
Cristian
Bettini
b,
Alberto
Zanelli
a,
Ilse
Manet
a,
Massimo
Gazzano
a,
Lucia
Maini
c,
Denis
Gentili
d,
Stefano
Toffanin
d,
Federico
Gallino
e,
Michele
Muccini
df,
Massimiliano
Cavallini
*d and
Manuela
Melucci
*a
aConsiglio Nazionale delle Ricerche, Istituto per la Sintesi Organica e la Fotoreattività, (CNR-ISOF), via P. Gobetti 101, 40129, Bologna, Italy. E-mail: manuela.melucci@isof.cnr.it
bMIST-ER Laboratory, via P. Gobetti 101, 40129, Bologna, Italy
cDipartimento di Chimica “G. Ciamician”, Università di Bologna, Via F. Selmi 2, 40126, Bologna, Italy
dConsiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati (CNR-ISMN), via P. Gobetti 101, 40129, Bologna, Italy. E-mail: m.cavallini@bo.ismn.cnr.it
eSAES-GETTERS S. p. A., Viale Italia 77, 20020, Lainate, MI, Italy
fE.T.C. s.r.l., via P. Gobetti 101, 40129, Bologna, Italy
First published on 20th October 2014
Abstract
2,3-Thieno(bis)imide (N) ended oligomers are emerging as valuable molecular materials for applications in organic electronics. Here, we report the synthesis and characterization of three new 2,3-thieno(bis)imide ended oligothiophenes (T) bearing unsaturated ethylene (E), azomethine (I) and ethinyl (A) inner bridges (NTE, NTI and NTA, respectively). The effect of the unsaturated bridge on the π-conjugation extent, molecular conformation and overall aromaticity is related to the functional optoelectronic and morphological properties and compared to the properties of the linear analogue (NTT) with a bithiophene inner moiety. Optical spectroscopy and cyclovoltammetry analysis show a strong red shift of the absorption and an increased energy band gap on going from NTI and NTE to NTA. The HOMO level decreases in the order NTE > NTI > NTA. Moreover, while the LUMO of NTE and NTA have almost the same energy, NTI has a LUMO energy about 0.1 eV lower, likely due to the electron withdrawing effect of the azomethine moiety. Morphological investigation of solution cast thin deposits shows that the unsaturated bridges promote the formation of concomitant polymorphs with the simultaneous presence of microcrystals with different morphology and fluorescence properties. Moreover, irreversible conversion of one polymorph to the other was achieved by thermal treatments for NTA and NTE and by exploiting this feature, we realized a time temperature integrator (TTI) device based on NTE material. This device allowed to monitor temperature evolutions in the range between RT and 200 °C by means of a red to yellow fluorescence switch that was detectable by optical microscopy.
Introduction
Multifunctional π-conjugated small molecules are experiencing a new revival as active components of field effect transistors (OFETs)1 and photovoltaic devices (OPVs)2 or as coadjutants of polymeric based devices.3A key issue of producing materials with optimized optoelectronic performances is molecular design allowing to achieve the desirable electronic structure, solid-state packing and physical properties.1–4 These features highly rely on the choice of proper π-conjugated building blocks, molecule size/shape and peripheric substituents. Among the recently emerged building blocks, the electron deficient 2,3-thienoimide group (N) is gaining increasing attention due to the promotion of n-type and ambipolar charge transport properties in the resulting N-ended oligomers.5 This new class of materials is being tested as active layer in ambipolar light emitting transistors (OLETs),6 field-effect transistors (OFETs),5 and both as donor and acceptor in photovoltaic cells (OPV).7 Due to its peculiar electron deficient feature, the thienoimide group strongly influences the optoelectronic properties of the final oligomers. In particular, 2,3-thienoimide impacts the LUMO energy level that results insensitive of the number of thienoimide moieties and mainly located on the oligomer periphery.5,6a Moreover, the interplay between intermolecular interactions enabled by the heteroatom rich 2,3-thienoimide chemical structure, and π–π stacking, strongly influences molecular conformation and solid state packing, two properties that are intimately related to optical and charge transport functionalities as well as to the final device performances. A highly crystalline morphology has been observed for thin films of 2,3-thieno(bis)imide derivatives.5,6 Several structure–property relationship studies have shown that the N-end substitution promotes π-stack or herringbone packing – the latter more typical for conventional oligothiophenes – depending on the molecular structure.6a,b Moreover, polymorphic behaviour evidences were found for some N derivatives in single crystal6b and thin films.8
Polymorphism9 is known for many commonly studied organic molecular semiconductors such as pentacene,10 rubrene,11 oligothiophenes12 and anthradithiophene13 among others. If on one hand, polymorphism allows a deeper comprehension of the structure–property relationships in organic semiconductors, it is also highly controversial since it can markedly affect the device performance and response reproducibility.
Despite this, we demonstrated the potential of polymorphic organic materials, such as a small trimeric 2,3-thieno(bis)imide derivative,8 for the application of time temperature integrators (TTI),14i.e. device capable of monitoring its thermal history through an irreversible variation of morphological and/or optical properties of a thin deposit.
Achieving control of polymorphism is therefore crucial not only for charge transport based devices, but also for novel applications based on the controlled conversion of functional polymorphs in thin films. In this respect, molecular design and structure–property relationship investigations play a key role.
Ethylene (E) bridge insertion in oligothiophenes has been associated to a reduced band gap derived from enhanced molecular planarity and a lower overall aromatic character than oligothiophenes containing the same number of sp2 carbons.15 Similarly, an azomethine (I) bridge promotes an even greater bathocromic effect and has also been investigated as a structural tool to tune the electronic properties of oligothiophenes.16 Contrarily, the main effect observed for acetylenic (A) bridged oligothiophenes with respect to their fully sp2 carbon based π-conjugated analogues is an increased band gap originating from the mismatch of the sp carbon orbitals with the π-delocalized system.17
On these bases, herein we focus on the insertion of ethylene, azomethine and acetylene bridges in N-ended materials as a strategy to alter a π-conjugated system, thus, to ultimately tune their optoelectronic and packing properties. The effect of the unsaturated bridges in NTE, NTI, NTA (Fig. 1) compounds on the functional properties has been evaluated and compared to those of conventional oligothiophenes or to a bridge-free 2,3-thieno(bis)imide isostructural derivative.6b During the thin film deposition, all compounds crystallized with concomitant polymorphs18 which were characterized by a different morphology and fluorescence emission. The polymorphic nature of these compounds prompted us to exploit the functional polymorphism of these materials combined with the possibility of thermal phase control to produce a NTE based TTI device.
 | ||
| Fig. 1 Molecular structure of compounds 1–3. | ||
Experimental
Synthesis
General information and the synthesis of all precursors are reported in the ESI file.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 to 100:0). The fractions containing the product were combined, the solvent evaporated, and the residue crystallized from hot toluene to give a dark red solid (100 mg, 57%). M.p. 255 °C; DE-MS (m/z): 662 (M˙+); absorption maximum, 465 nm, emission maximum, 588 nm in DCM; 1H NMR (CDCl3), δ [ppm]: 7.31 (s, 2H), 7.23 (d, 3J = 4.0 Hz, 2H), 7.04 (d, 3J = 4.0 Hz, 2H), 7.02 (s, 2H), 3.60 (t, 4H), 1.64 (m, 4H), 1.31 (m, 12H), 0.88 (m, 6H); 13C NMR (CDCl3), δ [ppm]: 163.9, 162.7, 149.8, 145.3, 143.7, 137.3, 134.3, 128.0, 126.7, 122.1, 116.5, 38.6, 31.4, 28.8, 26.5, 22.5, 14.0. Anal. calcd for C34H34N2O4S4 (662.90): C, 61.60; H, 5.17. Found: C, 61.54; H, 5.22.
:70 to 100:0). The fractions containing the product were combined, the solvent evaporated, and the residue crystallized from hot toluene to give a dark red solid (100 mg, 57%). M.p. 255 °C; DE-MS (m/z): 662 (M˙+); absorption maximum, 465 nm, emission maximum, 588 nm in DCM; 1H NMR (CDCl3), δ [ppm]: 7.31 (s, 2H), 7.23 (d, 3J = 4.0 Hz, 2H), 7.04 (d, 3J = 4.0 Hz, 2H), 7.02 (s, 2H), 3.60 (t, 4H), 1.64 (m, 4H), 1.31 (m, 12H), 0.88 (m, 6H); 13C NMR (CDCl3), δ [ppm]: 163.9, 162.7, 149.8, 145.3, 143.7, 137.3, 134.3, 128.0, 126.7, 122.1, 116.5, 38.6, 31.4, 28.8, 26.5, 22.5, 14.0. Anal. calcd for C34H34N2O4S4 (662.90): C, 61.60; H, 5.17. Found: C, 61.54; H, 5.22.
Optical spectroscopy
Quantum yield measurements of thin-deposits and solutions were carried out by a stand-alone and automatic absolute PL quantum yield measurements system (C9920-02, Hamamatsu). The measurements were performed in air, exciting the PL emission absorption maximum wavelength.Computational details
The molecular structure geometry of all the compounds has been optimized in their ground state in vacuum using the B3LYP19a,b exchange-correlation functional and 6-31+G* basis set as implemented in the Gamess code.19c,d TDDFT calculations of the lowest singlet–singlet excitation S0 → S1 have been performed to compute the optical gap Eg. LUMO energy levels were obtained by summing the optical gap to the HOMO levels. Overlap integrals Sh and Se have been obtained as Sij = 〈φi/φj〉 where φi,j is the HOMO or LUMO orbital of the i,j-molecule of the pair.Cyclic voltammetry
1 mmol L−1 solution of compounds 1–3 in Ar purged DCM (Aldrich, distilled over P2O5 and stored under Ar pressure) with 0.1 mol L−1 (C4H9)4NClO4 (Fluka, crystallized from CH3OH and vacuum dried). Analysis was performed in a home-made three-compartment glass cell under Ar pressure. The working electrode was semi-spherical Pt (area 0.05 cm2), the auxiliary electrode was a Pt wire in a separate compartment, and the reference electrode was an aqueous KCl saturated calomel electrode (E°ferrocene/ferricinium = 0.47 V vs. SCE) separated from the working electrode compartment by a liquid bridge containing the same solvent and support electrolyte.Thin deposits
Thin deposits were prepared by drop casting 20 μL cm−2 of a 1 g L−1 solution in toluene on Si/SiO2 wafers. The solvent was slowly evaporated at room temperature in a solvent-saturated atmosphere or in air. The substrates consist of a 10 × 10 mm2 piece of silicon covered by 200 nm of thermal oxides or pre-patterned Si/SiO2 substrates fabricated by photolithography. The substrates were washed by sonication for 2 min. in electronic-grade water (Milli-Q-pure quality), acetone and 2-propanol.Optical microscopy
Optical micrographs were recorded with a Nikon i-80 microscope equipped with epi-illuminator and cross polars (POM). All the images presented were recorded using objective: LU Plan ELWD 20X/0.40 objective.Laser scanning confocal fluorescence microscopy
Confocal imaging was performed on a Nikon A1 laser scanning confocal microscope equipped with a CW Argon ion laser (excitation at 488 nm, Melles Griot), and a 485 nm pulsed/CW diode laser (Picoquant, Germany). The system mounts an inverted Ti-E Nikon microscope. Confocal fluorescence imaging was carried out on the samples at RT. The 521 × 512 or 1024 × 1024 pixel images were collected using a Nikon PLAN APO VC 60× oil immersion objective with NA 1.40. With this imaging configuration, pixel side dimension ranged from 100 to 250 nm. Hexagonal pinhole dimension was set to 0.8 au corresponding to a dimension of 25 μm and optical thickness of 330 nm. In the scan head, a dichroic mirror reflecting 405 nm, 488 nm, 541 nm and 640 nm was used, as well as a 20/80 beam splitter. Fluorescence was collected in several spectral windows. In front of the photomultiplier, we used band-pass filters centered at 525 nm (50), 595 nm (50) and 700 nm (75). Spectral imaging has been performed using the Nikon A1 spectral detector consisting of a multi-anode photomultiplier with an array of 32 anodes. A wavelength range of 10 nm per anode has been applied.Fluorescence microscopy
Fluorescence images were recorded with a Nikon i-80 microscope equipped with epi-fluorescence (FM) using FM filter Nikon Ex 420, DM 435, BA 475 and Ex 535, DM 570, BA 590. FM images were recorded using a commercial CCD camera (Nikon CCD DS-2Mv). The illumination was performed by a 100 W halogen lamp at a fixed power (i.e. tension 12 V) and with a fixed time of acquisition of the CCD (500 ms). Image color composition was analyzed by Graphic Converter X software.Thermal annealing
Thermal annealing was performed under the optical microscope using a heating stage Linkham TMHS600 connected to a TP94 controller (control of 0.1 °C).X-ray diffraction analysis was carried out by means of a PANalytical X'Pert diffractometer equipped with a copper anode (λmean = 0.15418 nm) and a fast X'Celerator detector. Step 0.05° (2 theta scale), counting time 400 s.
Results and discussion
Synthesis
NTE (1) was prepared starting from (E)-1,2-di(thiophen-2-yl)ethene 4. This compound was synthesized according to an already described procedure leading to the trans derivative as majority product.20Trans-4 was then bis-stannylated (nBuLi, Bu3SnCl) to give 5 in 98% yield. Subsequent coupling with bromothienoimide 6 gives the target NTE in 57% isolated yield. The corresponding 1H-NMR spectrum confirms the exclusive presence of only one isomer.NTI (2) was obtained in nearly quantitative yield by forming the imine bridge between compound 12, with an amino end group and the formyl ended 14 (Scheme 2).
 | ||
| Scheme 1 Synthetic route to compound 1. | ||
 | ||
| Scheme 2 Synthetic route to compound 2. | ||
Firstly, 2-thiophenecarboxylic acid 7, was protected as methyl ester (7a) in 80% yield and then stannylated to give derivative 8 in 40% yield. A [Pd(0)] catalyzed Stille cross-coupling reaction between 8 and the brominated 2,3-thienoimide 6 gave the dimer 9 in 80% yield. The hydrolysis of the methyl ester occurred by treating compound 9 with NaOH, and by reacting the resulting crude with SOCl2. Besides hydrolyzing the ester, the strongly basic treatment also opened the thienoimide ring, so SOCl2 was used in order to dehydrate the intermediate and thus close again the thienoimide, giving compound 10 with a semiquantitative yield. By using diphenyl phosphoryl azide (DPPA), compound 10 was then converted into its azide derivative, which underwent Curtius rearrangement to the corresponding isocyanate. Since the reaction was carried out in the presence of tert-butanol, the final product of this synthetic step was the corresponding tert-butyl carbamate 11. This compound was obtained in 42% yield and converted to compound 12 by TFA treatment (99% yield).
Conversely, dimer 14 was prepared starting from 2-thiophenecarboxaldehyde that was first protected and then stannylated to give the organotin derivative 13 (details in ESI†). Then, Stille cross-coupling between 13 and brominated thienoimide 6 gave 14 in almost quantitative yield (98%, Scheme 2).
Finally, the two dimeric species 12 and 14 were coupled in iPrOH in the presence of catalytic amounts of TFA to give the target azomethine functionalized NTI in 90% yield. This synthetic route generally led to the trans isomer. Accordingly, DFT calculations show a good match between experimental and calculated HOMO and LUMO energy levels for the trans isomer (Table 1).‡
| Item | λabs (nm) | λema (nm) | E °ox (V) | E °red (V) | HOMO (eV) exp/th | LUMO (eV) exp/th | E g (eV) exp/th |
|---|---|---|---|---|---|---|---|
| a Excitation at the maximum absorption wavelength. b Taken from ref. 5a and corrected by CH2Cl2,/H2O liquid junction. c No difference between butyl and hexyl ends is expected. d Half-wave potential. e Peak potential at 0.1 V s−1. | |||||||
| NTT | 451 | 568 | — | — | −5.69c/−5.77 | −3.16c/−3.34 | 2.53/2.44 |
| NTE | 465 | 588 | 1.17, 1.41d | −1.21 | −5.54/−5.57 | −3.16/−3.21 | 2.38/2.36 |
| NTI | 471 | 544 | 1.28d, 1.43e | −1.09, −1.49 | −5.65/−5.73 | −3.28/−3.36 | 2.37/2.37 |
| NTA | 433 | 547 | 1.46e | −1.20 | −5.83/−5.74 | −3.17/−3.27 | 2.66/2.47 |
NTA, which is 3 with the ethinyl spacer, was synthesized by Sonogashira coupling between 2-iodothiophene and trimethylsilylacetylene followed by stannylation and Stille cross-coupling reaction between 15 (ref. 21) and the brominated thienoimide 6 provided the target material NTA in 68% yield (Scheme 3).
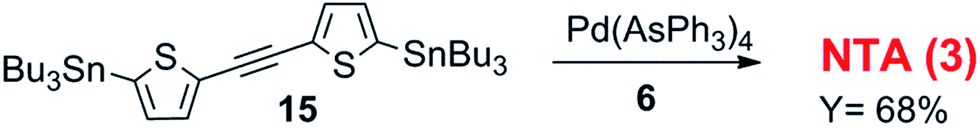 | ||
| Scheme 3 Synthetic route to compound 3. | ||
Optoelectronic properties
The electronic properties of the newly synthesized materials were investigated by combined optical, electrochemical and theoretical analysis and compared to those of a reference 2,3-thieno(bis)imide bridge-free system having the same number of thienyl rings, named NTT6b (Fig. 2a). The data are summarized in Table 1. | ||
| Fig. 2 (a) Molecular structure of reference bridge free 2,3-thieno(bis)imide system NTT6b (b) UV-vis and PL spectra of all compounds in CH2Cl2, (c) voltammograms in CH2Cl2 0.1 mol L−1 (C4H9)4NClO4 of the three molecules (1 mmol L−1). | ||
Absorption spectra (Fig. 2b) show a red shift of the absorption maxima from 433 nm to 465 nm and to 471 nm on going from NTA to NTE to NTI. On the other hand, the emission maximum of NTE (588 nm) is strongly red shifted with respect to those of NTA (547 nm) and NTI (544 nm). Similarly, a strong red shift of both absorption and emission is observed for NTE and NTI in comparison to NTT (465 nm, 471 nm vs. 451 nm for NTT) while a blue shift is observed for NTA with respect to NTT (433 nm vs. 451 nm). This can be likely ascribed to the lower overlap with the π-conjugated system of the (sp) C–C than that achieved by (sp2) CC, CN and TT moieties. Regarding emission spectra, NTE emission is red-shifted with respect to NTT (588 nm vs. 568 nm), NTI and NTA compound. The quantum efficiencies (PLQY) in solution were also measured. NTE and NTA displayed comparable PLQY, i.e. 19% and 20%, respectively, while a lower value was found for NTI (2%).
Cyclic voltammetry (CV) analysis results are reported in Fig. 2c and Table 1. The voltammogram of NTE (Fig. 2c) shows two quasi-reversible oxidation waves (1.17, 1.41 V vs. SCE) and a reduction one centred at −1.21 V (details in ESI†). The replacement of ethylene by azomethine induces the shift of the first oxidation wave that partially overlaps the second one (see slower scan rate in Fig. S1, ESI†) generating an irreversible process.
On the other hand, the reduction wave shifts to less negative potentials (−1.09 V) and a second reduction process appears into the electrochemical window (−1.49 V) indicating the possibility to host two electrons in the LUMO. Finally, NTA not only shows an irreversible oxidation wave shifted to the highest potential (1.46 V), but also a quasi-reversible reduction wave at the potential characteristic for the TBI moiety (about −1.2 V).
The HOMO5c energy levels decrease in the order NTE > NTI > NTA. On the other hand, the LUMOs of NTE and NTA have almost the same energy, but that of NTI is about 0.1 eV lower, likely due to the electron withdrawing effect of the azomethine moiety. In agreement with the spectroscopic data, the energy gap (Eg) of NTA is significantly larger than that of NTE and NTI.
To gain an insight on the optoelectronic properties, theoretical calculations were performed. Firstly, the most stable molecular conformation and the HOMO and LUMO energy levels and charge distribution of the new compounds were investigated by density functional theory (DFT) calculations (see Fig. 3).
 | ||
| Fig. 3 (a) Molecular structures with dihedral angles and (b) frontier orbitals isodensity plots and energy values of NTE, NTI, NTA. | ||
All the molecules adopt an almost planar conformation with dihedral angles between the N-moiety and the inner thiophene ring of about 160° and between the thiophene ring and the inner spacer of about 180° (NTE) and 165° (NTA). While NTE and NTA are centrosymmetric (Ci point group), NTI is non-centrosymmetric because of the azomethine spacer. This results in an asymmetric set of dihedral angles and in a charge distribution of frontier molecular orbitals. In NTE and NTA, the HOMO orbital is homogeneously distributed over the inner conjugated thiophene rings, while the LUMO orbital extends also to the electron deficient thienoimide units, in agreement with what is generally observed for N-ended systems as the reference NTT compound.6b On the contrary, for the non-symmetric NTI molecule, the LUMO is mainly localized on the thienoimide unit of the NC-bound molecular branch. DFT calculations, in agreement with experimental data, show a lower band gap for the NTE and NTI with respect to NTT. On the other hand, NTA shows a higher band gap and a significant shift of the HOMO level, likely due to a lower overlap of the sp carbons with the π-conjugated system, while the LUMO levels are much less affected by the unsaturated bridges since they are mainly located on the 2,3-thienoimide groups. This behaviour is in good accordance to that observed for ethylene,15a azomethine16 and acetylene17 bridged oligothiophenes without 2,3-thienoimide end groups.
Thin deposits morphology, polymorphism and fluorescence
Thin deposits of NTE, NTI and NTA, were grown by drop casting on silicon and glass surfaces in air.22 When deposited on a surface, all compounds exhibited a strong propensity to form large crystals. No continuous films were observed in between the crystals by AFM and laser confocal microscopy. As expected, thin deposits obtained from high boiling point solvents, such as toluene, show larger crystals than thin deposits obtained from low boiling point solvents, such as chloroform or dichloromethane. All thin deposits exhibit crystalline microstructure with strong birefringence under polarized optical microscopy (POM) and an intense photoluminescence (PL). Interestingly, concomitant polymorphs with markedly different fluorescence colour emission were clearly evident for all compounds.When deposited from toluene, NTE forms a mixture of rod-like and platelet-like crystals (Fig. 4a, red emitting and orange emitting polymorphs, respectively). Rod-like crystals appear as long crystallites (≤100 μm) randomly distributed on the surface with a width ranging from 5 μm to 10 μm. Platelet-like crystals show a regular shape with a long axis from 20 μm to 100 μm.§
 | ||
| Fig. 4 Fluorescence confocal images of the films of NTE, NTI, and NTA obtained upon evaporation of 0.5 mg mL−1 toluene solutions on glass. Top: images are a merge of the fluorescence collected on three PMT with bandpass filters 525/25 nm, 595/50 nm and 700/70 nm. Bottom: confocal fluorescence spectra collected for ROI indicated by the circles. | ||
Both polymorphs exhibit a strong birefringence under POM with the characteristic behaviour of optically anisotropic materials with extinction at precise orientations (rod-like crystals extinguish when the polarizers are oriented parallel or perpendicular to the long axis of the crystal, while platelets-like crystals extinguish when the polarizers are oriented along parallel or perpendicular directions to the border of the crystal). The evidence of light extinction at the same orientations in each crystal implies that they are either single crystals or formed by iso-oriented domains.
Fluorescence spectra performed by confocal microscopy (Fig. 4, bottom) show a luminescence spectrum peaking at 640 nm for the rod-polymorph and two emission peaks at 560 nm and 600 nm for the platelet polymorph. The polymorphic nature of platelets- and rod-like crystals was confirmed by XRD analysis carried out on samples specifically prepared in different conditions in order to strongly enhance the presence of one of the two forms. Rod- and platelets-like crystals show different patterns with the most intense peaks at 6.1°, 18.3°, 24.5° for the former and at 2.7°, 5.4° and 8.1° for the latter (the full list is reported as ESI†).
NTI deposits show needle shaped crystals (length < 500 μm) randomly distributed on the surface. Their width ranges from 10 μm to 100 μm resulting in an aspect ratio (length/width) between 1:5 and 1:50. Occasionally, some small aggregates (size < 2 μm) appear on the surfaces decorating the rod-like crystals (Fig. 4b). Crystals exhibit a strong birefringence under POM with the typical behaviour of optically anisotropic materials with extinction at precise orientations meaning that they are either single crystals or formed by domains oriented in the same direction.
The fluorescent image shows a strong red emission peaked at 660 nm from rod-like crystals, while the decorating aggregates emit in the green region.
Furthermore, NTA films show two polymorphs, i.e. fiber-like and rhombohedral crystals (Fig. 4c). The rhombohedral crystals show extinction when the long axis of the crystal is oriented 45° with respect to the polarizers, typical behaviour of optically anisotropic materials. The fiber-like crystals exhibit a strong birefringence, but they appear multicolored and do not extinguish at any directions, indicating their polycrystalline nature. Both polymorphs exhibit a strong fluorescence with two peaks at 535 and 575 nm for the crystals and two maxima at 560 and 600 nm for the bundle of fibers. Time-resolved fluorescence lifetime imaging measuring emission in the 505–535 nm, 565–605 nm and 635–675 nm ranges confirmed that the self-assembled fluorophores experienced locally different environments in the different morphologies (details in ESI†).
The emission of all compounds in thin deposits was also investigated by conventional PL spectroscopy indicative of the macroscopic film (Fig. S6, ESI†). According to the space resolved PL spectra (Fig. 4, bottom), red shifted emission was observed for NTE and NTI with respect to NTA. The PLQY for NTE and NTA thin deposits was 6% and 10%, respectively, i.e. lower than those observed in solution, and 4% for NTI.
DFT calculations were exploited to tentatively explain the origin of the observed polymorphism.19 In particular, the possibility of conformational polymorphism was considered. However, the conformation energy plot computed by varying the dihedral angle (θ) between the thienoimide moiety and the inner thiophene ring (Fig. S2, ESI†) shows two minima corresponding to the ANTI and the SYN conformers with a small energy difference (≈2 kcal mol−1). This, in combination with the already observed crystallization of N-based molecules in both ANTI and SYN forms, suggests that the observed concomitant polymorphs might be due to the crystallization of both conformers.
Moreover, calculations were exploited to investigate the effect of the aromaticity variation, induced by the unsaturated bridge substitution, on molecular π-stacking, through the computation of the overlap integrals Sh and Se for HOMO and LUMO orbitals, respectively. In Table 2, the overlap integrals computed for a molecular pair, 4Å-spaced perpendicular to molecular axis, are reported for NTT and the bridge substituted derivatives. A dramatic decrease in the LUMOs overlap is observed for all the substituted molecules with respect to NTT. In particular, as expected, NTA, having the acetylenic spacer and the most reduced aromatic character, shows the worst LUMO overlap. A reduced effect is observed for HOMOs overlaps that, however, are lower with respect to NTT. These preliminary calculations confirm that decreasing the aromaticity of the molecules may cause a decreasing of the intermolecular interactions. Since all the structures observed for symmetric N-based oligomers show strong π–π interactions, we will further investigate if the decreased aromaticity could lead to a different packing motif.
| Item | S h | S e |
|---|---|---|
| NTT | 707 | 767 |
| NTE | 702 | 381 |
| NTI | 599 | 588 |
| NTA | 688 | 283 |
Thermal effects in thin deposits
The behaviour of thin deposits upon thermal annealing was explored for all compounds.Fig. 6a and b show the fluorescence microscopy images of NTE films before and after the thermal cycle RT → 200 °C → RT.
 | ||
| Fig. 5 XRD profile for rod-like (red) and platelet-like crystals (orange). | ||
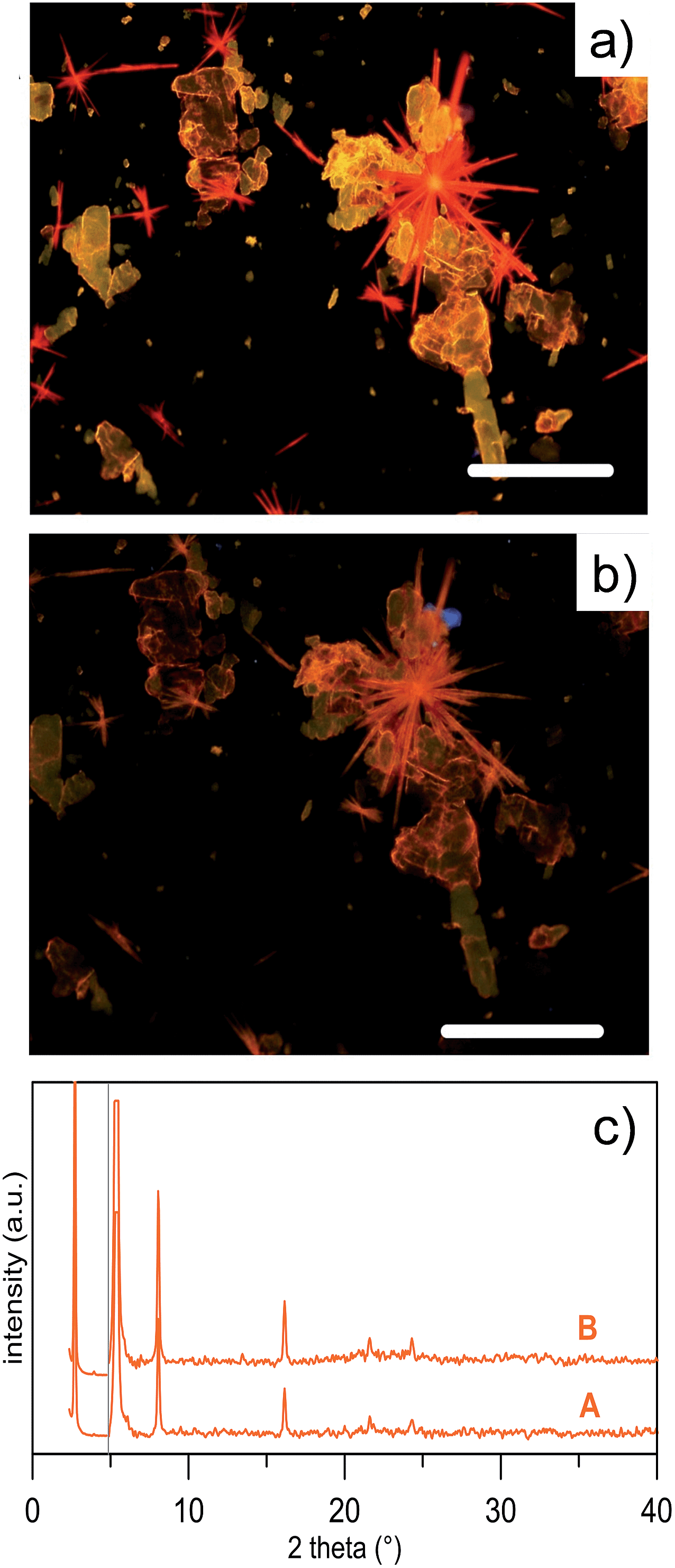 | ||
| Fig. 6 Thin deposits observed by fluorescence microscopy for NTE. (a) Fluorescence microscopy image of a drop cast thin deposit of NTE (toluene 1 g L−1 on Si/SiO2 wafer); the bar scale is 100 μm. (b) The same image after thermal treatment at 200 °C for 30 s. (c) XRD profile of the same sample before (A) and after thermal cycle (B). The B curve is that of the platelet-like crystals shown in Fig. 5. | ||
Upon thermal annealing, the two polymorphs of NTE exhibit a different behaviour. While the platelet-like crystals persisted till the melting point (≈260 °C, DSC, ESI Fig. S7†), the rod-like crystals showed an irreversible change of the emission, that progressively changed from red to orange, which corresponded to the emission of platelet-like crystals. The transition is observed also in the DSC curve of the rod-like crystals which present an endothermic peak at ∼120 °C, corresponding to the solid-solid transition at the platelet-like crystal phase. The transition was irreversible and no morphological changes were observed in the shape of the crystals but the POM was consistent with the presence of a polycrystalline compound (Fig. 6b). The structural evolution associated to the thermal treatment was investigated by XRD that confirmed the evolution from one polymorph into the other (Fig. 6c).
The same approach was also used for a NTA thin deposit which was heated up to 200 °C and then cooled down to room temperature. Upon this treatment, while the morphology and emission features of rhombohedral crystals remained apparently almost unaltered, the fiber-like crystals exhibited an irreversible change of the PL, which switched irreversibly from yellow to green (Fig. 7).
 | ||
| Fig. 7 Thin deposits observed by fluorescence microscopy of NTA. (a) Fluorescence microscopy image of a drop cast thin deposit of NTA (toluene 1 g L−1 on Si/SiO2 wafer); the bar scale is 100 μm. (b) The same image after thermal treatment at 200 °C for 30 s. | ||
Although we were not able to obtain thin deposits with only one form, XRD were performed on NTA thin deposits at room temperature and after the thermal cycle RT → 200 °C → RT (Fig. S9, ESI†). The XRD profiles show significant differences, in particular, the disappearing of some reflections after the thermal cycle support, the polymorphic conversion observed by optical microscopy.
Finally, azomethine NTI did not show detectable fluorescence differences upon thermal treatment before the melting point. However in POM, a solid state transition is observed for the rod-like crystals, which is consistent with the solid-solid transition at 170 °C, observed in the DSC curve.
Time temperature integrator (TTI)
The polymorphism of NTE was exploited for the fabrication of a TTI device,8,14 monitoring the evolution of temperature-dependent fluorescence properties.A commercial CCD recorded fluorescence images during the thermal treatment. Fig. 8 shows the number of counts (NCs) of the red, green and blue component recorded by the CCD as a function of the temperature at which the device was exposed for at least 30 s. Each image was recorded fixing the intensity of illumination and the time of acquisition of the CCD (see details in the method section). Upon heating the thin deposit at different temperatures (the devices were tested upon several cycles), the crystals were stable and changed their emission colour at the highest temperature experienced for more than 30 s. This colour was retained after cooling down the thin deposit to room temperature (Fig. 8).
 | ||
| Fig. 8 Quantitative analysis of the fluorescence images. A commercial CCD recorded fluorescence images. The colours of the graphs correspond to the colour components of the CCD (red, blue, green). The figure shows the number of counts versus temperature obtained using a filter Ex 420, DM 435, BA 475, which enables the transmission of all visible components except a slight portion of the blue. | ||
The quantitative analysis of the CCD counts of PL images shows that for each temperature, the colour recorded by the CCD is formed by a unique and defined combination of red, green and blue (RGB) components. Therefore, the temperature at which the device was exposed could be analytically derived from the colour composition of the PL image. This is the principal requirement of a TTI based on colour changes. It should be noted that for NTE in the range of RT-120 °C (i.e. below the transition temperature between the two polymorphs, therefore independent from polymorphism), higher sensitivity is obtained from the red component with an accuracy of ±5 °C. On the other hand, for temperatures higher than 120 °C, i.e. above the transition temperature of the polymorphs, the best accuracy can be extracted from the blue component with an accuracy of <±2 °C, i.e. 2 times better than the interval RT-120 °C. This behaviour suggests that the sensitivity to temperature exposure in TTI devices based on this class of materials can be improved by polymorphic transition and therefore tailored by chemical design.
Conclusions
In conclusion, we have reported the synthesis and the application in a TTI device of new thieno(bis)imide materials bearing unsaturated ethylene, azomethine and acetylene inner bridges. The optoelectronic and morphological properties were investigated and compared to those of a bridge-free isostructural N-derivative. Combined DFT calculations and CV analysis show that the energy gap of ethylene and azomethine based compound NTE and NTI is lower than that of acetylene based NTA. This was ascribed to the lower overlap of the C–C sp bond with the π-conjugated system, with respect to the C–C sp2 bonds and is in good accordance to what was observed for N-free systems. While the HOMO levels decrease in the order NTE > NTI > NTA, the LUMOs of NTE and NTA have almost the same energy. However, a slightly lower LUMO energy was found for NTI likely due to the electron withdrawing effect of the azomethine moiety.Thin deposits obtained by solution casting present high crystallinity for all compounds showing concomitant polymorphs. Indeed, the simultaneous presence of crystals with different morphology and fluorescence was detected by optical microscopy for NTE and NTA samples. XRD confirms the polymorphic nature of the different crystals. Crystallization attempts are currently under way for defining the single crystal structure. Nevertheless, successful control of the crystal phase was achieved by thermal treatments of the films. Indeed, space resolved fluorescence spectra, DSC and XRD analysis confirm that thermal heating allows us to irreversibly convert one polymorph into the other. Exploiting such features, we produced a TTI device able to monitor thermal changes of the film by optical microscopy.
This work introduces a chemical design strategy to tune the optoelectronic properties and polymorphism attitude of small molecules usable for TTI devices. Concomitant polymorph formation combined with the possibility of controlling the polymorph type and solid state fluorescence, may enable the development of new organic devices and sensors based on functional polymorph conversion.
Notes and references
- (a) H. Sirringhaus, Adv. Mater., 2014, 26, 1319 CrossRef CAS PubMed; (b) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef CAS PubMed; (c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (d) A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548 CrossRef CAS PubMed.
- (a) J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821 CrossRef CAS PubMed; (b) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 CrossRef CAS PubMed; (c) Z. Lu, B. Jiang, X. Zhang, A. Tang, L. Chen, C. Zhan and J. Yao, Chem. Mater., 2014, 26, 2907 CrossRef CAS; (d) Y. Huang, E. G. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006 CrossRef CAS PubMed.
- (a) S.-S. Cheng, P.-Y. Huang, M. Ramesh, H.-C. Chang, L.-M. Chen, C.-M. Yeh, C.-L. Fung, M.-C. Wu, C.-C. Liu, C. Kim, H.-C. Lin, M.-C. Chen and C.-W. Chu, Adv. Funct. Mater., 2014, 24, 2057 CrossRef CAS; (b) J. Smith, W. Zhang, R. Sougrat, K. Zhao, R. Li, K. Cha, K. A. Amassian, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2012, 24, 2441 CrossRef CAS PubMed; (c) E. Orgiu, A. Merari Masillamani, J.-O. Vogel, E. Treossi, A. Kiersnowski, M. Kastler, W. Pisula, F. Dotz, V. Palermo and P. Samorı, Chem. Commun., 2012, 48, 1562 RSC; (d) R. Hamilton, J. Smith, S. Ogier, M. Heeney, J. E. Anthony, I. McCulloch, J. Veres, D. D. C. Bradley and T. D. Anthopoulos, Adv. Mater., 2009, 21, 1166 CrossRef CAS.
- (a) P. Kumar, K. N. Shivananda, W. Zajaczkowski, W. Pisula, Y. Eichen and N. Tessler, Adv. Funct. Mater., 2014, 24, 2530 CrossRef CAS; (b) K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2014, 26, 587 CrossRef CAS; (c) T. Lei, J.-V. Wang and J. Pei, Chem. Mater., 2014, 26, 594 CrossRef CAS; (d) S. C. B. Mannsfeld, Nat. Mater., 2012, 11, 489 CrossRef CAS PubMed.
- (a) M. Melucci, M. Zambianchi, L. Favaretto, M. Gazzano, A. Zanelli, M. Monari, R. Capelli, S. Troisi, S. Toffanin and M. Muccini, Chem. Commun., 2011, 47, 11840 RSC; (b) M. Durso, C. Bettini, A. Zanelli, M. Gazzano, M. G. Lobello, F. De Angelis, V. Biondo, D. Gentili, R. Capelli, M. Cavallini, S. Toffanin, M. Muccini and M. Melucci, Org. Electron., 2013, 14, 3089 CrossRef CAS PubMed; (c) M. Durso, D. Gentili, C. Bettini, A. Zanelli, M. Cavallini, F. De Angelis, M. G. Lobello, V. Biondo, M. Muccini, R. Capelli and M. Melucci, Chem. Commun., 2013, 49, 4298 RSC.
- (a) M. Melucci, L. Favaretto, M. Zambianchi, M. Durso, M. Gazzano, A. Zanelli, M. Monari, M. G. Lobello, F. De Angelis, V. Biondo, G. Generali, S. Troisi, W. Koopman, S. Toffanin, R. Capelli and M. Muccini, Chem. Mater., 2013, 25, 668 CrossRef CAS; (b) M. Melucci, M. Durso, C. Bettini, M. Gazzano, L. Maini, S. Toffanin, S. Cavallini, M. Cavallini, D. Gentili, V. Biondo, G. Generali, F. Gallino, R. Capelli and M. Muccini, J. Mater. Chem. C, 2014, 2, 3448 RSC.
- (a) L. G. Mercier, A. Mishra, Y. Ishigaki, F. Henne, G. Schulz and P. Bäuerle, Org. Lett., 2014, 16, 2642 CrossRef CAS PubMed; (b) J. D. Douglas, M. S. Chen, J. R. Niskala, O. P. Lee, A. T. Yiu, E. P. Young and J. M. J. Fréchet, Adv. Mater., 2014, 26, 4606 CrossRef CAS; (c) L. Fu, H. Pan, T. T. Larsen-Olsen, T. R. Andersen, E. Bundgaard, F. C. Krebs and H.-Z. Chen, Dyes Pigm., 2013, 97, 141 CrossRef CAS PubMed.
- D. Gentili, M. Durso, C. Bettini, I. Manet, M. Gazzano, R. Capelli, M. Muccini, M. Melucci and M. Cavallini, Sci. Rep., 2013, 3, 2581 Search PubMed.
- (a) J. Bernstein, Polymorphism in Molecular Crystals, Claren-don Press, Oxford, 2002 Search PubMed; (b) J. Bernstein, Cryst. Growth Des., 2011, 11, 632 CrossRef CAS.
- (a) C. C. Mattheus, A. B. Dros, J. Baas, A. Meetsma, J. L. de Boer and T. T. M. Palstra, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 939 CAS; (b) T. Siegrist, C. Besnard, S. Haas, M. Schiltz, P. Pattison, D. Chernyshov, B. Batlogg and C. Kloc, Adv. Mater., 2007, 19, 2079 CrossRef CAS; (c) G. Giri, E. Verploegen, S. C. B. Mannsfeld, S. Atahan-Evrenk, D. H. Kim, S. Y. Lee, H. A. Becerril, A. Aspuru-Guzik, M. F. Toney and Z. Bao, Nature, 2011, 480, 504 CrossRef CAS PubMed.
- (a) D. E. Henn, W. G. Williams and D. J. J. Gibbons, Appl. Crystallogr., 1971, 4, 256 CrossRef CAS; (b) O. D. Jurchescu, A. Meetsma and T. T. M. Palstra, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 330 Search PubMed.
- (a) S. T. Salammal, J.-Y. Balandier, J.-B. Arlin, Y. Olivier, V. Lemaur, L. Wang, D. Beljonne, J. Cornil, A. R. Kennedy, Y. H. Geerts and B. Chattopadhyay, J. Phys. Chem. C, 2014, 118, 669 CrossRef; (b) A. Moser, I. Salzmann, M. Oehzelt, A. Neuhold, H.-G. Flesch, J. Ivanco, S. Pop, T. Toader, D. R. T. Zahn, D.-M. Smilgies and R. Resel, Chem. Phys. Lett., 2013, 574, 51 CrossRef CAS PubMed; (c) T. Siegrist, C. Kloc, R. A. Laudise, H. E. Katz and R. C. Haddon, Adv. Mater., 1998, 10, 379 CrossRef CAS.
- O. D. Jurchescu, D. A. Mourey, S. Subramanian, S. R. Parkin, B. M. Vogel, J. E. Anthony, T. N. Jackson and D. Gundlach, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085201 CrossRef.
- (a) A. Calo, P. Stoliar, F. C. Matacotta, M. Cavallini and F. Biscarini, Langmuir, 2010, 26, 5312 CrossRef CAS PubMed; (b) M. Cavallini, A. Calo, P. Stoliar, J. C. Kengne, S. Martins, F. C. Matacotta, F. Quist, G. Gbabode, N. Dumont, Y. H. Geerts and F. Biscarini, Adv. Mat., 2009, 21, 4688 CAS.
- (a) P. Frère, J.-M. Raimundo, P. Blanchard, J. Delaunay, P. Richomme, J.-L. Sauvajol, J. Orduna, J. Garin and J. Roncali, J. Org. Chem., 2003, 68, 7254 CrossRef PubMed; (b) G. Neculqueo, V. Rojas Fuentes, A. Lopez, R. Matute, S. O. Vasquez and F. Martinez, Struct. Chem., 2012, 23, 1751 CrossRef CAS PubMed.
- (a) S. Dufresne, M. Bourgeaux and W. G. Skene, J. Mater. Chem., 2007, 17, 1166 RSC; (b) N. Kiriy, V. Bocharova, A. Kiriy, M. Stamm, F. C. Krebs and H.-J. Adler, Chem. Mater., 2004, 16, 4765 CrossRef CAS; (c) Y. Dong, D. Navarathne, A. Bolduc, N. McGregor and W. G. Skene, J. Org. Chem., 2012, 77, 5429 CrossRef CAS PubMed.
- M. Melucci, G. Barbarella, M. Zambianchi, P. Di Pietro and A. Bongini, J. Org. Chem., 2004, 69, 4821 CrossRef CAS PubMed.
- J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3440 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS; (d) C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Theory and Applications of Computational Chemistry, the first forty years, Amsterdam, Elsevier, 2005 Search PubMed.
- K. Starcevic, D. W. Boykin and G. Karminski-Zamola, Heteroat. Chem., 2003, 14, 218 CrossRef CAS.
- E.T.C. S.r.l., M., Melucci; L. Favaretto, M. Zambianchi, R. Capelli, M. Patent: WO2014076662 A1, 2014; Pg. 24.
- (a) M. Surin, P. Sonar, A. C. Grimsdale, K. Mullen, S. De Feyter, S. Habuchi, S. Sarzi, E. Braeken, A. V. Heyen, M. Van der Auweraer, F. C. De Schryver, M. Cavallini, J. F. Moulin, F. Biscarini, C. Femoni, R. Lazzaroni and P. Leclere, J. Mater. Chem., 2007, 17, 728 RSC; (b) M. Cavallini, P. Stoliar, J. F. Moulin, M. Surin, P. Leclere, R. Lazzaroni, D. W. Breiby, J. W. Andreasen, M. M. Nielsen, P. Sonar, A. C. Grimsdale, K. Mullen and F. Biscarini, Nano Lett., 2005, 5, 2422 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Comprehensive experimental details on the synthesis and characterization of the intermediate compounds in Schemes 1–3, cyclic voltammetry at different scan rates, fluorescence lifetime imaging, photoluminescence in thin deposits, differential scanning calorimetry, theoretical calculations, X-ray diffraction. See DOI: 10.1039/c4tc01792g |
| ‡ HOMO and LUMO energy levels for the cis-isomer are blue shifted by about 0.10 eV (−5.81 eV and −3.46 eV). |
| § Rod-like majority from the saturated solution, platelets-like majority from the diluted solution. |
| This journal is © The Royal Society of Chemistry 2015 |