
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid-bilayer coated nanosized bimodal mesoporous carbon spheres for controlled release applications†
Benjamin
Mandlmeier‡
,
Stefan
Niedermayer‡
,
Alexandra
Schmidt
,
Jörg
Schuster
and
Thomas
Bein
*
Department of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstraße 5-13 (Gerhard-Ertl Building), 81377 Munich, Germany. E-mail: bein@lmu.de
First published on 12th November 2015
Abstract
Highly mesoporous nanosized carbon spheres (MCS) equipped with an active lipid bilayer demonstrate pronounced molecular release behavior, and excellent potential for drug delivery applications. We report a facile synthesis route for the creation of colloidal MCS with a bimodal pore size distribution, featuring a high BET surface area combined with high pore volume. Bimodal mesoporosity was achieved by a simultaneous co-assembly of a polymer resin (resol), tetraethyl orthosilicate (TEOS) and a block copolymer (Pluronic F127). The spherical geometry originates from casting the precursor mixture into a macroporous silica hard template, having a mean pore size of 60 nm, followed by thermopolymerization and final carbonization at 900 °C in nitrogen atmosphere. The final bimodal mesoporous MCS were obtained after removal of inorganic compounds by etching with hydrofluoric acid. Colloidal suspensions of MCS were prepared by oxidation with ammonium persulfate. MCS were loaded with calcein as a model drug. Efficient sealing of the MCS was achieved with a supported lipid bilayer (SLB). The SLB acts as a diffusion barrier against the uncontrolled release of encapsulated dye molecules until the release is triggered via the addition of a surface active agent. The high surface area and pore volume and the excellent release characteristics make the SLB-coated MCS a promising release-on-demand system.
Introduction
Colloidal mesoporous carbon particles feature several properties that make them attractive candidates for controlled release and possibly drug delivery applications. These properties include high surface areas and pore volumes resulting in high loading capacities and good dispersibility. Additionally, carbon can be equipped with various chemical functionalities, which are often required to create more complex systems. Up to now, important systems explored in targeted drug delivery are based on mesoporous silica,1–3 liposomes,4,5 protein-based nanocarriers6 and polymers.7–10 Different porous carbon morphologies, such as activated carbon particles,11,12 carbon nanotubes (CNTs)12–16 and ordered mesoporous carbon (CMK-3),17 loaded with certain compounds and exhibiting good biocompatibility18,19 have been reported in the context of drug delivery. Combining magnetic properties with carbon nanostructures is also being discussed for medical applications.20 Nevertheless, certain carbon nanomaterials such as multi-walled CNT, carbon fibers and certain types of particles (carbon black) can lead to cytotoxic effects depending on their size, aspect ratio and surface chemistry.21 Another study showed that functionalization of CNTs reduced toxic effects to the cytosol.22 Several reports describe the formation of mesoporous carbon for drug delivery by replicating silica hard templates. Zhu et al. showed the functionalization of mesoporous carbon with a thermo-responsive polymer.17 Based on this work, spherical mesoporous carbon in the size range of 500–800 nm was recently prepared, exhibiting a BET surface area of 1069 m2 g−1 and high pore volume of 1.49 cc g−1, with a pore diameter of 6.0 nm.22 Many approaches for the preparation of mesoporous carbons are known,23 and the morphology can be controlled by the dimensions of hard templates. For example, MCM-48 silica particles were used as template for ordered mesoporous carbon (CMK-1).24 Furthermore, mesoporous carbon nanoparticles (MCNs) providing a high BET surface area of 2034 m2 g−1 and a pore volume of 1.2 cc g−1 were applied in drug delivery experiments.25 Hydrophilic amino-functionalized MCNs in the range of 100 to 150 nm were also obtained, and their high drug storage capacity was attributed to a BET surface area of 1309 m2 g−1 and a pore volume of 0.9 cc g−1 (with small pores below 3 nm).26 High biocompatibility was shown for mesoporous carbon nanoparticles hydrothermally derived from resol as carbon source and the block-copolymer Pluronic F127.19 With this approach, particle sizes ranging from ca. 20 to 140 nm were obtained, but the highest BET surface area of 1131 m2 g−1 was accompanied by a small pore size of 2.6 nm for 90 nm large particles. The influence of the particle size on endocytosis was demonstrated with MCNs having small diameters of around 90 nm.27 These samples were prepared by a hydrothermal route, using the block co-polymer Pluronic F127 as soft-template, resulting in particles with a BET surface area of 746 m2 g−1, a pore volume of 0.76 cc g−1, and a pore size of 2.7 nm.A novel general route for ordered mesoporous carbon (OMC) with high porosity involves a concerted co-assembly of carbon and silica precursors with Pluronic F127 and successive template removal after full carbonization, providing a bimodal pore size distribution.28 Based on this method, we recently reported the synthesis of dispersible and porous carbon spheres (sized about 330 nm) using macroporous silica as hard template.29 Those MCNs provided the highest inner pore volumes for mesoporous carbon nanoparticles of 2.32 cc g−1 and also one of the highest surface areas of 2445 m2 g−1 with a bimodal pore size distribution of large and small mesopores of 6 nm and 3.1 nm. The above-mentioned properties make these particles attractive candidates for drug delivery applications.
Here we report the synthesis of very small mesoporous carbon nanospheres with extremely high porosity and present the first efficient cap system for colloidal mesoporous carbon nanospheres (Fig. 1).
 | ||
| Fig. 1 Surfactant induced release of calcein (green) from mesoporous carbon nanoparticles encapsulated by a supported lipid bilayer. | ||
We synthesized very small mesoporous carbon nanospheres with diameters ranging from 45 to 70 nm using a macroporous silica hard template providing appropriate dimensions. Our particles feature a very high BET surface area of 2003 m2 g−1 and a high pore volume of 1.95 cc g−1. To the best of our knowledge, this is the highest specific pore volume for mesoporous carbon nanoparticles at a size below 100 nm and also one of the highest surface areas. Additionally, a bimodal porosity with maxima at 5.9 and 3.1 nm was achieved by removal of F127 from the pores and silica from the walls. In this study we combine an extremely high loading capacity and very suitable particle sizes for improved cell uptake. The use of supported lipid bilayers (SLBs) as cap systems has been successfully demonstrated for mesoporous silica nanoparticles.29–35 Being a very simple imitation of a cell membrane, SLBs can enhance circulation times and accumulation in tumor tissue as reported for the delivery of doxorubicin.36 Compared to liposomes, the solid nanoparticle core offers two major advantages. First, narrow size distributions and second, enhanced stability in comparison with liposomes can be achieved.32,33,37,38 Based on these attractive features, we transferred the concept of SLBs to mesoporous carbon and present the first efficient cap system for colloidal mesoporous carbon nanospheres. Efficient sealing of the mesopores could be achieved by coating each carbon sphere with an SLB. Regarding the lipid formulation, we used a mixture of cationic and neutral lipids (DOTAP and DOPC) and 60 nm MCS. To demonstrate the effectiveness of our cap-system, the triggered release of calcein was studied in vitro by fluorescence spectroscopy.
Experimental section
Materials
Sodium dodecyl sulfate, potassium persulfate, ammonium persulfate, methyl methacrylate (99%), tetraethyl orthosilicate (technical grade), concentrated HCl (37% in water), sodium hydroxide, Pluronic F127, calcein, Triton X-100, sulfuric acid and organic solvents were obtained from Aldrich and used without further purification. Formalin (37 wt% formaldehyde in water) and phenol was purchased from Merck KGaA and Pluronic F127 from BASF AG. Used lipids: DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids), DOTAP (1,2-dioleoyl-3-trimethylammonium-propane, Avanti Polar Lipids).Synthesis of PMMA spheres
PMMA spheres of about 60 nm were prepared by a modified route reported elsewhere.39 The emulsion polymerization was started by adding a solution of 227.2 mg potassium persulfate (K2S2O8, 0.84 mmol) in 2 mL water to a mixture of 27.56 g methyl methacrylate (MMA, 275.3 mmol), 196 mg sodium dodecyl sulfate (SDS, 0.68 mmol) in 98 mL water at 70 °C. The water was degassed by flushing with nitrogen for 1 h, before SDS and MMA were added and the mixture was then stirred under refluxing conditions for 1 h at 70 °C. After 2 h of polymerization the reaction was stopped by external cooling and applying air to the three-neck round bottom flask (250 mL). The resulting colloidal solution was washed three times with water by centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm (50228 rcf) for 30 min. This solution was dried in plastic petri dishes for several days and the resulting flakes of several mm in size were dried at 80 °C for 2 h.
500 rpm (50228 rcf) for 30 min. This solution was dried in plastic petri dishes for several days and the resulting flakes of several mm in size were dried at 80 °C for 2 h.
Synthesis of macroporous silica
For the synthesis of the macroporous silica the obtained PMMA flakes were collected on a filter paper and cast multiple times with a hydrolyzed silica precursor solution. The residual solution was removed by applying vacuum to the Büchner funnel. For the silica solution, 12 mL tetraethyl orthosilicate (TEOS, 53.8 mmol) were added to 8 mL ethanol under stirring. To the clear solution 6 mL water and 1 mL concentrated hydrochloric acid (HCl, 37%) were dropped and stirred for another 10 min at room temperature.29 Finally, the PMMA/SiOx composite was calcined in air at 550 °C for 5 h (ramp: 1 °C min−1).Synthesis of mesoporous carbon spheres
For this purpose, a mixture of resol precursor as carbon source and silica precursor were used for casting the previously prepared macroporous silica template. The resol precursor was prepared according to Meng et al.40 Here, 21 g formalin (37 wt% formaldehyde in water, 0.259 mol) was added dropwise at 50 °C to a solution of 12.2 g phenol (0.128 mol) and 2.6 g of 20 wt% sodium hydroxide solution (0.013 mol). The final solution was then heated for 1 h at 75 °C and cooled to room temperature. After neutralizing the basic solution using hydrochloric acid (1 M), water was removed by rotary evaporation. The resulting product was diluted in absolute ethanol to achieve a solution of 20 wt% (resol precursor).For the final casting solution, 2.0 g of Pluronic F127, 20.0 g of ethanol, and 2.0 g of 0.2 M HCl were mixed well at 40 °C. Then, 4.16 g TEOS and 5.0 g of the 20 wt% resol solution were added.28 The resulting yellow solution was stirred for another 5 h at the same temperature. Typically, 10 g of the solution were cast on 100 mg of the macroporous silica template, and the evaporation of the solvents and self-assembly of the F127 occurred at room temperature in an open Petri dish during 2–3 days. The resulting and filled flakes were collected and thermopolymerized in air at 100 °C for 24 h, followed by carbonization at 900 °C in a stream of nitrogen. Here, the material was heated first to 350 °C (rate: 1 °C min−1; dwell time: 3 h), then to 600 °C (rate: 1 °C min−1) and finally to 900 °C (rate: 5 °C min−1; dwell time: 2 h). The resulting shiny black bulk material was additionally treated with aqueous hydrofluoric acid (10 wt%), usually 100 mg solid material in 20 mL solution, for 2 days, to remove silica from the macroporous template and from the walls of the mesoporous carbon phase. This treatment was followed by filtration and threefold washing with water (MilliporeQ) and absolute ethanol.
Oxidation of nanosized mesoporous carbon spheres
To obtain a colloidal suspension of carbon spheres, the bulk material was oxidized in a mixture of ammonium persulfate and sulfuric acid.41 Specifically, 30 mg MCS were added to a mixture of 2.4 g ammonium persulfate in 4 mL sulfuric acid (1 M). The resulting suspension was stirred for 24 h at room temperature. Subsequently, the particles were washed 5 times by centrifugation (14000 rpm, 16873 rcf, 8 min) and redispersion in 5 mL MilliporeQ water.
Preparation of SLB@MCS loaded with calcein
The amount of 1 mg oxidized colloidal mesoporous carbon was dispersed in 500 μL of an aqueous solution of calcein (1 mM) and stirred (500 rpm) for 2 h at room temperature in the dark. After centrifugation (14000 rpm, 16873 rcf, 8 min), 100 μL of a premixed lipid solution (70 μL DOPC + 30 μL DOTAP, each 2.5 mg mL−1 in a mixture of 40 vol% EtOH/60 vol% water) were added. Upon addition of 700 μL of MilliporeQ water, the formation of a supported lipid bilayer on the external surface of MCS was induced. The SLB@MCS were then centrifuged and redispersed in 800 μL MilliporeQ water, to eliminate unsupported lipids and ethanol.
In vitro release experiment
The amount of 200 μL of the aqueous suspension (pH = 7.2) containing SLB@MCS loaded with calcein was transferred into a tube which could be closed with a dialysis membrane (with a molecular-weight cutoff of 16000 g mol−1).42 This custom made cap fits on the opening of a fluorescence cuvette, filled with MilliporeQ water (pH = 7.2). Only the fluorescent molecules are able to diffuse through the dialysis membrane, whereas the MCS material stays entrapped in the cap and is not able to enter the cuvette, where the dye molecules are detected by fluorescence spectroscopy. For the delivery experiment, a time-based measurement with an excitation wavelength of 495 nm and an emission wavelength of 517 nm was recorded. The dye-loaded sample was monitored in the closed state up to 4 h, showing no significant release of the dye molecules. After the addition of 5 μL Triton X-100 (1:1000 v/v in H2O) into the tube containing the SLB@MCS, the removal of the lipids from the MCS allowed for the diffusion of calcein out of the pores through the dialysis membrane and their detection in the cuvette. The release in the open state was monitored for an additional 3 h.
Characterization
Scanning electron microscopy (SEM) was performed on a JEOL JSM-6500F operating at 3–5 kV (field emission).Transmission electron microscopy (TEM) images were obtained with an FEI Titan 80–300 equipped with a field-emission gun (300 kV) using the high-angle annular dark-field mode (STEM-HAADF).
Dynamic light scattering (DLS) of aqueous solutions was recorded on a Malvern Zetasizer-Nano equipped with a 4 mW He–Ne laser (633 nm) and avalanche photodiode detector.
Nitrogen sorption measurements were performed on a NOVA 4000e at −196 °C (Quantachrome) after outgassing the sample at 120 °C under vacuum. A QSDFT model of N2 on carbon (slit/cylindr./sphere pores, adsorption branch) was used to calculate the pore volume, and the surface area was calculated with the BET model (p/p0 = 0.05–0.20).
Small-angle X-ray scattering was performed on a Bruker D8 Discover (Cu-Kα1 = 1.5406 Å, theta-theta geometry).
IR spectra were recorded on a Bruker Equinox 55 FT-IR, using KBr pellets.
Fluorescence spectra were recorded on a PTI spectrofluorometer with a photomultiplier detection system (model 810/814). The 1 cm quartz cuvette (QG Hellma) was placed in a cuvette holder with magnetic stirring.
UV-Vis spectra were recorded on a NanoDrop ND-2000 with 1 μL samples.
Results and discussion
Three-dimensionally ordered macroporous (3DOM) silica, which was further used as hard template, was prepared by replicating an array of close-packed PMMA spheres. PMMA spheres were prepared by emulsion polymerization. The washed particles were dried in petri dishes at room temperature to obtain millimeter sized bulk material. Fig. 2a represents an SEM image of an ordered domain of packed PMMA spheres with diameters of around 75–80 nm. These PMMA templates were collected, and cast several times with a pre-hydrolyzed TEOS precursor solution, to achieve a complete filling of the interstitial voids. The resulting PMMA/SiOx composite was dried for several hours and calcined at 550 °C to solidify the silica precursor and to remove the PMMA template. The resulting pore diameter ranges from about 60–70 nm, which is smaller than the initial size of the PMMA spheres, attributed to heating-induced shrinkage.43,44 The bulk material shows good accessibility of the pores through random interpore connections in the range of about 20 nm. A rough silica surface was attributed to the loose initial packing of the PMMA spheres. The random orientation is also indicated by a diffuse ring, obtained by the Fourier Transform (FFT, inset) of the rectangular area (white). The diffuse ring indicates a d-spacing of around 70 nm, which is slightly smaller than the lattice spacings of the PMMA template (Fig. 2b). | ||
| Fig. 2 SEM images of (a) the PMMA colloidal crystal template before TEOS impregnation, (b) the corresponding randomly ordered (FFT inset) macroporous silica replicate after calcination at 550 °C (inset: a 2D fast Fourier transform (FFT) of the white square) and (c) bimodal mesoporous carbon spheres after carbonization at 900 °C in nitrogen and final removal of silica residues by HF treatment. (d) DLS and photograph (inset) depict a homogeneous size distribution of the spheres in solution. | ||
This macroporous silica was further used as hard template for the synthesis of bimodal mesoporous carbon spheres. As carbon source we used resol, which was co-assembled with two porogen compounds, TEOS and Pluronic F127. This precursor mixture enables a homogeneous dispersion of TEOS within the walls of the carbon mesostructure during an evaporation-induced self-assembly (EISA), occurring during the casting and drying of the precursor within the empty voids of the macroporous silica hard template. Subsequent thermopolymerization at 100 °C for 24 h and carbonization at 900 °C in nitrogen results in full carbonization and conversion of TEOS into silica, which is dispersed in the walls of the mesoporous network. Simultaneously the total removal of Pluronic F127 is responsible for mesopore formation. All silica residues were successfully etched with hydrofluoric acid (10 wt%), which creates a loose packing of nanosized mesoporous carbon particles (MCS) in the range of 45 to 68 nm (Fig. 2c). The particles can be separated either by oxidation with ammonium persulfate or ultrasound treatment, leading to well-dispersed colloidal suspensions (Fig. 2d, photograph). The DLS measurement shows a maximum size of 59 nm, which is consistent with the SEM data.
The obtained MCS were also characterized by STEM-HAADF (Fig. 3a and b), demonstrating their uniform particle size. The very small carbon spheres feature a rough surface with accessible mesopores and smaller ones that are present in the walls. The pore structure seems to be ordered and regarding the small-angle X-ray scattering data (Fig. S1, ESI†) a d-spacing of around 8.8 nm for the MCS was found. The broad reflection is attributed to a relatively small confined size within this macroporous template. In contrast, we previously found a pronounced highly ordered hexagonal phase within carbon spheres having diameters of around 330 nm.29 From nitrogen sorption measurements (Fig. 3c) a type IV isotherm was obtained, showing a wide hysteresis and no defined saturation at high relative pressures. We attribute this to textural porosity between the particles. A high BET surface area of 2003 m2 g−1 and a high total pore volume of 1.95 cc g−1 was achieved.
 | ||
| Fig. 3 (a and b) STEM-HAADF images of MCS at different magnifications, obtained after HF treatment. Nitrogen sorption measurements reveal a type IV isotherm (c) and a bimodal pore size distribution (d). | ||
The bimodal porosity, resulting from the removal of F127 and silica from the walls, exhibits two maxima at 5.9 nm and 3.1 nm, respectively. Taken together, the above properties suggest that these small mesoporous particles are attractive candidates for introducing chemical functionality and for applications as potential drug delivery system. For the creation of a SLB@MCS system, the hydrophobic particle surface had to be oxidized. Here we used a procedure known in the literature.41 The successful oxidation of MCS in a solution of ammonium persulfate in sulfuric acid is shown by IR spectroscopy (Fig. 4). The emerging peaks (at 1725, 1618, 1385, 1210 and 1030 cm−1), can be attributed to carbonyl (carboxylic acid, keto-groups, and esters) and hydroxyl groups formed during the oxidation process (black curve). This oxidized surface is expected to be negatively charged at neutral pH, thus enhancing interactions with the positively charged head groups of the phospholipids and facilitating the formation of an SLB around the MCS. In addition to the above-mentioned benefits, DOPC features a double bond in the chain, and hence this lipid offers the possibility to enable a triggered release with so-called photosensitizers that generate reactive oxygen species upon light activation.45–47
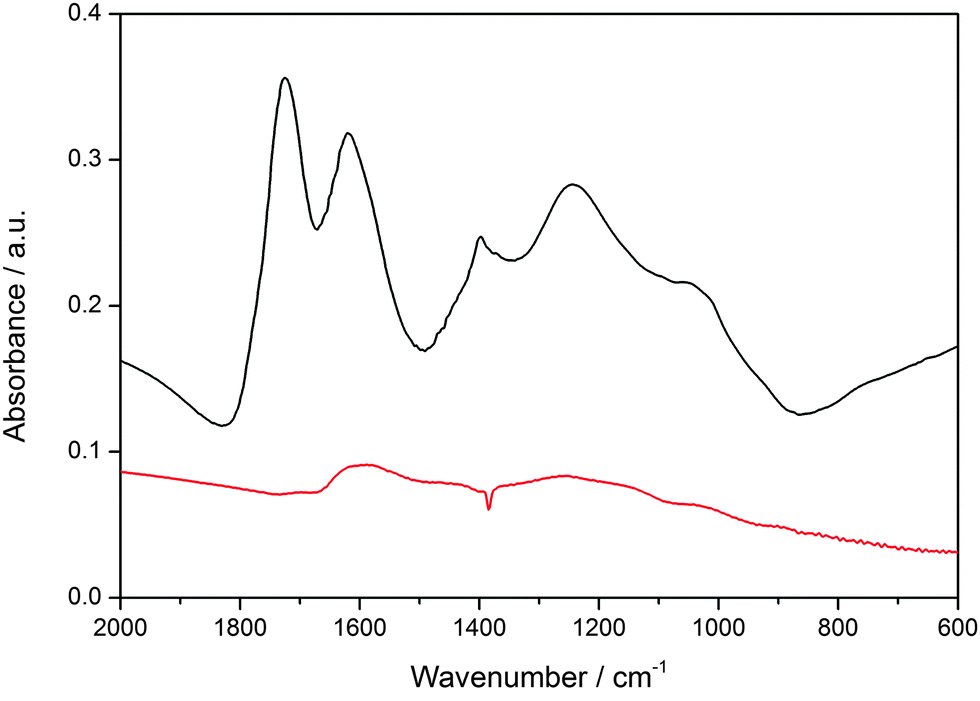 | ||
| Fig. 4 Infrared spectroscopy of as synthesized (red) and oxidized MCS (black). | ||
To show the potential of SLB@MCS as a release-on-demand system, we demonstrate that the SLB is able to prevent the premature release of dye molecules from the mesostructure at body temperature (37 °C).
For this purpose, we loaded a fluorescent dye (calcein) as a model for drug molecules into the mesopores of MCS and capped the nanospheres with a lipid bilayer containing DOPC and DOTAP (Fig. S2, ESI†). Afterwards, the calcein-loaded SLB@MCS were transferred into a tube sealed with a dialysis membrane (Fig. 5a) and mounted on top of a fluorescence cuvette filled with water (pH = 7.2). The membrane only allows the dye molecules to diffuse into the cuvette, whereas the MCS stay entrapped in the tube. As long as the cap system blocks the release of the dye, no fluorescence signal can be detected (Fig. 5b).31,42 In this experiment, even after 4 h, no significant release was observed. Only after rupture of the SLB through the addition of the surfactant Triton X-100, dye molecules were able to diffuse out of the bimodal porous carbon particles across the membrane into the cuvette. The entire amount of calcein was released within 3 h (Fig. S3, ESI†). The amount of 121 μg calcein per mg MCS illustrates the high loading capacity of these highly porous carbon spheres (Fig. S4, ESI†).
 | ||
| Fig. 5 (a) Schematic presentation and photo of the release setup.31,42 (b) Release profile of calcein in SLB@MCS at 37 °C. | ||
Conclusions
We present a strategy for the synthesis of 60 nm large and colloidal nanosized carbon spheres (MCS). Their bimodal porosity features a high BET surface area as well as a high pore volume, which makes them attractive candidates for drug delivery agents with high loading capacity. The synthesis is based on the replication of small macroporous silica hard templates, by concerted co-assembly of a mixture of resol, TEOS and Pluronic F127. After successive thermopolymerization, carbonization and silica removal, the bimodal mesoporous carbon spheres can be separated into colloidal MCS by oxidative treatment. The mesoporous nanoparticles were loaded from solution with fluorescent calcein as model drug and efficiently sealed with a stable supported lipid bilayer (SLB) cap system. Molecular release experiments demonstrate the effective closure and release of this system, providing a tight seal until the addition of Triton X-100, which destabilizes the lipid bilayer and initiates a complete and triggered release of the fluorescent dye from the highly porous carbon. Therefore, our results show the ability of SLB@MCS as effective cap system around colloidal mesoporous carbon nanoparticles with enhanced loading capacities for triggered release applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
The authors gratefully thank the Excellence Initiative of the Deutsche Forschungsgemeinschaft (DFG), SFB 749 and SFB 1032, the Nanosystems Initiative Munich (NIM) cluster, and the Center for NanoScience (CeNS) for financial support. S.N. thanks the “Verband der Chemischen Industrie” (VCI) for his Kekulé grant.pport.Abbreviations
| BET | Brunauer–Emmett–Teller |
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| DOTAP | 1,2-Dioleoyl-3-trimethylammonium-propane |
| FFT | Fast Fourier transform |
| MCS | Nanosized mesoporous carbon spheres |
| HAADF | High-angle annular dark-field |
| PMMA | Poly(methyl methacrylate) |
| SLB | Supported lipid bilayer |
| STEM | Scanning transmission electron microscope |
| TEOS | Tetraethyl orthosilicate. |
Acknowledgements
We thank Dr Steffen Schmidt for TEM characterization, Tina Reuther for N2-sorption measurements and Dr Bastian Rühle for graphics design.References
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- Y. Yang and J. Li, Adv. Colloid Interface Sci., 2014, 207, 155–163 CrossRef CAS PubMed.
- J. Shen, H. C. Kim, H. Su, F. Wang, J. Wolfram, D. Kirui, J. Mai, C. Mu, L. N. Ji, Z. W. Mao and H. Shen, Theranostics, 2014, 4, 487–497 CrossRef CAS.
- F. Yang, C. Jin, Y. Jiang, J. Li, Y. Di, Q. Ni and D. Fu, Cancer Treat. Rev., 2011, 37, 633–642 CrossRef CAS PubMed.
- M. A. Tran, R. J. Watts and G. P. Robertson, Pigm. Cell Melanoma Res., 2009, 22, 388–399 CrossRef CAS PubMed.
- F. Zhao, G. Shen, C. Chen, R. Xing, Q. Zou, G. Ma and X. Yan, Chem. – Eur. J., 2014, 20, 6880–6887 CrossRef CAS PubMed.
- E. Wagner, Acc. Chem. Res., 2012, 45, 1005–1013 CrossRef CAS PubMed.
- M. Meyer, C. Dohmen, A. Philipp, D. Kiener, G. Maiwald, C. Scheu, M. Ogris and E. Wagner, Mol. Pharmaceutics, 2009, 6, 752–762 CrossRef CAS PubMed.
- S. Matsumoto, R. J. Christie, N. Nishiyama, K. Miyata, A. Ishii, M. Oba, H. Koyama, Y. Yamasaki and K. Kataoka, Biomacromolecules, 2009, 10, 119–127 CrossRef CAS PubMed.
- K. Miyata, M. Oba, M. Nakanishi, S. Fukushima, Y. Yamasaki, H. Koyama, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287–16294 CrossRef CAS.
- A. Hagiwara, T. Takahashi, K. Sawai, C. Sakakura, M. Yamaguchi, K. Osaki, H. Tsujimoto, T. Ohyama, H. Tomoda and A. Yamamoto, et al. , Anticancer Drugs, 1994, 5, 194–198 CrossRef CAS.
- A. Hagiwara, T. Takahashi, K. Sawai, C. Sakakura, M. Hoshima, T. Ohyama, M. Ohgaki, T. Imanishi, A. Yamamoto and S. Muranishi, Cancer, 1996, 78, 2199–2209 CrossRef CAS.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- J. Ren, S. Shen, D. Wang, Z. Xi, L. Guo, Z. Pang, Y. Qian, X. Sun and X. Jiang, Biomaterials, 2012, 33, 3324–3333 CrossRef CAS PubMed.
- Z. Liu, K. Chen, C. Davis, S. Sherlock, Q. Cao, X. Chen and H. Dai, Cancer Res., 2008, 68, 6652–6660 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, S. M. Tabakman, K. Yang and H. Dai, Mater. Today, 2011, 14, 316–323 CrossRef CAS.
- S. Zhu, C. Chen, Z. Chen, X. Liu, Y. Li, Y. Shi and D. Zhang, Mater. Chem. Phys., 2011, 126, 357–363 CrossRef CAS.
- J. C. Carrero-Sanchez, A. L. Elias, R. Mancilla, G. Arrellin, H. Terrones, J. P. Laclette and M. Terrones, Nano Lett., 2006, 6, 1609–1616 CrossRef CAS.
- Y. Fang, D. Gu, Y. Zou, Z. Wu, F. Li, R. Che, Y. Deng, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7987–7991 CrossRef CAS PubMed.
- S. Boncel, A. P. Herman and K. Z. Walczak, J. Mater. Chem., 2012, 22, 31–37 RSC.
- A. Magrez, S. Kasas, V. Salicio, N. Pasquier, J. W. Seo, M. Celio, S. Catsicas, B. Schwaller and L. Forro, Nano Lett., 2006, 6, 1121–1125 CrossRef CAS PubMed.
- H. Dumortier, S. Lacotte, G. Pastorin, R. Marega, W. Wu, D. Bonifazi, J. P. Briand, M. Prato, S. Muller and A. Bianco, Nano Lett., 2006, 6, 1522–1528 CrossRef CAS PubMed.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- T.-W. Kim, P.-W. Chung, I. I. Slowing, M. Tsunoda, E. S. Yeung and V. S. Y. Lin, Nano Lett., 2008, 8, 3724–3727 CrossRef CAS PubMed.
- J. Gu, S. Su, Y. Li, Q. He and J. Shi, Chem. Commun., 2011, 47, 2101–2103 RSC.
- J. Zhu, L. Liao, X. Bian, J. Kong, P. Yang and B. Liu, Small, 2012, 8, 2715–2720 CrossRef CAS PubMed.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef CAS PubMed.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS.
- V. Cauda, H. Engelke, A. Sauer, D. Arcizet, C. Bräuchle, J. Rädler and T. Bein, Nano Lett., 2010, 10, 2484–2492 CrossRef CAS PubMed.
- J. Liu, X. Jiang, C. Ashley and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 7567–7569 CrossRef CAS PubMed.
- J. Liu, A. Stace-Naughton, X. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 1354–1355 CrossRef CAS PubMed.
- G. Nordlund, J. B. Sing Ng, L. Bergstrom and P. Brzezinski, ACS Nano, 2009, 3, 2639–2646 CrossRef CAS PubMed.
- A. Schlossbauer, A. M. Sauer, V. Cauda, A. Schmidt, H. Engelke, U. Rothbauer, K. Zolghadr, H. Leonhardt, C. Bräuchle and T. Bein, Adv. Healthcare Mater., 2012, 1, 316–320 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- T. M. Bayerl and M. Bloom, Biophys. J., 1990, 58, 357–362 CrossRef CAS PubMed.
- W. J. Duncanson, M. A. Figa, K. Hallock, S. Zalipsky, J. A. Hamilton and J. Y. Wong, Biomaterials, 2007, 28, 4991–4999 CrossRef CAS PubMed.
- B. Mandlmeier, J. M. Szeifert, D. Fattakhova-Rohlfing, H. Amenitsch and T. Bein, J. Am. Chem. Soc., 2011, 133, 17274–17282 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- A. Vinu, K. Z. Hossian, P. Srinivasu, M. Miyahara, S. Anandan, N. Gokulakrishnan, T. Mori, K. Ariga and V. V. Balasubramanian, J. Mater. Chem., 2007, 17, 1819–1825 RSC.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS PubMed.
- B. T. Holland, C. F. Blanford and A. Stein, Science, 1998, 281, 538–540 CrossRef CAS.
- B. T. Holland, C. F. Blanford, T. Do and A. Stein, Chem. Mater., 1999, 11, 795–805 CrossRef CAS.
- D. Ling, B. C. Bae, W. Park and K. Na, Biomaterials, 2012, 33, 5478–5486 CrossRef CAS PubMed.
- Y. Yang, W. Song, A. Wang, P. Zhu, J. Fei and J. Li, Phys. Chem. Chem. Phys., 2010, 12, 4418–4422 RSC.
- S. A. Mackowiak, A. Schmidt, V. Weiss, C. Argyo, C. von Schirnding, T. Bein and C. Bräuchle, Nano Lett., 2013, 13, 2576–2583 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Small-angle X-ray scattering, chemical structures of phospholipids and lipids, calculation on calcein loading. See DOI: 10.1039/c5tb01635e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |