
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualisation of dual radiolabelled poly(lactide-co-glycolide) nanoparticle degradation in vivo using energy-discriminant SPECT
J.
Llop
*a,
P.
Jiang
b,
M.
Marradi
c,
V.
Gómez-Vallejo
d,
M.
Echeverría
c,
S.
Yu
b,
M.
Puigivila
a,
Z.
Baz
a,
B.
Szczupak
e,
C.
Pérez-Campaña
a,
Z.
Mao
b,
C.
Gao
*b and
S. E.
Moya
*c
aRadiochemistry and Nuclear Imaging, CIC biomaGUNE, Paseo Miramón 182, 20009, San Sebastián, Guipúzcoa, Spain. E-mail: jllop@cicbiomagune.es; Tel: +34 943 00 53 33
bMOE Key Laboratory of Macromolecular Synthesis, Functionalization, Department of Polymer Science, Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: cygao@zju.edu.cn; Tel: +86 571 8795 1108
cSoft Mater Nanotechnology Laboratory, CIC biomaGUNE, Paseo Miramón 182, 20009, San Sebastián, Guipúzcoa, Spain. E-mail: smoya@cicbiomagune.es; Tel: +34 943 00 53 11
dRadiochemistry Platform, CIC biomaGUNE, Paseo Miramón 182, 20009, San Sebastián, Guipúzcoa, Spain
eNuclear Imaging Platform, CIC biomaGUNE, Paseo Miramón 182, 20009, San Sebastián, Guipúzcoa, Spain
First published on 25th June 2015
Abstract
The determination of nanoparticle (NP) stability and degradation in vivo is essential for the accurate evaluation of NP biodistribution in medical applications and for understanding their toxicological effects. Such determination is particularly challenging because NPs are extremely difficult to detect and quantify once distributed in a biological system. Radiolabelling with positron or gamma emitters and subsequent imaging studies using positron emission tomography (PET) or single-photon emission computerised tomography (SPECT) are some of the few valid alternatives. However, NPs that degrade or radionuclides that detach or are released from the NPs can cause artefact. Here, submicron-sized poly(lactide-co-glycolide) nanoparticles (PLGA-NPs) stabilised with bovine serum albumin (BSA) were dual radiolabelled using gamma emitters with different energy spectra incorporated into the core and coating. To label the core, 111In-doped iron oxide NPs were encapsulated inside PLGA-NPs during NP preparation, and the BSA coating was labelled by electrophilic substitution using 125I. After intravenous administration into rats, energy-discriminant SPECT resolved each radioisotope independently. Imaging revealed different fates for the core and coating, with a fraction of the two radionuclides co-localising in the liver and lungs for long periods of time after administration, suggesting that NPs are stable in these organs. Organ harvesting followed by gamma counting corroborated the SPECT results. The general methodology reported here represents an excellent alternative for visualising the degradation process of multi-labelled NPs in vivo and can be extended to a wide range of engineered NPs.
Introduction
The pharmacokinetics (PK) and biological fate of nanoparticles (NPs) need to be known to accurately assess their toxicological effects1 and therapeutic efficacy2 in biomedical applications. However, NPs are extremely difficult to detect and quantify once distributed in a biological system. In preclinical studies, organs are often harvested at pre-determined time points after administration or incorporation of the NPs and, depending on their nature, are then directly visualised by imaging techniques such as transmission electron microscopy (TEM)3 and confocal Raman microscopy4 or indirectly quantified by ion beam microscopy5 or inductively coupled plasma mass spectrometry (ICP-MS).6 These techniques require the sacrifice of large numbers of experimental animals to ensure statistical power, can only be applied to certain NPs, and not all of them are quantitative. New in situ methodologies are required for PK and fate studies in vivo.NPs can be labelled with radionuclides (e.g., positron or gamma emitters) to reduce animal use and generate quantitative data in fate studies. Radiolabelled NPs are then commonly detected in vivo using ultra-high sensitivity imaging modalities such as positron emission tomography (PET) or single-photon emission computerised tomography (SPECT).7 A wide variety of procedures have been developed for the incorporation of positron or gamma emitters into NPs including direct ion8–10 or neutron activation,11–13 the attachment of bifunctional chelators (BFCs) followed by complexation with a radiometal,14–16 the incorporation of a pre-labelled tag into NPs,17–19 or the incorporation of a labelled precursor during NP synthesis.20 Although the spatiotemporal data gathered by radiolabelling and subsequent imaging are useful, the radiochemical integrity or chemical stability of the NPs is not reported: NPs that degrade or radionuclides that detach or are released from the NPs can cause artefact.
Ideally, in vivo radiochemical integrity and NP stability should be determined when investigating NP biodistribution. Moreover, knowledge of NP stability or degradation is fundamental when assessing the biological fate of NPs, since NP degradation can significantly alter toxicological endpoints.21 For example, the degradation of an organic coating around a metal NP may lead to the exposure of the metal core to the cell interior. As a consequence, the metal cations release could catalyse biochemical reactions or increase intracellular ion concentrations which might negatively affect cell homeostasis. Additionally, NP degradation is directly related to the efficiency of NPs in carrying therapeutics and releasing them at their desired location if they are designed to deliver drugs, and the evaluation of their in vivo stability is critical for the successful translation of drug candidates into the clinic.
The assessment of the in vivo stability of small radiolabelled organic molecules is usually achieved by blood sampling followed by plasma isolation and analysis using instrumental analytical techniques.22,23 Although this strategy is also suitable for the assessment of the in vivo stability of radiolabelled NPs, they are not easily isolated from blood samples. Therefore, the problem is often simplified by assessing NP stability in vitro using model media such as water, buffers, or biological fluids24–26 and extrapolating the results to in vivo conditions. Despite being valid in some cases, this strategy has severe limitations due to the inherent complexity of biological systems.
The incorporation of multiple imaging agents into a single NP can provide complementary information regarding their stability when used with a combination of imaging modalities. In one of the few reported examples of this approach, oleate-stabilised magnetic NPs with a DSPE-PEG coating were labelled with 64Cu and investigated by combined PET and magnetic resonance imaging (MRI).27 However, simultaneous PET-MRI acquisition remains a challenge in the preclinical setting, with consecutive or parallel studies using both modalities required. Additionally, both modalities have very different intrinsic sensitivity, which complicates experimental set up and data analysis. More recently, iron oxide NPs stabilised with oleic acid and phospholipids were radiolabelled with 59Fe, 14C, and 111In and the biodistribution of all three radioisotopes investigated in mice following sacrifice and organ harvesting.28 Although this study generated useful information about NP stability, 59Fe and 14C are inappropriate imaging agents for in vivo studies and animal sacrifice was required.
Very recently, strategies for the preparation of double-labelled functionalised gold NPs have been reported.29,30 Following a similar approach, we present here a strategy for the in vivo determination of NP stability, also based on dual radiolabelling using two gamma emitters with different emission energies. In our approach, the NP core and the surface were independently labelled. We exploited the favourable properties of poly(lactide-co-glycolide) NPs (PLGA-NPs) which, due to their submicron size, display excellent biodegradability and biocompatibility. Additionally, PLGA copolymers were approved by the Food and Drug Administration (FDA) for clinical drug delivery.31 Furthermore, their size and the use of emulsion techniques in their preparation allowed our novel approach to be implemented. First, we synthesised iron oxide NPs containing 111In and stabilised with oleic acid. Iron oxide NPs were then encapsulated in the PLGA-NP core by mixing them with the PLGA phase during emulsification, overcoming the drawback of chemically modifying PLGA molecules for radiolabelling, which may alter their hydrophobicity and affect NP synthesis. Bovine serum albumin (BSA) was used as a stabilising agent for the emulsion droplets, facilitating the incorporation of 125I, the second radioisotope, by electrophilic substitution on the tyrosine residues of the protein. Subsequent SPECT imaging with energy discrimination allowed quantitative assessment of the in vivo stability of NPs, minimising the use of experimental animals. This was achieved by monitoring the change of the intensities of 111In and 125I, after appropriate decay correction taking into consideration the half-lives for each isotope. Dissection through gamma counting experiments was conducted and correlated with SPECT images for validation. This general strategy can be extended to a wide variety of engineered NPs and is a valuable tool for in vivo evaluation of NP stability.
Results and discussion
Synthesis, radiolabelling and characterization of NPs
The synthesis of dual labelled NPs ([125I/111In]NP1, see Fig. 1) consisted of two steps. In the first step, the PLGA core was labelled by including 111In-doped iron oxide NPs stabilised with oleic acid during NP fabrication; in the second step, the BSA used as a surface stabiliser was labelled with 125I. Radiolabelling of the NP core might be envisioned via attachment of a radiotag to lactic-co-glycolic polymers. This process is, however, challenging. Hence, the entrapment of pre-labelled iron oxide NPs was considered as a convenient strategy. | ||
| Fig. 1 Schematic of the preparation of dual-labelled [125I/111In]NP1 NPs. | ||
Iron oxide NPs were prepared by a slight modification of a previously published co-precipitation method.32 The addition of 111InCl3 during co-precipitation yielded mono-disperse iron oxide magnetic NPs coated with oleic acid, with a diameter of 12 ± 3 nm as determined by TEM (Fig. 2a). NPs were further dispersed in an organic PLGA solution using a well-established protocol.33 An aqueous BSA solution was transferred to the organic phase, and the solution was sonicated to form organic phase droplets stabilised with BSA and containing iron oxide-loaded PLGA-NPs ([111In]NP1). Finally, the incorporation of 125I by electrophilic aromatic substitution was carried out to yield [125I/111In]NP1.
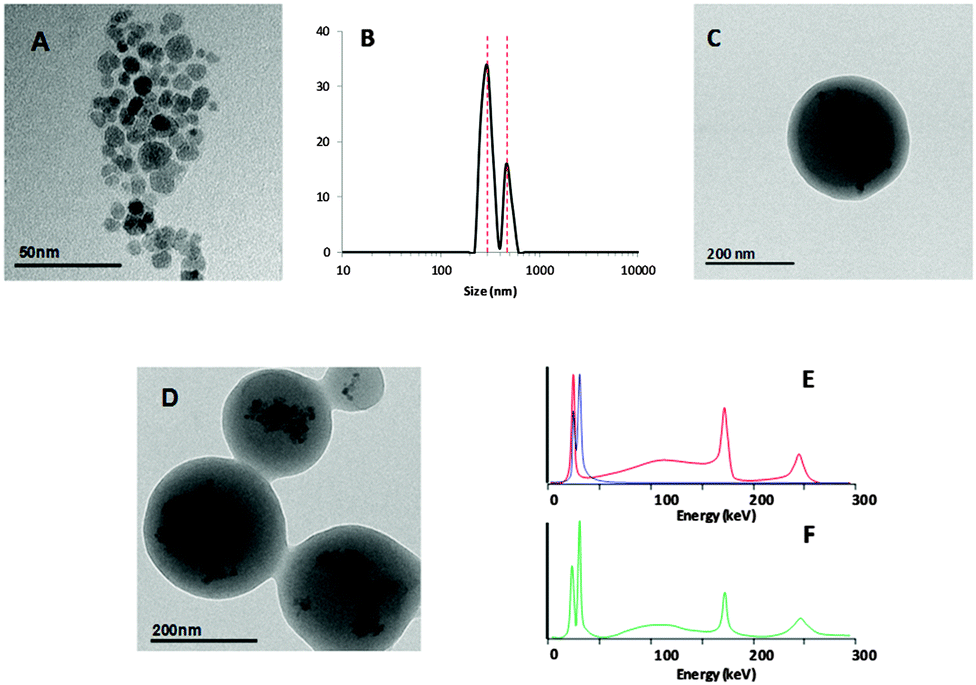 | ||
| Fig. 2 Characterisation of dual-labelled NPs: (a) TEM images of oleic acid-stabilised iron oxide NPs; (b) intensity distribution for NP1 in water as determined by dynamic light scattering; (c and d) TEM images of NP1; (e) gamma spectra corresponding to 111In (red line) and 125I (blue line); (f) gamma spectrum of NP1. | ||
The selection of the correct combination of radioisotopes was of paramount importance to enable subsequent imaging studies using energy discriminant SPECT. Ideally, the energy spectra should not overlap, and the energy peaks should fall within the energy range of the SPECT system used (in our case 25–250 keV). Additionally, both radioisotopes should have a sufficient half-life to investigate biodistribution for long periods of time after administration. 111In (T1/2 = 2.8 days, maximum energy emission at 171 and 245 keV) is a radioisotope with good in vivo imaging characteristics, which has been widely used in both the pre-clinical and clinical settings. It is usually attached to the molecule of interest by the formation of a radiometal–chelator complex; however, previous studies have suggested that it can be readily incorporated into the crystal lattice of iron oxide NPs,34 as here. For BSA labelling, 125I was chosen for several reasons: first, it has been widely used for the radiolabelling of peptides, proteins, and antibodies via electrophilic substitution by taking advantage of the presence of tyrosine residues; second, 125I has an energy maximum at 35.5 keV and a long half-life (close to 60 days) and is reasonably cheap and widely available. Hence, both isotopes have a relatively long half-life, appropriate emission characteristics, and established radiolabelling routes.
The hydrodynamic diameter of [125I/111In]NP1 NPs was determined by dynamic light scattering (DLS): the mean NP size was 300 ± 40 nm (∼75%), with a minor population (∼25%) of 460 ± 70 nm (Fig. 2b) also observed in the intensity distribution. TEM analysis performed after radioactive decay showed that iron oxide NPs were properly entrapped in the polymeric matrix core (Fig. 2c and d). TEM imaging under dry conditions revealed PLGA-NP sizes of about 250 ± 50 nm. The PLGA-NPs had a negative ξ-potential of −15 mV at neutral pH. Gamma spectrometry of the final NPs showed the presence of peaks corresponding to 111In and 125I (Fig. 2e and f). The ratio of both isotopes was always maintained in the range 125I/111In = 1.3–1.6.
The stability of [125I/111In]NP1 was measured in physiological saline (0.9% NaCl) and mouse plasma at 37 °C. The NPs showed good stability in physiological saline (Fig. 3), with both isotopes remaining attached to the NPs up to 48 hours. At t = 6 days, significant release of 125I and 111In was observed, although 64% and 75% of the radioactivity, respectively, remained attached to the NPs.
 | ||
| Fig. 3 Stability of [125I/111In]NP1 in physiological saline (top) and mouse plasma (bottom) expressed as % radioactivity attached to the NP at different time points of incubation (T = 37 °C). Results are expressed as mean ± standard deviation (n = 3 per time point). | ||
Interestingly, incubation in plasma resulted in the progressive release of 125I. At 48 h of incubation, approximately 50% of the 125I was released from the NPs. This value increased up to 85% at t = 6 days. The decrease in the 125I signal observed in mouse serum hints at the removal of labelled BSA from the NPs. BSA can be replaced by unlabelled proteins from the serum or simply released as a consequence of their interaction with serum components. BSA molecules are linked to the PLGA only by hydrophobic interactions, which may not be strong enough to guarantee their attachment to the NP surface in the presence of serum components. On the other hand, the 111In percentage remained almost constant up to 2 days, with a progressive decrease afterwards to reach values around 65% at t = 6 days. These values suggest that iron oxide NPs remain PLGA encapsulated over the first 48 h of the experiment, and the release occurs very slowly and only at long times after incubation. This was expected, since degradation (weight loss) of PLGA in vitro is reported to only occur after at least one week in phosphate-buffered saline (pH 7.4).35,36
In vivo imaging studies
Stability studies were followed by in vivo SPECT imaging; static images were acquired for each radioisotope at different time points after administration using a SPECT camera with energy discrimination (Fig. 4a); pure 125I-labelled BSA was used as a control (Fig. 4b). | ||
| Fig. 4 Coronal SPECT images of the biodistribution of (a) [125I/111In]NP1 and (b) [125I]BSA after intravenous administration into mice; red colour: 125I; green colour: 111In. Images were co-registered with a CT image of the same animal to anatomically localize the SPECT signal. CT images were adjusted in the Y axis at each time point for appropriate fitting with the tracer distribution image. (c) A schematic of the different organs. | ||
SPECT imaging after administration of the NPs showed that the biodistribution of the two radiotags could be followed independently. Three hours after administration, the majority of the radioactivity of both radioisotopes (red for 125I, green for 111In) was located in the liver and lungs with 125I/111In ratios of 1.12 ± 0.21 and 0.92 ± 0.32, respectively (Fig. 4a), suggesting that the majority of the NPs remained intact in these organs. Accumulation in the lung was probably due to the size of the NPs, which may form micro-emboli by occlusion of the small lung capillaries.37 Sequestration by the reticulo-endothelial system (RES) may also contribute to lung accumulation. At this time point, there was selective accumulation of 125I in the bladder and thyroid glands, suggesting that the NPs had already undergone some degradation. One and two days after administration, a fraction of 125I and 111In still co-localised in the liver and lungs (125I/111In ratio = 1.07 ± 0.14 and 0.92 ± 0.18 in the liver at t = 24 and 48 h, respectively; 125I/111In ratio = 0.67 ± 0.22 and 0.42 ± 0.16 in the lungs at t = 24 and 48 h, respectively), but the 125I signal in the thyroid gland progressively increased with time. Six days after NP administration, 125I was almost cleared from the body (except the thyroid gland), with the majority of detected radioactivity originating from 111In in the liver. This suggests that the NP core had not fully degraded.
In order to obtain comparative data, pure 125I-labelled BSA was also administered to the animals and SPECT scans were conducted at three, 24, and 48 hours (Fig. 4b). Pure 125I-labelled BSA rapidly accumulated in the lungs, liver, thyroid gland, and urine. 125I was detected in the liver after 24 h and was almost cleared from this organ by 48 h, at which point radioactivity was only detected in the bladder and thyroid gland.
Altogether, the results suggest that [125I/111In]NP1 biodistributes due to the attachment of the protein to the NPs, and at initial time points the BSA follows the biodistribution of the core. It has previously been reported that 125I-labelled BSA undergoes rapid clearance from mouse livers after intravenous administration (<0.2% of injected dose after 24), with the labelled metabolite [125I]mono(di-)iodotyrosin detected in hepatocytes.38 This radiometabolite can easily cross biological membranes, which might explain the low uptake at later time points. The elimination of 125I was much less dramatic from dual labelled NPs (125I remained detectable in the liver six days after administration), suggesting that the radiolabelled BSA remained attached to NPs in the liver.
Ex vivo studies
The biodistribution and chemical stability of the NPs were also investigated by dissection and gamma counting, which also provided accurate, organ-level biodistribution information and corroborated the in vivo data. The accumulation of 125I and 111In in different organs expressed as the % of injected dose per gram of tissue (%ID g−1) is shown in Fig. 5. | ||
| Fig. 5 Accumulation of 125I and 111In in different organs as determined by dissection and gamma counting and expressed as the % of injected dose per gram of tissue (%ID g−1). Results are expressed as mean ± standard deviation (n = 3 per time point). | ||
For both radioisotopes, the highest radioactivity concentrations were detected in the lungs at early time points after administration (%ID g−1 ∼40 at t < 2 h). The liver had the next highest concentrations (%ID g−1 between 20 and 30%), with a non-significant increasing trend observed between 5 and 30 min and a progressive decrease thereafter. After 48 h, the %ID g−1 were 11.6 ± 3.2 and 3.3 ± 1.8 for 125I and 16.6 ± 4.6 and 9.6 ± 3.2 for 111In in the liver and lungs, respectively. The accumulation of 125I in the thyroid gland increased over time, reaching values close to 100%ID g−1 at 48 h. Interestingly, the accumulation of 111In in the thyroid glands was very low (<3%) at all time points, which can be considered insignificant given the very small size of the thyroid gland (between 4–5 mg). The accumulation of 125I was extremely high in the urine soon after administration (131 ± 33% ID g−1 at t = 30 min), with accumulation of 111In below 5% throughout the experiment. Values below 0.1% ID g−1 were obtained for both radioisotopes in the brain (results not shown). Blood radioactivity was similar for both isotopes, with initial %ID g−1 values in the range of 1.5–2%, which progressively decreased to below 1% at t > 2 h.
125I/111In %ID g−1 ratios at different time points for different organs after intravenous administration of the dual-labelled NPs are shown in Fig. 6: values close to 1 denote that the amounts of both isotopes (expressed as %ID g−1) are equivalent, suggesting that the two labels remain together and that the NPs have not degraded. At early time points after administration, the 125I/111In ratio was always close to 1, irrespective of the organ analysed. This ratio was maintained in the liver up to 24 h, with a slight, non-significant decrease thereafter (0.70 ± 0.27 at t = 48 h). A similar trend was observed in the lungs, with values very close to 1 at early time points after administration. However, a decreasing trend was readily observed by 24 h (0.52 ± 0.24 at t = 24 h) that became more pronounced at t = 48 h (0.34 ± 0.22). These values are concordant with those obtained in vivo using SPECT and energy discrimination. The 125I/111In ratios in the urine, intestine, and thyroid glands were very high even at early time points after NP administration, confirming selective accumulation of radioiodine in these tissues. As observed in the in vitro stability studies and in vivo SPECT experiments, these results suggest that labelled proteins slowly detached from the NP core in the presence of plasma proteins and this phenomenon was likely to have been even quicker in the in vivo circulation.
 | ||
| Fig. 6 125I/111In ratios in different organs as determined by dissection and gamma counting. Results are expressed as mean ± standard deviation (n = 3 per time point). | ||
The abovementioned degradation of the protein coating which might have been replaced by other proteins during circulation was previously reported in in vitro studies,39 and is an important issue since it can alter the biodistribution of NPs. Most coatings are not only used to protect the NP core or stabilise them in aqueous media, but also to direct the NPs to specific organs or tissues or to carry specific functions. The loss of these functionalities may lead to modification of the properties of NPs. In the case considered here, BSA is not covalently bonded to the NPs but interacts with PLGA by hydrophobic interactions, since it was used as a surfactant to stabilise the PLGA emulsion that led to the formation of the NPs. Under these conditions, the NP-PLGA core interaction is weak, which probably makes the removal of BSA easy. Covalently attaching the protein to the NP would most likely increase protein stability. This knowledge about the stability of the BSA coating is helpful for the design of surface coatings of NPs and how to engineer them to produce longer circulation times or improve their stability.
Experimental section
Reagents
All chemical reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and were of analytical grade purity unless otherwise specified. Nanopure water (18.2 MΩ cm) was obtained using a Thermo Scientific Barnstead NANOpure Diamond Water System (Thermo Fisher Scientific, Waltham, MA, USA). Na[125I]I (solution in 0.1 M NaOH) was obtained from Perkin Elmer (Beaconsfield, UK), and 111InCl3 (solution in HCl) was purchased from Covidien (Dublin, Ireland).Preparation of 111In-labelled iron oxide NPs
Iron(II) chloride tetrahydrate (50 mg) and iron(III) chloride hexahydrate (60 mg) were dissolved in ultrapure water (25 mL). A fraction of this solution (0.5 mL) was withdrawn using a pipette and diluted with ultrapure water (4.5 mL) in a vial. 111InCl3 solution (780 μL, 512 MBq) was immediately added, and the vial was capped and flushed with nitrogen gas (solution A). In parallel, ammonia (29% aqueous solution, 2.34 mL) was added to a 100 mL flask and diluted with ultrapure water to a final volume of 20 mL. The solution was stirred for 30 min under nitrogen gas flow (solution B). Solution A was then slowly added to solution B with constant stirring (500 rpm) at room temperature. The solution turned yellow-black due to the formation of iron oxide NPs. After 30 min, the NPs were collected using a magnet, washed with water three times, and finally dispersed in purified water (5 mL).Preparation of oleic acid-modified 111In-labelled iron oxide NPs
Radiolabelled iron oxide NPs prepared as above were sonicated (20% power, 2 s + 2 s cycles) for 30 min (total running time). Oleic acid (100 μL) was then added dropwise under sonication. To prevent a temperature rise in the reaction mixture, a water bath (20 °C) was used. After 2 h, the NPs were collected using a magnet. To recover NPs attached to the sonication tip and to the walls of the vial, the aqueous solution was removed by decantation, hexane (1 mL) was added, and the mixture was further sonicated. Finally, the collected NPs were dispersed into hexane and the solution was concentrated to a final volume of 25 μL.Preparation of PLGA-BSA particles
Oleic acid-modified 111In-labelled iron oxide NPs as prepared above (25 μL) were added to a PLGA (PLGA 85:15) solution (2% in dichloromethane, 125 μL) and the resulting mixture was sonicated for 10 min. BSA solution (2% in purified water, 600 μL) was added, and the mixture was again sonicated for 20 s. The resulting solution was immediately poured into purified water (15 mL) and stirred for 2 h. PLGA NPs were then collected by centrifugation and washed twice with purified water. The resulting NPs were finally dispersed in PBS (50 μL).Radiolabelling of PLGA-BSA NPs with 125I
Iodination was carried out using Pierce® iodination beads. The beads were washed with PBS (1 mL), dried with filter paper, and introduced to a vial with PBS (100 μL, pH = 7.2) and Na125I (ca. 20 μL, 37 MBq). The mixture was incubated at room temperature for 5 min with occasional vigorous shaking. PLGA-BSA NPs (PBS solution, see above) were added to this solution, and the mixture was incubated for 15 min at room temperature. The NPs were finally isolated by centrifugation and washed twice with PBS.NP characterisation
Size distributions and ξ-potentials were determined for all samples. Transmission electron microscopy (TEM) was performed using a JEOL JEM-1400 plus microscope (Jeol, Tokyo, Japan) working at 120 kV. For TEM examinations, a single drop (10 μL) of the aqueous solution (∼0.1 mg mL−1 in milliQ water) of the NPs was placed onto a carbon film on 400 square mesh copper grids (CF-400-Cu, Electron Microscopy Sciences, Hatfield, PA, USA). The grid was left to dry in air for several hours at room temperature.DLS and ξ-potential measurements were performed using a Malvern Zetasizer Nano ZS system (Malvern Instruments, Malvern, UK). For particle size determination, ten measurements for 10 s were carried out at a temperature of 25 °C at a 173° scattering angle. A refractive index of 1.52 for PLGA was used. For ξ-potential measurements, a minimum of 15 runs were carried out. NP effective surface charge in 10 mM PBS was measured at pH 7.4. Measurements were performed in quintuplicate. The Smoluchowski approximation was used to convert the electrophoretic mobility to ξ-potential values.
Radiolabelled NPs were also characterised by γ-spectrometry using a multi-channel analyser γ-spectrometer (MUCHA, Raytest, Straubenhardt, Germany) calibrated with standard energy calibration sources. A small fraction of the NPs was placed into a vial and the energy spectrum was recorded. The areas under the peaks at 35 kV (maximum emission for 125I) and 171 keV (energy peak for 111In) were used to determine the absolute amounts of both radioisotopes.
Radiochemical integrity of labelled NPs
To assess the radiochemical stability, NPs were prepared and purified as described above and subsequently resuspended in 300 μL of PBS or mouse plasma. The suspension was then divided into five different aliquots containing 50 μL of the NPs each. The aliquots were kept at 37 °C for five and 30 min, and two, 24, 48 and 144 hours, respectively. The samples were then centrifuged and the NPs were separated by decantation and washed twice with PBS. The energy spectra of the supernatant and the NPs were then recorded, and the absolute amount of 125I and 111In in both fractions was determined.Animal experiments
Three hours prior to examination, mice (three per labelled species) received an intravenous administration of either (i) [125I/111In]NP1 or (ii)125I-labelled BSA. With the mouse under isoflurane anaesthesia (1.5–2% in oxygen), whole-body SPECT/CT scans were acquired at 3 h, 24 h, 48 h, and 6 days post-injection (p.i.). Using a full ring detector, 360° of data were acquired by rotating the collimator at 45° (45 steps, 1° per step). Data were collected in an energy window of 30–200 keV, and the duration of the scans ranged between two and four hours. As a general rule, the image acquisition time was chosen based on the number of counts detected; thus, longer imaging times were required for later p.i. scans. During image acquisition, mice were kept under normothermic conditions using a heating blanket (Homeothermic Blanket Control Unit; Bruker BioSpin GmbH, Karlsruhe, Germany). After each SPECT scan, CT acquisitions were performed to provide anatomical information about each animal. The CT acquisition consisted of 220 views acquired in 0.88° increments around the animal with 16 ms exposure per view. The X-ray tube settings were 70 kV and 32 mA.
The SPECT images were reconstructed using the 2D OSEM iterative algorithm with 10 iterations/1 subsets for 111In in the 154–188 keV energy window, and 5 iterations/1 subsets for 125I in the 27–35 keV energy window. The images were reconstructed into 128 × 128 × 32 arrays with a voxel size of 0.4 × 0.4 × 2.46 mm and were corrected for scatter but not attenuation. The CT images were reconstructed using a cone beam filtered back-projection Feldkamp algorithm into 437 × 437 × 523 arrays with a voxel size of 0.2 × 0.2 × 0.2 mm.
Dissection and gamma counting: animals (n = 30) were anesthetised with isofluorane and a solution of dual-labelled NPs (containing ca. 0.8 MBq of 125I and 0.5 MBq of 111In) injected through the tail vein. After injections, animals were allowed to recover. Just before sacrifice by cervical dislocation (five and 30 min and two, 24, and 48 after dose administration), animals were anesthetised again and blood samples were collected by cardiac puncture. After sacrifice, liver, lungs, brain, cerebellum, intestine, and thyroid glands were quickly removed and rinsed twice with deionised water; urine samples were also obtained. The gamma spectra for organs and fluids were obtained in the multi-channel analyser γ-spectrometer (MUCHA, Raytest).
Conclusions
In conclusion, we presented a novel methodology for the synthesis of dual-radiolabelled BSA-coated PLGA-NPs with two gamma emitters without overlapping energy spectra located in the core and at the surface. The biodistribution and biological fate of the labels at the surface coating and in the core could be determined independently by SPECT imaging and corroborated by dissection and gamma counting. Imaging showed that NPs accumulated in the lungs and liver at early time points, at which point the 111In/125I ratio remained constant; however, 125I progressively accumulated in the thyroid glands and urine, resulting in a decrease in co-localisation with the signal corresponding to 111In located at the core. After six days, there was almost complete clearance of 125I, but the 111In signal was still detectable in the liver. These results indicate that 125I-labelled BSA is progressively removed from the core to follow a different pattern of biodistribution and clearance than the core. These results help in explaining the fate and process of degradation of the surface coatings of PLGA-NPs, and this methodology represents an excellent alternative for visualising the degradation process of multi-labelled NPs in vivo.Acknowledgements
The authors would like to thank the Ministerio de Economía y Competitividad (MINECO, project MAT2013-48169-R), the Departamento de Educación, Política Lingüística y Cultura del Gobierno Vasco (project PI-2014-1-90), the FP7-Infrastructures project QNANO (Project reference: 262163), and the Natural Science Foundation of China (51120135001) for financial support. The authors also would like to thank Eleftheria Diamanti for assistance in Figure design.Notes and references
- A. Lankoff, W. J. Sandberg, A. Wegierek-Ciuk, H. Lisowska, M. Refsnes, B. Sartowska, P. E. Schwarze, S. Meczynska-Wielgosz, M. Wojewodzka and M. Kruszewski, Toxicol. Lett., 2012, 208, 197–213 CrossRef CAS PubMed.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomed.: Nanotechnol. Biol. Med., 2012, 8, 147–166 CrossRef CAS PubMed.
- W. H. De Jong, M. C. Burger, M. A. Verheijen and R. E. Geertsma, Materials, 2010, 3, 4681–4694 CrossRef CAS PubMed.
- J. Moger, B. D. Johnston and C. R. Tyler, Opt. Express, 2008, 16, 3408–3419 CrossRef CAS.
- X. Zhou, M. Dorn, J. Vogt, D. Spemann, W. Yu, Z. Mao, I. Estrela-Lopis, E. Donath and C. Gao, Nanoscale, 2014, 6, 8535–8542 RSC.
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. A. M. Sip and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 CrossRef CAS PubMed.
- K. Stockhofe, J. M. Postema, H. Schieferstein and T. L. Ross, Pharmaceuticals, 2014, 7, 392–418 CrossRef CAS PubMed (and references therein).
- C. Pérez-Campaña, V. Gómez-Vallejo, M. Puigivila, A. Martín, T. Calvo-Fernández, S. E. Moya, R. F. Ziolo, T. Reese and J. Llop, ACS Nano, 2013, 7, 3498–3505 CrossRef PubMed.
- K. Abbas, I. Cydzik, R. del Torchio, M. Farina, E. Forti, N. Gibson, U. Holzwarth, F. Simonelli and W. Kreyling, J. Nanopart. Res., 2010, 12, 2435–2443 CrossRef CAS.
- F. Simonelli, P. Marmorato, K. Abbas, J. Ponti, J. Kozempel, U. Holzwarth, F. Franchini and F. Rossi, IEEE Trans. NanoBiosci., 2011, 10, 44–50 CrossRef PubMed.
- S. H. Jung, K. I. Kim, J. H. Ryu, S. H. Choi, J. B. Kim, J. H. Moon and J. H. Jin, Appl. Radiat. Isot., 2010, 68, 1025–1029 CrossRef CAS PubMed.
- J. Roy and S. Lahiri, Green Chem., 2006, 8, 1063–1066 RSC.
- S. Hirn, M. Semmler-Behnke, C. Schleh, A. Wenk, J. Lipka, M. Schaffler, S. Takenaka, W. Moller, G. Schmid, U. Simon and W. G. Kreyling, Eur. J. Pharm. Biopharm., 2011, 77, 407–416 CrossRef CAS PubMed.
- J. Frigell, I. Garcia, V. Gómez-Vallejo, J. Llop and S. Penades, J. Am. Chem. Soc., 2014, 136, 449–457 CrossRef CAS PubMed.
- B. R. Jarrett, B. Gustafsson, D. L. Kukis and A. Y. Louie, Bioconjugate Chem., 2008, 19, 1496–1504 CrossRef CAS PubMed.
- E. J. Keliher, J. Yoo, M. Nahrendorf, J. Lewis, B. Marinelli, A. Newton, M. Pittet and R. Weissleder, Bioconjugate Chem., 2011, 22, 2383–2389 CrossRef CAS PubMed.
- S. Guerrero, J. R. Herance, S. Rojas, J. F. Mena, J. D. Gispert, G. A. Acosta, F. Albericio and M. J. Kogan, Bioconjugate Chem., 2012, 23, 399–408 CrossRef CAS PubMed.
- S. Rojas, J. D. Gispert, R. Martín, S. Abad, C. Menchón, D. Pareto, M. V. Víctor, M. álvaro, H. García and J. R. Herance, ACS Nano, 2011, 5, 5552–5559 CrossRef CAS PubMed.
- J. J. Willman, Z. Cheng, C. Davis, A. M. Lutz, M. L. Schipper, C. H. Nielsen and S. S. Gambhir, Radiology, 2006, 249, 212–219 CrossRef PubMed.
- P. P. Di Mauro, V. Gómez-Vallejo, Z. Baz, J. Llop and S. Borros, Bioconjugate Chem., 2015, 26, 582–592 CrossRef CAS PubMed.
- S. J. Soenen, J. M. Montenegro, A. M. Abdelmonem, B. B. Manshian, S. H. Doak, W. J. Parak, S. C. de smedt and K. Braeckmans, Acta Biomater., 2014, 10, 732–741 CrossRef CAS PubMed.
- I. Odano, C. Halldin, P. Karlsson, A. Varrone, A. J. Airaksinen and R. N. Farde, NeuroImage, 2009, 45, 891–902 CrossRef PubMed.
- V. Gómez-Vallejo, A. Martín, M. Aginalde, E. San Sebastián, D. Padro, F. Cossio and J. Llop, Appl. Radiat. Isot., 2012, 70, 2545–2551 CrossRef PubMed.
- R. Sharma, Y. Xu, S. W. Kim, M. J. Schueller, D. Alexoff, S. D. Smith, W. Wang and D. Schlyer, Nanoscale, 2013, 5, 7476–7483 RSC.
- A. Polyák, I. Hajdu, M. Bodnan, G. Trencsenyi, Z. Postenyi, V. Haasz, G. Janoki, L. Balogh and J. Borbely, Int. J. Pharm., 2013, 449, 10–17 CrossRef PubMed.
- B. E. Ocampo-García, F. de M. Ramírez, G. Ferro-Flores, L. M. de León-Rodríguez, C. L. Santos-Cuevas, E. Morales-ávila, C. Arteaga de Murphy, M. Pedraza-López, L. A. Medina and M. A. Camacho-López, Nucl. Med. Biol., 2010, 38, 1–11 CrossRef PubMed.
- C. Glaus, R. Rossin, M. J. Welch and G. Bao, Bioconjugate Chem., 2010, 21, 715–722 CrossRef CAS PubMed.
- H. Wang, R. Kumar, D. Nagesha, R. I. Duclos Jr, S. Sridhar and S. J. Gatley, Nucl. Med. Biol., 2015, 42, 65–70 CrossRef CAS PubMed.
- K. C. L. Black, W. J. Akers, G. Sudlow, B. Xu, R. Laforest and S. Achilefu, Nanoscale, 2015, 7, 440–444 RSC.
- W. G. Kreyling, A. M. Abdelmonem, Z. Ali, F. Alves, M. Geiser, N. Haberl, R. Hartmann, S. Hirn, D. Jimenez de Aberasturi, K. Kantner, G. Khadem-Saba, J.-M. Montenegro, J. Rejman, T. Rojo, I. Ruiz de Larramendi, R. Ufartes, A. Wenk and W. J. Parak, Nat. Nanotechnol., 2015 DOI:10.1038/nnano.2015.111.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- D. Horák, N. Semenyuk and F. Lednický, J. Polym. Sci., Part A-1: Polym. Chem., 2003, 41, 1848–1863 CrossRef PubMed.
- Y. Qiu, R. Palankar, M. Echeverría, N. Medvede, S. E. Moya and M. Delcea, Nanoscale, 2013, 5, 12624–12632 RSC.
- J. Zeng, B. Jia, R. Qiao, C. Wang, L. Jing, F. Wang and M. Gao, Chem. Commun., 2014, 50, 2170–2172 RSC.
- B. Li, J. Yang, L. Ma, F. Li, Z. Tu and C. Gao, Tissue Eng., Part A, 2014, 20, 1–11 CrossRef PubMed.
- W. Wang, B. Li, J. Yang, L. Xin, Y. Li, H. Yin, Y. Qi, Y. Jiang, H. Ouyang and C. Gao, Biomaterials, 2010, 31, 8964–8973 CrossRef CAS PubMed.
- P. G. Waser, U. Muller, J. Kreuter, S. Berger, K. Munz, E. Kaiser and B. Pfluger, Int. J. Pharm., 1987, 39, 213–227 CrossRef CAS.
- M. Nishikawa, F. Staud, S. Takemura, Y. Takakura and M. Hashida, Biol. Pharm. Bull., 1999, 22, 214–218 CAS.
- M. Chanana, P. Rivera Gil, M. A. Correa-Duarte, L. M. Liz-Marzán and W. J. Parak, Angew. Chem., Int. Ed., 2013, 52, 4179–4183 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |