
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Receptor-targeting fluorescence imaging and theranostics using a graphene oxide based supramolecular glycocomposite†
Ding-Kun
Ji‡
a,
Yue
Zhang‡
ab,
Yi
Zang
*b,
Wang
Liu
c,
Xiongwen
Zhang
c,
Jia
Li
*b,
Guo-Rong
Chen
a,
Tony D.
James
d and
Xiao-Peng
He
*a
aKey Laboratory for Advanced Materials & Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, P. R. China. E-mail: xphe@ecust.edu.cn; Fax: +86-21-64252758; Tel: +86-21-64253016
bNational Center for Drug Screening, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: yzang@simm.ac.cn; jli@simm.ac.cn
cInstitute of Drug Discovery and Development, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University (ECNU), 3663 N. Zhongshan Rd., Shanghai 200062, P. R. China
dDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
First published on 6th November 2015
Abstract
Intercellular glycoligand-receptor interactions are implicated in a number of disease-related processes. Effective tools that target these receptors may facilitate disease theranostics. However, owing to their low binding affinity, multivalent presentation of glycoligands is needed to increase the avidity with transmembrane receptors. While previous strategies focus on the covalent coupling of glycoligands to a synthetic backbone, we show here that the use of graphene oxide (GO) greatly enhances the cellular and tissue imaging ability of a small-molecule fluorescence glycoprobe. We determine that GO with an optimum size may serve as a clustering platform to reinforce the interaction of the glycoprobe with its selective receptor on a cancer cell. This phenomenon has been consistently observed with the xenograft tissue of a tumor-bearing mouse. Using this principle we have further constructed a supramolecular glycocomposite by co-assembling the glycoprobe and an anticancer drug onto a single GO surface. In addition to imaging ability, this material displays improved toxicity for liver cancer cells that over express the glycoprotein receptor, when compared to the control cells.
Introduction
Intercellular ligand–receptor interactions are crucial for the initiation of numerous physiological and pathological events. For example, the galectins, which recognize galactose-terminated glycoconjugates, are over-secreted during the progression of cancer, inflammation and heart disease. In the process of influenza virus invasion, sialic acid-terminated oligosaccharides of human cells are recognized by the hemagglutinins expressed on the viral particle surface, facilitating virus invasion. Another class of transmembrane receptors are the C-type lectins, which are distributed on specific types of cells.1–3 They can interact with selective glycoligands (glycans) to promote human diseases.4,5 As a consequence, effective methods to probe these dynamic events directly on the cellular level are important for the advancement of chemical glycobiology and disease theranostics.Considering the low affinity between a glycoligand and the receptor, the ligands require multivalent presentation to increase the binding avidity. Many elegant methods have been developed to construct multivalent glycopolymers6,7 and glycoconjugates8 to address the avidity. However, these methods mainly focus on the covalent coupling of glycoligands to a synthetic backbone, which might increase the complexity of probe preparation. Recently, we9–13 and others14–21 have shown that fluorogenic composite materials (FCMs) that feature a material substrate (especially graphene oxide [GO]) on which a fluorophore-tagged ligand is self-assembled are emerging as promising biosensing materials owing to their ease of preparation and manipulation, low cost and ability to capture receptor expression of live cells.
Here, we uncover that the addition of GO greatly enhances the receptor-targeting cellular imaging effect of a small-molecule fluorescence glycoprobe. We determined that GO with an optimum size may act as a clustering platform to reinforce the selective interaction between the glycoprobe and receptor on a cancer cell. This phenomenon has been consistently observed with the xenograft tissue of a tumor-bearing mouse. Using this principle we have further constructed a supramolecular glycocomposite by co-assembling the glycoprobe and an anticancer drug onto a single GO surface. In addition to imaging ability, this material displays improved toxicity for liver cancer cells that over express the glycoprotein receptor, when compared to the control cells without receptor expression (Fig. 1).
 | ||
| Fig. 1 (a) Conditions and reagents: (I) CuSO4·5H2O, sodium ascorbate in CH2Cl2/H2O (10/1, v/v), and then Et3N. (b) Schematic illustration of the formation of the GO-based supramolecular glycocomposite for receptor-targeting theranostics. GO = graphene oxide; CPT = camptothecin. | ||
Results and discussion
The glycoprobe (DK1, Fig. 1a) was synthesized by a click reaction between alkynyl galactoside 1 and an azido dicyanomethylene 2 (Fig. 1a), according to a previous protocol.22 The galactosyl moiety can be recognized by the transmembrane asialoglycoprotein receptor (ASGPr) selectively expressed on hepatocytes (we used Hep-G2, a human hepatoma cell line), while the fluorophore can produce an optical signal and self-assemble to the surface of GO via π-stacking and other forces of interaction such as van der Waals and hydrogen-bonding.9–14 The fluorescence imaging of DK1 for ASGPr-rich Hep-G223 was carried out and optimized in the absence and presence of a series of GOs with different sizes (G0: 0–10 nm; G1: 50–200 nm; G2: 200–500 nm; G3: 500–1000 nm). The atomic force microscopic characterization of the GOs is shown in Fig. S1 (ESI†).We observed that, among the GOs used, the presence of G1 and G2 produced an increased cellular fluorescence compared to DK1 alone (Fig. 2a and c). In contrast, the presence of G0 and G3 did not cause the same level of fluorescence enhancement. To confirm that the observed fluorescence imaging results were based on selective galactose-ASGPr interactions, we used a modified Hep-G2 cell line with a reduced expression level of ASGPr (sh-ASGPr)10,24 as well as other cancer cell lines derived from different tissues (human cervix cancer Hela, human lung cancer A549, human colon cancer HCT-116 and human breast cancer MCF-7) as negative controls. With the optimal imaging condition (40 μM/50 μg mL−1), we observed that DK1@G2 produced minimal fluorescence upon incubation with sh-ASGPr and other cancer cells (Fig. 2b and d). Moreover, pre-incubation of increasing free D-galactose gradually lowered the fluorescence generated in Hep-G2 cells (Fig. S2, ESI†). We also determined that the fluorescence intensity produced by DK1@G2 (Fig. 2d) was in good agreement with the ASGPr expression level of the different cells (Fig. 2e), and that the presence of increasing DK1 and DK1@G2 barely suppressed the viability of Hep-G2 cells determined by two independent cell viability counting assays (Fig. S3, ESI†). All these observations suggest that the fluorescence imaging results observed at the cellular level are predominantly based on galactose-ASGPr recognition. To test the cellular localization of the glycoprobe, confocal microscopy was used. The imaging result suggested that DK1 was endocytosed and trafficked to the lysosome, which is confirmed by co-localization with a lysotracker (Fig. S4, ESI†).
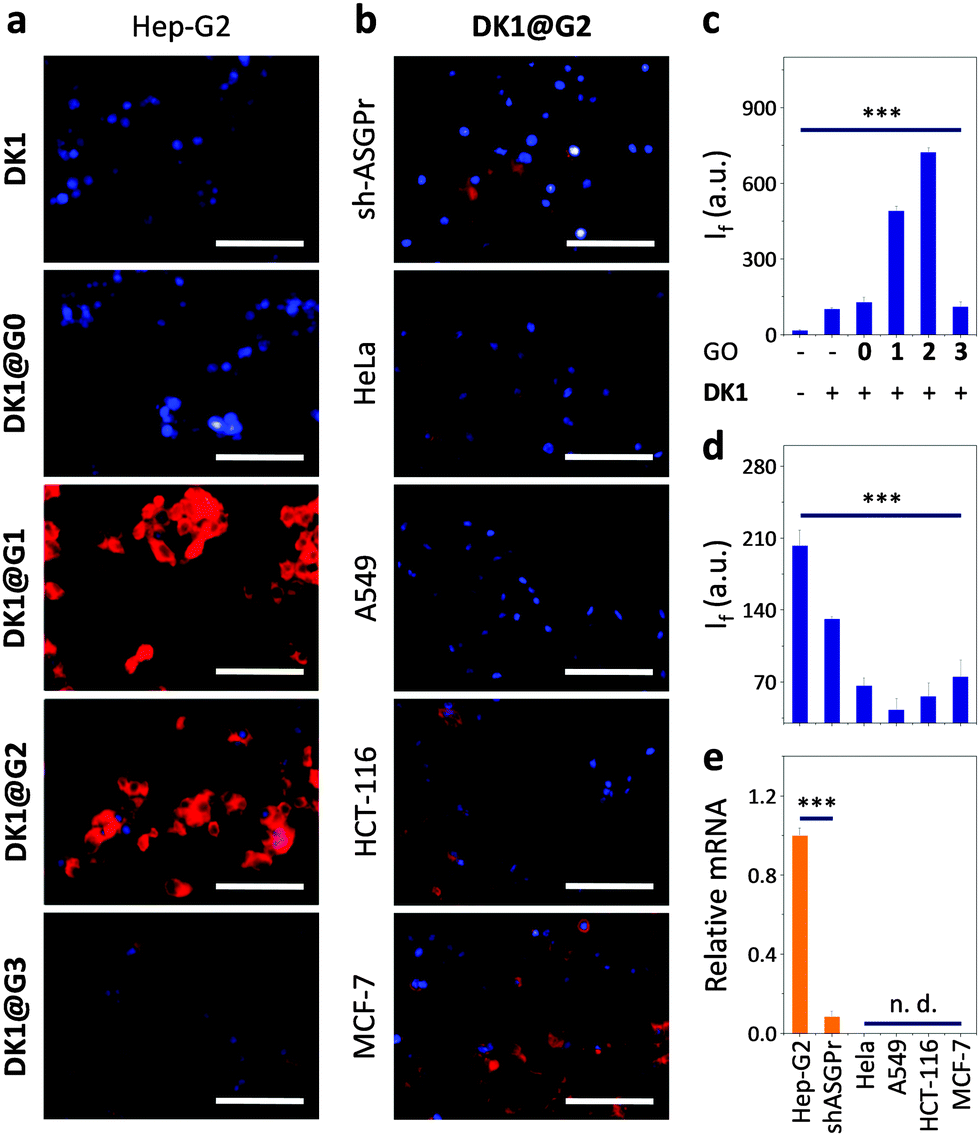 | ||
| Fig. 2 Fluorescence imaging (a) and quantification (c) of Hep-G2 cells with DK1 (40 μM) in the absence or presence of G0, G1, G2 and G3 (50 μg mL−1). Fluorescence imaging (b) and quantification (d) of different human cancer cell lines using DK1@G2 (40 μM/50 μg mL−1) (Hep-G2: human liver cancer; sh-ASGPr: Hep-G2 cells with reduced expression level of the asialoglycoprotein receptor (ASGPr); Hela: human cervix cancer; A549: human lung cancer; HCT-116: human colon cancer; MCF-7: human breast cancer). (e) Relative mRNA level of ASGPr of different cells determined by real-time quantitative polymerase chain reaction (***P < 0.001; n. d. means not detectable). Excitation channel: 520–550 nm; emission: 580–650 nm. Scale bar: 100 μm. | ||
With these results in hand, we then used a mouse model with a liver tumor to test the GO effect at the tissue level. Female athymic BALB/c nu/nu mice with Hep-G2 tumor xenograft were established according to a previous report,25 and the tumor, liver, lung, heart and spleen were removed to prepare tissue sections. By incubation of DK1 with the tumor sections in the absence or presence of GO, we determined that DK1@G2 (40 μM/50 μg mL−1, the optimal concentration used for cellular imaging) resulted in a stronger fluorescence imaging of the tumor than DK1 alone and other DK1@GOs (Fig. 3a and c). We also observed that DK1@G2 selectively stained the Hep-G2 tumor xenograft and liver among other sections from the animal model (Fig. 3b and d). This observation accords with the fact that the galactose-selective ASGPr is only over-expressed on the tumor and liver, but not on heart, spleen and lung (Fig. S5, ESI†).26 These results suggest that, similar to the observation on the cellular level, the selective galactose ligand–ASGP receptor based fluorescence imaging is enhanced by GO probably due to the multivalent display of DK1.
 | ||
| Fig. 3 Fluorescence imaging (a) and quantification (c) of Hep-G2 xenograft slice of female athymic BALB/c nu/nu mice with DK1 (40 μM) in the absence and presence of G0, G1, G2 and G3 (50 μg mL−1). Fluorescence imaging (b) and quantification (d) of different tissue slices including liver, heart, spleen and lung with DK1@G2 (40 μM/50 μg mL−1). Excitation channel: 520–550 nm; emission: 580–650 nm. Scale bar: 200 μm. | ||
From our previous work, we have demonstrated that amongst GOs of different sizes, G2 enhances the interaction between DK1 and a selective lectin receptor to the greatest extent.22 Spectroscopic measurements suggest that both the size and oxygen content of the GO have an impact on the ligand–receptor interactions. On the one hand, increasing the size, reduces the oxygen content of a GO, leading to a stronger adsorption of dye molecules but weaker adsorption of proteins. Therefore, GO with larger sizes (such as G3 with 500–1000 nm size range) might bind more strongly with dye molecules, whilst compromising the binding with a lectin receptor. However, a decrease of size increases the oxygen content and this might enhance the association between the GO (such as G0) and a protein, whilst decreasing the amount of glycoprobe–GO assembly, thus lowering the ability of the GO to augment the protein–ligand binding. Therefore, we deduced that the medium-sized GOs (such as G1 and G2) have an optimal size as well as oxygen content to promote the multivalent glycoligand–receptor interactions.22 Similarly, these observations may be applicable for glycoprobe–transmembrane receptor interactions. However, due to the complexity of the cellular environment, more sophisticated experiments and structurally more defined GOs are required to fully elaborate the observed GO size effect.
With this GO-based multivalent strategy, we sought to further construct a supramolecular glycocomposite by the co-assembly of DK1 and an anticancer drug (camptothecin – CPT) to a single GO surface for receptor-targeting theranostics. G2 with the best imaging effect was used to assemble with DK1 and CPT at two different concentrations. We observed that the glycocomposites retained their fluorescence imaging ability with selectively for Hep-G2 cells over the other control cells tested (Fig. 4a and b). Then, we measured the toxicity of the glycocomposites for the cancer cells (Fig. 4). While CPT alone showed insignificant (1 μM – low concentration group) or moderate (10 μM – high concentration group) toxicity upon a short-time incubation (15 min) with the cancer cells, the presence of G2 enhanced the toxicity of the low-concentration group. This is in agreement with the reported drug delivering ability of graphene to improve cancer therapy.27 Interestingly, in both groups, the presence of DK1 selectively enhanced the toxicity for Hep-G2 that highly express ASGPr, but not for HeLa and A549 without receptor expression, in a concentration-dependent manner. These data suggest the suitability of using the supramolecular glycocomposites for receptor-targeting cancer theranostics. Our supramolecular strategy could also be extended to other biomolecular systems for specific cellular targeting.28
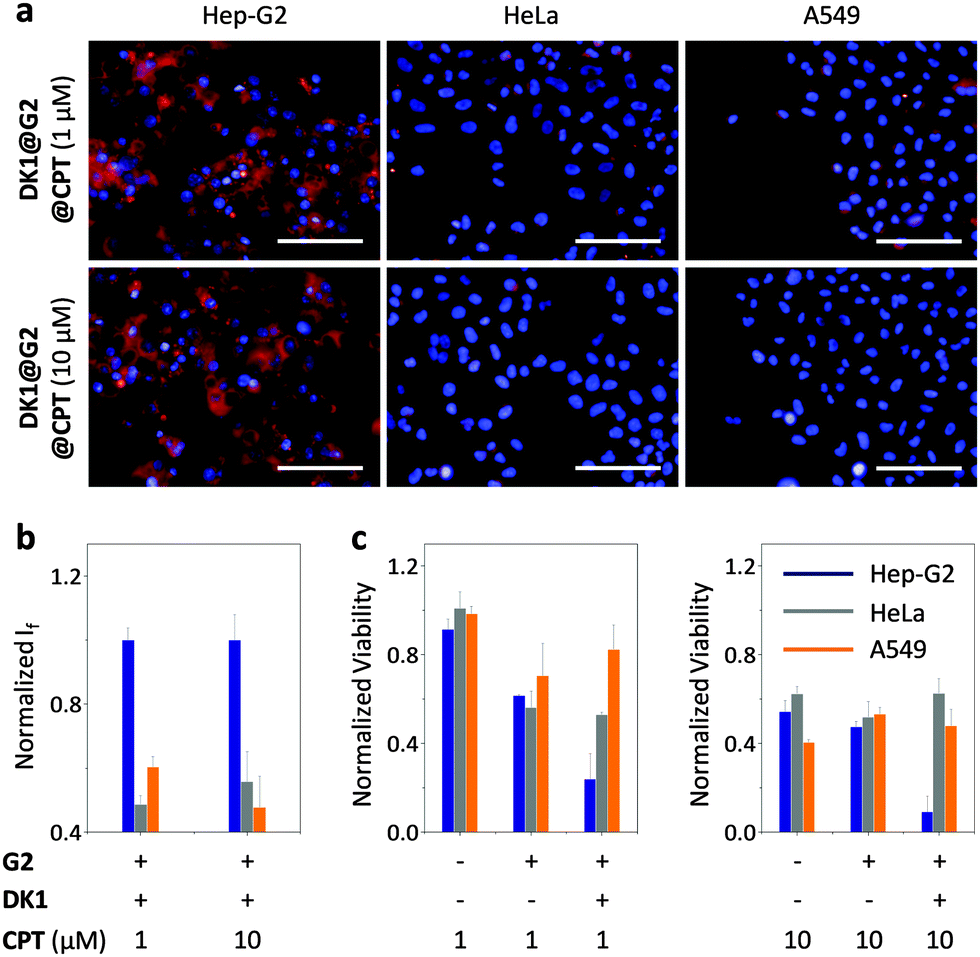 | ||
| Fig. 4 Fluorescence imaging (a) and quantification (b) of different cells with the supramolecular DK1@G2@CPT (DK1: 40 μM; G2: 50 μg mL−1). (c) Viability of different cells upon treatment with CPT, CPT@G2 and DK1@G2@CPT. Excitation channel: 520–550 nm; emission: 580–650 nm. Scale bar: 100 μm. | ||
Conclusions
We have determined that GO can enhance the cellular and tissue imaging effect of a small-molecule fluorescence glycoprobe. This phenomenon could probably be reasoned by the clustering effect of GO for the fluorophore-labelled glycoligands, increasing the avidity with transmembrane receptors. Moreover, this non-covalent, multivalent strategy has proven suitable for the construction of a unique supramolecular glycocomposite comprising both the glycoprobe and an anticancer drug. With retained fluorescence imaging ability, this glycocomposite material displayed enhanced toxicity for liver cancer cells with ASGPr expression, but not for the control cells without the receptor expression. This research suggests a unique GO-based supramolecular strategy for receptor-targeting disease theranostics.Acknowledgements
We thank the 973 project (2013CB733700), the Fundamental Research Funds for Central Universities (222201414010), the Key Project of Shanghai Science and Technology Commission (13NM1400900) and the National Science Foundation of China (21572058, 21576088 and 91213303). The Catalysis And Sensing for our Environment (CASE) network is thanked for research exchange opportunities. Prof. He Tian is thanked for his helpful discussion. Prof. Juan Xie (ENS de Cachan) is thanked for kindly providing synthetic intermediates.Notes and references
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- T. Feizi and W. Chai, Nat. Rev. Mol. Cell Biol., 2004, 5, 582–588 CrossRef CAS PubMed.
- G. W. Hart and R. J. Copeland, Cell, 2010, 143, 672–676 CrossRef CAS PubMed.
- F.-T. Liu and G. A. Rabinovich, Nat. Rev. Cancer, 2005, 5, 29–41 CrossRef CAS PubMed.
- J. Mukherjee, H. J. de Haas, A. D. Petrov, A. Tawakol, N. Haider, A. Tahara, C. C. Constantinescu, J. Zhou, H. H. Boersma, T. Imaizumi, M. Nakano, A. Finn, Z. Fayad, R. Virmani, V. Fuster, L. Bosca and J. Narula, Nat. Med., 2014, 20, 215–219 CrossRef PubMed.
- L. L. Kiessling and R. A. Splain, Annu. Rev. Biochem., 2010, 79, 619–653 CrossRef CAS PubMed.
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC.
- (a) S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed; (b) Z. Yu, R. Paul, C. Bhattacharya, T. C. Bozeman, M. J. Rishel and S. M. Hecht, Biochemistry, 2015, 54, 3100–3109 CrossRef CAS PubMed; (c) Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- X.-P. He, Y. Zang, T. D. James, J. Li and G.-R. Chen, Chem. Soc. Rev., 2015, 44, 4239 RSC.
- H.-L. Zhang, X.-L. Wei, Y. Zang, J.-Y. Cao, S. Liu, X.-P. He, Q. Chen, Y.-T. Long, J. Li, G.-R. Chen and K. Chen, Adv. Mater., 2013, 25, 4097–4101 CrossRef CAS PubMed.
- D.-K. Ji, G.-R. Chen, X.-P. He and H. Tian, Adv. Funct. Mater., 2015, 25, 3483 CrossRef CAS.
- X.-P. He, Q. Deng, L. Cai, C.-Z. Wang, Y. Zang, J. Li, G.-R. Chen and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 5379–5382 CAS.
- X. Sun, B. Zhu, D.-K. Ji, Q. Chen, X.-P. He, G.-R. Chen and T. D. James, ACS Appl. Mater. Interfaces, 2014, 6, 10078–10082 CAS.
- L. Wang, K.-Y. Pu, J. Li, X. Qi, H. Li, H. Zhang, C. Fan and B. Liu, Adv. Mater., 2011, 23, 4386–4391 CrossRef CAS PubMed.
- F. Li, H. Pei, L. Wang, J. Lu, J. Gao, B. Jiang, X. Zhao and C. Fan, Adv. Funct. Mater., 2013, 23, 4140–4148 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- E. Morales-Narváez and A. Merkoçi, Adv. Mater., 2012, 24, 3298–3308 CrossRef PubMed.
- L. Feng, L. Wu and X. Qu, Adv. Mater., 2013, 25, 168–186 CrossRef CAS PubMed.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- H. Jang, J. Lee and D.-H. Min, J. Mater. Chem. B, 2014, 2, 2452–2460 RSC.
- D.-K. Ji, Y. Zhang, X.-P. He and G.-R. Chen, J. Mater. Chem. B, 2015, 3, 6656–6661 RSC.
- E. I. Rigopoulou, D. Roggenbuck, D. S. Smyk, C. Liaskos, M. G. Mytillnaiou, E. Feist, K. Conrad and D. P. Bogdanos, Autoimmun. Rev., 2012, 12, 260–269 CrossRef CAS PubMed.
- Z. Li, S.-S. Deng, Y. Zang, Z. Gu, X.-P. He, G.-R. Chen, K. Chen, T. D. James, J. Li and Y.-T. Long, Sci. Rep., 2013, 3, 2293 Search PubMed.
- S. Mukhopadhyay, P. K. Panda, D. S. Das, N. Sinha, B. Behera, T. K. Maiti and S. K. Bhutia, Acta Pharmacol. Sin., 2014, 35, 814–824 CrossRef CAS PubMed.
- F. Lv, X. He, L. Wu and T. Liu, Bioorg. Med. Chem. Lett., 2013, 23, 1878–1882 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Y. Wen, W. Liu, C. Bagia, S. Zhang, M. Bai, J. M. Janjic, N. Giannoukakis, E. S. Gawalt and W. S. Meng, Acta Biomater., 2014, 10, 4759–4767 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, experimental section and additional references. See DOI: 10.1039/c5tb02057c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2015 |