
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of double macromolecular network (DMMN) hydrogels based on hyaluronan and high molecular weight poly(ethylene glycol)†
Changjiang
Fan
ab,
Chao
Zhang
c,
Liqiong
Liao
*a,
Sheng
Li
a,
Weiping
Gan
a,
Jinping
Zhou
a,
Dong-An
Wang
*b and
Lijian
Liu
a
aKey Laboratory of Biomedical Polymers of Ministry of Education & College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: liqiongliao@whu.edu.cn
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Blk N1.3-B2-13, Singapore 637457, Singapore. E-mail: DAWang@ntu.edu.sg; Fax: +65 6791 1761; Tel: +65 6316 8890
cSchool of Engineering, Sun Yat-Sen University, Guangzhou, Guangdong 510006, P. R. China
First published on 9th July 2015
Abstract
Abundant research efforts have been devoted to meet the demands for high-strength hydrogels in biomedical applications. Double-network (DN) hydrogels and homogeneous hydrogels are two typical samples. In this study, a novel ultra-strong and resilient double macromolecular network (DMMN) hydrogel system has been developed via a two-step sequential cross-linking process using hyaluronan (HA) and high molecular weight poly(ethylene glycol) (PEG) for the first and second network, respectively. A lower concentration of the HA precursor solution and a higher concentration of the PEG precursor solution as well as a higher molecular weight of the PEG precursor are beneficial to produce high-strength DMMN gels. Dynamic light scattering measurements demonstrate that DMMN gels possess the more evenly distributed polymer networks; the distinctive double and relatively evenly distributed networks of the DMMN gel make it combine the current DN and homogeneous network strategies for preparing robust hydrogels. The optimized DMMN gel is capable of sustaining up to 50 MPa of compressive stress. Besides, DMMN gels exhibit excellent cytocompatibility. This study expands the DN principle in designing and fabricating high-strength hydrogels with biocompatible macromolecules that show a promising prospect for biomedical applications.
1 Introduction
Inferior mechanical strength of hydrogels has severely limited their applications as drug release matrices, tissue engineering scaffolds, and biosensors.1–3 Recently, high-strength hydrogels have attracted increasing attention.4–9 The failure of hydrogels under stress mainly includes two processes named initial “crack formation” and “crack propagation”.10 One strategy to improve the mechanical strength of hydrogels is to fabricate hydrogels with homogeneous networks, which can reduce the probability of initial “crack formation”.11,12 On the other hand, lowering the “crack propagation” in hydrogels may also help achieve a high strength. The double-network (DN) concept is an excellent and attractive strategy to prepare strong hydrogels by increasing the resistance of the network against “crack propagation”.6,10 The DN gel is generally fabricated via a two-step sequential cross-linking process. In the fabrication of a typical DN gel, a densely cross-linked and rigid network is first synthesized, which swells to equilibrium in a solution containing a crosslinker, an initiator, and a neutral monomer for the second network; and then polymerization of the neutral monomer takes place in the first network to generate the loosely cross-linked and flexible second network.6 The network structure of the DN gel and mechanisms of the mechanical enhancement have been extensively studied in recent years.13–16 The flexible polymer chains of the second network entangle with each other as well as with the rigid chains of the first network.13,15 The failure first occurs in the tightly cross-linked first network under stress, and the resulting cracks can be bridged by the second network which acts as not only an absorber of elastic energy but also a de facto molecular crack stopper to resist the crack propagation into a macroscopic scale.15 The formation and propagation of numerous cracks can help dissipate a considerable amount of fracture energy, and result in a high mechanical strength.10,16 A series of DN gels have been reported and demonstrated the applicability of the DN principle.17–20Based on the fracture mechanisms of DN gels, energy absorption and dissipation of the ductile second network plays a critical role in the drastic enhancement of the mechanical strength of DN gels. According to our previous study,21,22 the hydrogel fabricated with poly(ethylene glycol) 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 diacrylate (PEG20K-DA, Mn = 20000 g mol−1) could store and dissipate much more fracture energy in the compression process compared with the hydrogel synthesized from lower molecular weight poly(ethylene glycol) 4000 diacrylate (PEG4K-DA, Mn = 4000 g mol−1). In this study, double macromolecular network (DMMN) gels have been designed and fabricated, based on the DN principle, using a two-step photocrosslinking with methacrylated hyaluronan (HA–MA) for the first network and PEG20K-DA or PEG4K-DA for the second network. The influence of the molecular weight and initial solution concentration of poly(ethylene glycol) diacrylate (PEG-DA) as well as the initial concentration of the HA–MA solution on the mechanical strength of DMMN has been evaluated in detail. The homogeneities of DMMN hydrogels are examined with a dynamic light scattering technique.
000 diacrylate (PEG20K-DA, Mn = 20000 g mol−1) could store and dissipate much more fracture energy in the compression process compared with the hydrogel synthesized from lower molecular weight poly(ethylene glycol) 4000 diacrylate (PEG4K-DA, Mn = 4000 g mol−1). In this study, double macromolecular network (DMMN) gels have been designed and fabricated, based on the DN principle, using a two-step photocrosslinking with methacrylated hyaluronan (HA–MA) for the first network and PEG20K-DA or PEG4K-DA for the second network. The influence of the molecular weight and initial solution concentration of poly(ethylene glycol) diacrylate (PEG-DA) as well as the initial concentration of the HA–MA solution on the mechanical strength of DMMN has been evaluated in detail. The homogeneities of DMMN hydrogels are examined with a dynamic light scattering technique.
2 Materials and methods
2.1 Materials
Hyaluronic acid sodium (HA, Mn = 5 × 105 g mol−1) is purchased from Shengqiang Biotech Co., Ltd (Liuzhou, China) and used as received. Poly(ethylene glycol) (PEG) with a molecular weight of 4000 g mol−1 (PEG4K) and 20000 g mol−1 (PEG20K) and N,N-dimethylformamide (DMF) are purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Glycidyl methacrylate (GMA) is purchased from Sigma-Aldrich. A 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) photoinitiator is purchased from Ciba Specialty Chemicals. Acryloyl chloride (Aladdin Reagents Co., Ltd, Shanghai, China) is freshly distilled before use. Triethylamine (Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) is refluxed with phthalic anhydride for 12 hours, distilled and refluxed with calcium hydride for another 12 hours, and distilled before use. Toluene and tetrahydrofuran (THF) are refluxed with CaH2 and distilled before use.
2.2 Synthesis of precursors
PEGs, including PEG20K and PEG4K, are end-capped with acrylate groups to form polymerizable PEG-DA precursors according to our previous report.21 Briefly, 0.5 mmol of PEG is dissolved in 200 mL of anhydrous toluene at 140 °C and azeotropically distilled to remove trace amounts of water. After cooling to room temperature, 3.0 mmol of anhydrous triethylamine is added into the solution, and subsequently the mixture solution is placed in a low temperature bath (<0 °C) under an argon atmosphere for 30 minutes. 5 mL of THF containing acryloyl chloride (3.0 mmol) is added dropwise within one hour, followed by continuous stirring for 2 hours. The reaction solution is stirred in an oil bath at 45 °C overnight, filtered through diatomite, concentrated with rotary evaporation, precipitated dropwise in anhydrous diethyl ether under stirring, and dried under vacuum at room temperature. The resultant PEG-DA precursor is dialyzed in deionized (DI) water for three days, and freeze-dried.A hyaluronan–methacrylate conjugate (HA–MA) is synthesized by referring to the previous study after slight modification.23 Briefly, HA (1.0 g, 2.4 mmol) is dissolved in 250 mL of the phosphate-buffered saline (PBS, pH 7.4) solution, followed by the addition of 80 mL of DMF under vigorous stirring. GMA (0.1 mol) and triethylamine (0.05 mol) are added into the HA solution. The reaction mixture is stirred for five days at room temperature, and concentrated with rotary evaporation. The concentrated solution is dialyzed for three days in DI water, and then freeze-dried.
2.3 Fabrication of hydrogels
The PEG-DA or HA–MA precursor solution containing 0.05% (g mL−1) Irgacure 2959 is transferred to cylindrical molds (diameter 4 mm) using a pipette, and then exposed to 365 nm ultraviolet (UV) light (30 mW cm−2) for 5 minutes to obtain PEG or HA hydrogels. The hydrogels synthesized from 10% and 20% (g mL−1) of the PEG20K-DA solution are named as PEG10 and PEG20, respectively. The hydrogels fabricated from 2%, 2.8%, and 3.5% (g mL−1) of the HA–MA solution are named as the HA2.0, HA2.8, and HA3.5 gel, respectively.For the preparation of DMMN hydrogels, the HA gels, synthesized from 2%, 2.8%, or 3.5% (g mL−1) of the HA–MA solution, are immediately immersed into PEG20K-DA solutions (10%, 15%, or 20%, g mL−1) containing 0.05% (g mL−1) of Irgacure 2959. After 72 hours of immersion, the fully swelled HA gels are subject to crosslinking under UV light for 5 minutes after wiping off the surface solution to obtain DMMN gels. A series of DMMN gels are prepared with HA–MA and PEG20K-DA as parent precursors, and referred to as the DMMN-x–y gel (x and y stand for the initial percentage concentration of HA–MA and PEG20K-DA precursors, respectively). Besides, the DMMN-2-4K gel is fabricated from 2% (g mL−1) of the HA–MA solution and 20% (g mL−1) of the PEG4K-DA precursor solution.
2.4 1H NMR characterization
The 1H NMR spectra of PEG-DA and HA–MA precursors are recorded on a Mercury VX-300 spectrometer (Varian, USA) using D2O as a solvent, in which the relative integral intensities are used to calculate the acrylation of PEG-DA and methacrylation of HA–MA.2.5 Dynamic light scattering measurements
All dynamic light scattering (DLS) tests are carried out on an ALV/DLS/SLS-5000E light scattering goniometer (ALV/CGS-8F, ALV, Germany) with vertically polarized incident light (632.8 nm) from a He–Ne laser equipped with an ALV/LSE-5003 light scattering electronics unit and a multiple tau digital correlator. The measurements are conducted at a 90° angle, keeping the temperature at 25 °C.24,25 The samples of DLS measurements include PEG20K-DA solutions and hydrogels. PEG20K-DA solutions are directly filtered into the sample cells using a filter (PALL 4614), having a pore size of 0.45 μm. For the gels, the 20% (g mL−1) PEG20K-DA and 2% (g mL−1) HA–MA solutions containing Irgacure 2959 (0.05%, g mL−1) are added into the sample cells, respectively, by filtering through a 0.45 μm pore size filter and subjected to photopolymerization under 365 nm UV light at 30 mW cm−2 for 5 minutes. After the DLS measurements, 20% (g mL−1) PEG-DA solution containing Irgacure 2959 (0.05%, g mL−1) is filtered into the sample cells containing HA gels. After 96 hours of incubation, the system is exposed to UV light for 5 minutes to obtain DMMN gels for DLS measurements.2.6 Compression test
An unconfined compression test of fully swollen gels is carried out on a LLYOD material testing machine equipped with a 100 N or 10 kN force transducer. One cylindrical gel sample is placed on the lower compression plate, and subjected to compression at the rate of 1.0 mm min−1. The elastic modulus of the gel is calculated from the slope of the initial linear range of stress–strain curve according to the Hookean model.21 The value of fracture stress and fracture strain is obtained from the stress–strain curve at the fracture point. Fracture energy is defined as the integral area of the stress–strain curve till the fracture point.2.7 Swelling of the HA gel in PEG-DA solution
Freshly synthesized HA gels from 40 μL of the HA–MA solution are immersed in PEG-DA solution. At the predetermined time, four samples are collected and weighed (Ws0), respectively, after wiping off the surface solution, and then weighted (Wd0) after drying at 50 °C under vacuum for three days. The polymer concentration in HA gels is calculated using the following equation:| Concentration (%) = Wd0/(Ws0 − Wd0) × 100% |
2.8 Mass ratio of DMMN hydrogels
HA gels, synthesized from 40 μL of the HA–MA solution, and corresponding DMMN gels are prepared. These gels are placed in DI water at ambient temperature for three days to reach equilibrium swelling, and dried for three days under vacuum at 50 °C. The dry weights of the HA gel (WHA) and the corresponding DMMN gel (WDMMN) are measured using an electronic balance. The mass ratio of the second PEG network to the first HA network is estimated as follows:| Mass ratio = (WDMMN − WHA)/WHA |
2.9 Water content
Freshly prepared gels are placed in DI water at room temperature, where the water is changed every day. After three days of immersion, the samples are weighed (Ws) after wiping off the surface water, and then dried at 50 °C under vacuum for three days. The dry samples are weighted (Wd). The water content of the gel is calculated using the following equation:| Water content (%) = (Ws − Wd)/Ws × 100% |
2.10 Statistical analysis
The results are presented as mean ± standard deviation with at least three samples. Student's t-test is used to estimate statistical significance (p ≤ 0.05) between two groups.3 Results and discussion
HA is one major component of the extracellular matrix of skin, cartilage, and the vitreous humor. It is a linear, negatively charged, high molecular weight mucopolysaccharide, and an important biopolymer to synthesize hydrogels for biomedical applications.26,27 HA–MA is synthesized by grafting glycidyl methacrylate onto HA, which is used as a precursor for the first network of DMMN gels. Biocompatible PEG4K-DA and PEG20K-DA, instead of a small molecular monomer commonly used in the preparation of traditional DN gels,6 are selected as precursors for the second network. The typical 1H-NMR spectra of HA–MA and PEG20K-DA are shown in Fig. S1 (ESI†).DMMN gels are fabricated by a two-step photocrosslinking. As shown in Fig. 1A, HA gels are synthesized from UV-light induced gelation of HA–MA solutions (Fig. 1A-a), and then immersed in PEG-DA solutions (Fig. 1A-b). HA gels swell gradually (Fig. 1B), at the same time, PEG-DA precursors and photoinitiators diffuse into HA gels (Fig. 1A-c). The fully swelled HA gels are taken out and exposed to UV light for the second crosslinking to obtain DMMN gels (Fig. 1A-d).
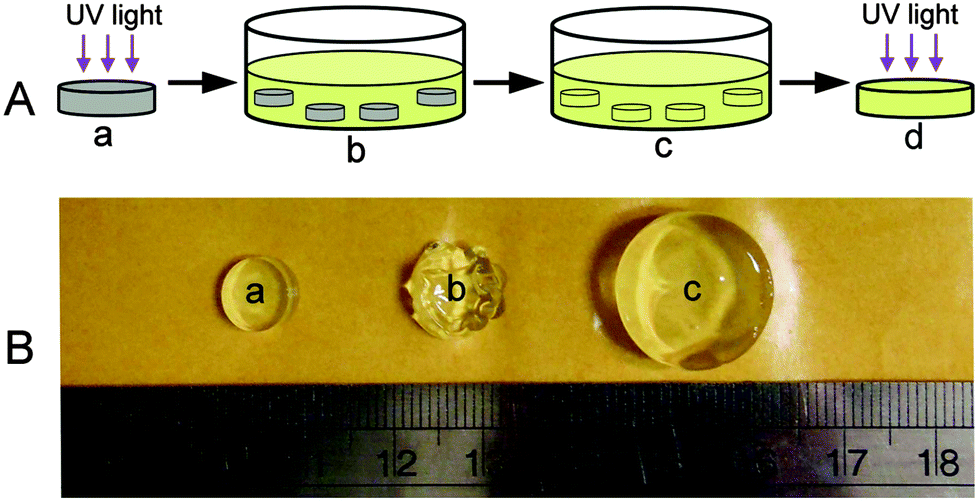 | ||
| Fig. 1 (A) Fabrication of the DMMN gel: (a) preparation of the HA gel by UV light induced crosslinking of HA–MA solution, (b) immersion of HA gels in PEG-DA solution, (c) reaching equilibrium swelling of HA gels, and (d) exposure to UV light for the second crosslinking to obtain the DMMN gel. (B) Images of the HA2.0 gel during swelling in 20% (g mL−1) of PEG20K-DA solution: (a) freshly synthesized HA2.0 gel, (b) after 2 hours of immersion, and (c) after three days of immersion. | ||
The diffusion process of the second precursor into the first network is crucial to determine the polymer content and mass ratio of the second and the first networks that have a great influence on the mechanical properties of the resultant DN gels.10 In particular, the macromolecular PEG-DA is different from the small monomer that is usually used to fabricate DN gels.6 For example, flexible PEG-DA precursors mainly exist in the form of chain aggregates in (semi-)concentrated solution and have a lower diffusion rate than the small monomer.28,35 Therefore, the diffusion of PEG20K-DA into the HA2.0 gel is typically monitored as a function of immersion time. As shown in Fig. 2, the polymer concentration in the HA2.0 gel rapidly increases from about 2% to 24% (w/w) within the first hour of immersion, which can be attributed to the de-swelling of HA2.0 gels due to the osmotic pressure caused by a great difference in polymer concentrations between the inside and outside of the HA2.0 gel. The similar phenomenon is also reported by Khademhosseini et al.29 After five hours of immersion, the polymer concentration in the HA2.0 gel reaches up to 31.5 ± 1.0% (w/w), whereas, the increase in the rate (slope of the curve) from the first to the 5th hour becomes smaller than that within the first hour. The polymer concentration in the HA2.0 gel decreases from the 5th to the 24th hour, which indicates that the re-swelling of the HA2.0 gel occurs. After 72 hours of incubation, the polymer concentration in the HA2.0 gel stabilizes at 22 ± 0.5% (w/w), which suggests that the concentration of the PEG20K-DA precursor reaches equilibrium between the inside and outside of the HA2.0 gel. This result demonstrates that three day immersion is enough for the diffusion of the PEG20K-DA precursor into HA gels.
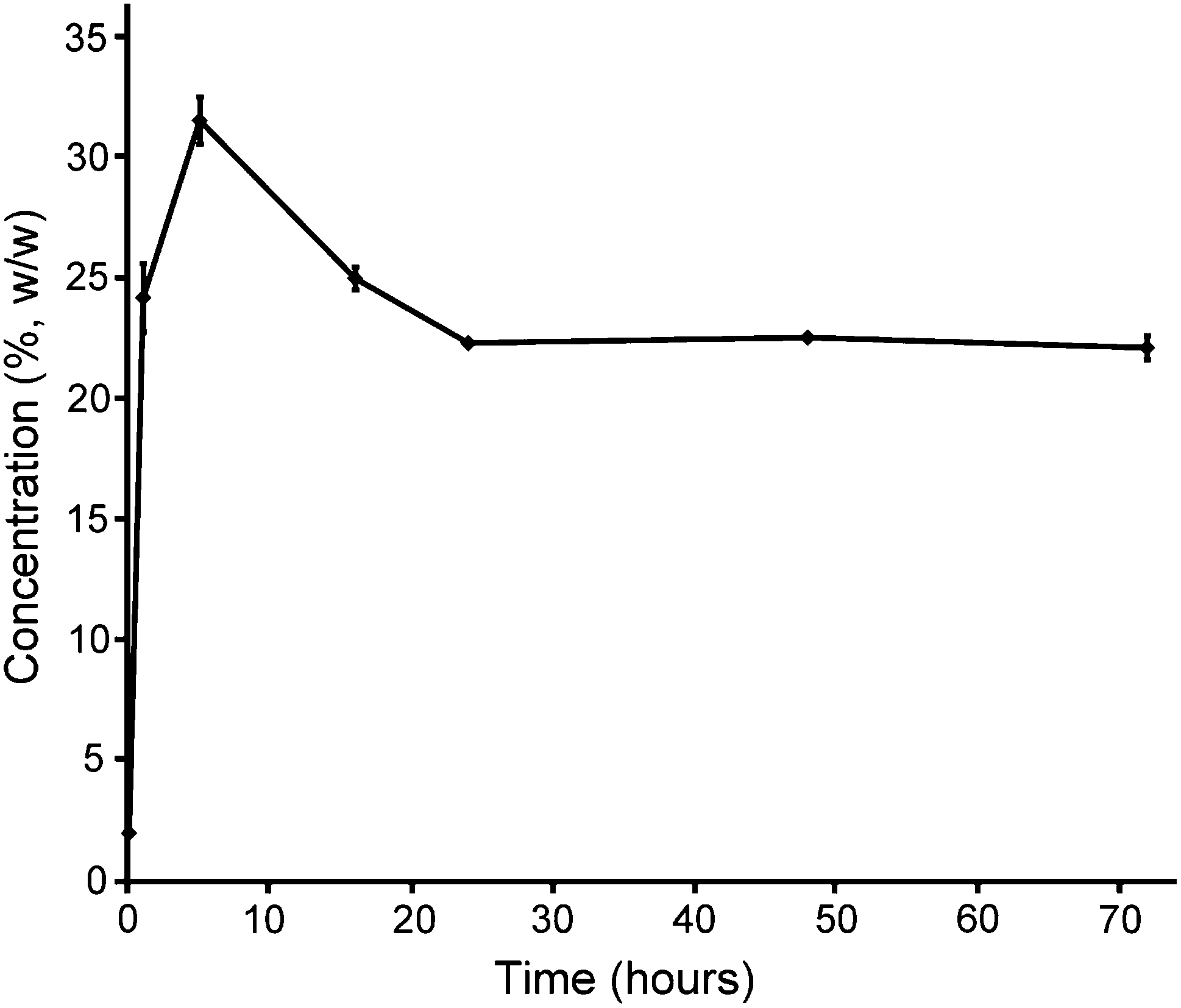 | ||
| Fig. 2 Polymer concentration in the HA2.0 gel as a function of immersion time in 20% (g mL−1) PEG20K-DA solution. | ||
A series of hydrogels, including PEG gels, HA gels, and DMMN gels, are fabricated, and the compression tests are performed to study their mechanical properties with varied concentrations or molecular weights of precursors (Table 1). Interestingly, the PEG10 gel is stronger than the PEG20 gel; the fracture stress and fracture energy of the PEG10 gel are significantly higher than those of the PEG20 gel. This result does not accord with the classical Lake–Thomas theory that is followed by HA gels whose fracture stress and fracture energy are increased from the HA2.0 to HA2.8 and HA3.5 gel.30 Actually, we have observed a similar phenomenon in our previous study,21 and the unusual result should be attributed to the PEG network structures that are derived from the precursor solutions. Dynamic light scattering (DLS) measurements are performed to investigate the chain structures in PEG20K-DA solutions. As shown in Fig. 3A, in a dilute PEG20K-DA solution (0.5%, g mL−1), major PEG20K-DA precursors exist as a single chain, and their hydrodynamic radii (4.2 ± 0.6 nm) are close to the theoretical value (3.3 nm).31 In a (semi-)concentrated solution, more flexible PEG20K-DA chains entangle with each other to form chain aggregates. The hydrodynamic radius of PEG20K-DA chain aggregates increases from 58 ± 20 nm to 380 ± 97 nm with the increase of the concentration of the PEG20K-DA solution from 0.5% to 10% (g mL−1). When the concentration of the PEG20K-DA solution is 20% (g mL−1), the PEG20K-DA single chain disappears, and the hydrodynamic radius of chain aggregates reaches up to 1018 ± 124 nm. The increase in the size of chain aggregates from 10% to 20% (g mL−1) of PEG20K-DA solutions leads to a significant increase in spatial inhomogeneities that is directly demonstrated by a 5-fold increase in ensemble average scattering intensity (〈I〉E) (Fig. 3B).11,32 The spatial chain inhomogeneities can be frozen during the cross-linking process.34,35 Therefore, the networks of the PEG20 gel are more inhomogeneous than those of the PEG10 gel, typically including more network defects between chain aggregates and unevenly distributed cross-linking points as well as more inhomogeneous polyacrylate kinetic chains. The more inhomogeneities of PEG20 gels than PEG10 gels can cause severe stress concentration under stress,11 resulting in lower fracture strain, fracture stress, and fracture energy (Table 1).
| Samples | Fracture stress (MPa) | Fracture strain (%) | Fracture energy (kJ m−3) | Mass ratio | Water content (%) |
|---|---|---|---|---|---|
| PEG10 | 5.78 ± 0.85 | 97.4 ± 1.2 | 275 ± 48 | — | 97.4 ± 0.2 |
| PEG20 | 1.14 ± 0.37 | 86.3 ± 1.5 | 101 ± 30 | — | 95.7 ± 0.1 |
| HA2.0 | 0.02 ± 0.01 | 45.6 ± 6.6 | 1.6 ± 0.4 | — | 99.9 ± 0.0 |
| HA2.8 | 0.07 ± 0.02 | 38.5 ± 1.4 | 5.1 ± 1.1 | — | 99.7 ± 0.1 |
| HA3.5 | 0.11 ± 0.01 | 39.9 ± 0.9 | 8.2 ± 0.4 | — | 99.5 ± 0.1 |
| DMMN-2-10 | 17.7 ± 3.9 | 95.8 ± 1.2 | 678 ± 128 | 28.6 ± 3.7 | 97.4 ± 0.2 |
| DMMN-2-15 | 30.5 ± 6.3 | 95.6 ± 2.4 | 1277 ± 110 | 40.7 ± 1.4 | 96.6 ± 0.1 |
| DMMN-2-20 | 50.1 ± 4.4 | 94.6 ± 2.9 | 2374 ± 372 | 46.5 ± 2.0 | 95.9 ± 0.1 |
| DMMN-2.8-20 | 8.22 ± 3.7 | 88.4 ± 1.6 | 475 ± 119 | 26.3 ± 4.0 | 94.3 ± 0.7 |
| DMMN-3.5-20 | 1.03 ± 0.16 | 76.2 ± 1.5 | 215 ± 42 | 14.5 ± 0.7 | 92.2 ± 0.2 |
| DMMN-2-4K | 2.73 ± 0.77 | 66.6 ± 0.9 | 323 ± 62 | 183.9 ± 11.9 | 88.7 ± 0.6 |
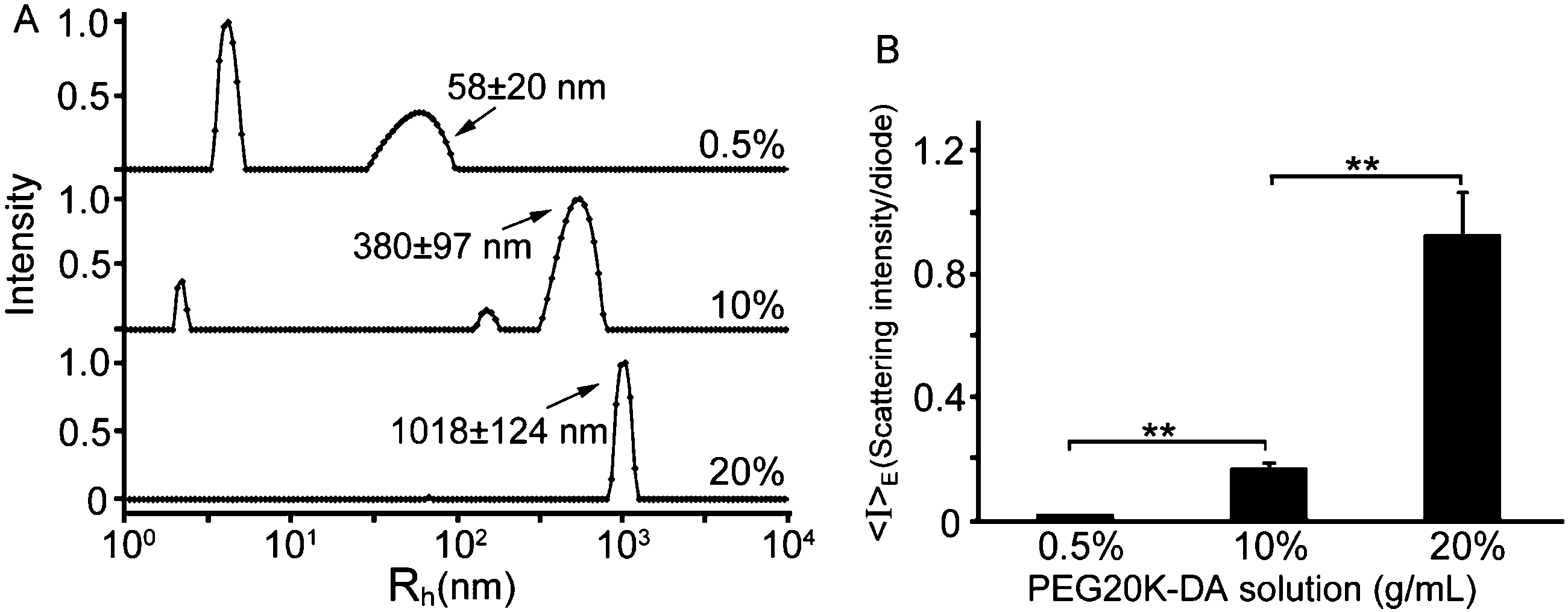 | ||
| Fig. 3 (A) Hydrodynamic radius distribution of PEG20K-DA chain aggregates in 0.5%, 10%, and 20% (g mL−1) of PEG20K-DA solution. (B) The ensemble average scattering intensity (〈I〉E) of 0.5%, 10%, and 20% (g mL−1) PEG20K-DA solution. Statistical significance is indicated with **(p ≤ 0.01). | ||
However, the fracture stress and fracture energy of the DMMN gels, fabricated with 2% (g mL−1) of the HA–MA solution for the first network, gradually increase with the increase of PEG20K-DA concentration from 10% to 15% and 20% (g mL−1); at the same time, their fracture strains are comparable (Table 1). In other words, the second PEG20K networks in the DMMN-2-20 gel are capable of absorbing and storing more fracture energy than those in DMMN-2-15 and DMMN-2-10 gels under stress. Surprisingly, this result does not agree with aforementioned PEG10 and PEG20 gels.
The elastic modulus of the HA2.0 gel is too small to be accurately measured; the elastic modulus of the DMMN-2-20 gel (41.3 ± 3.2 kPa) is not significantly different from that of the PEG20 gel (39.9 ± 9.5 kPa). The comparable elastic modulus indicates that the PEG20K networks both in the DMMN-2-20 gel and the PEG20 gel sustain the stress, but the fracture strain of the DMMN-2-20 gel (94.6 ± 2.9%) is significantly larger than that of the PEG20 gel (86.3 ± 1.5%). The homogeneity of the HA2.0 gel (the first network for DMMN-2-20 gel synthesis), DMMN-2-20 gel, and PEG20 gel is studied using DLS tests. As shown in Fig. 4b, compared with the PEG20 gel, the HA2.0 gel shows much lower average scattering intensity (〈I〉E), and as expected, 〈I〉E of the DMMN-2-20 gel is significantly lower than that of the PEG20 gel. These data demonstrate that the DMMN-2-20 gel, including PEG20K networks and HA networks, is composed of more evenly distributed polymer chains. The fracture patterns of the PEG20 gel and DMMN-2-20 gel are carefully observed and compared (Fig. 4a). The fracture of the PEG20 gel occurs in the middle and produces big pieces of fragments. This fracture pattern can be attributed to the evident stress concentration caused by network inhomogeneities,32,33 just like the above-mentioned unevenly distributed cross-linking points and inhomogeneous polyacrylate kinetic chains, as well as network defects. However, the entire DMMN-2-20 gel breaks into tiny pieces, suggesting that the load is evenly applied on the gel networks. These phenomena again demonstrate the homogeneity of the DMMN-2-20 gel. Therefore, the more evenly distributed PEG20K networks in the DMMN-2-20 gel, containing more PEG20K chains, can absorb and store more energy than those in the DMMN-2-15 gel and DMMN-2-10 gel, when they suffer the same compression deformation (namely strain).21 Compared with the inhomogeneous PEG20 gel, the more evenly distributed PEG20K networks in the DMMN-2-20 gel may have resulted from the disaggregation of PEG20K-DA precursors from chain aggregates in the diffusion process into the HA2.0 gel.35–37Fig. 5 illustrates the proposed formation mechanisms of the PEG gel and DMMN gel. The flexible PEG20K-DA precursors take random coil conformation and entangle with each other to form chain aggregates, which are frozen during the cross-linking process and results in an inhomogeneous PEG gel (Route 1).40,41 In Route 2, the HA2.0 gel serves as a template for the diffusion of PEG20K-DA precursors. PEG20K-DA chains dissociate them from the large chain aggregates in solution and diffuse into the HA2.0 gel, and then DMMN gels with more evenly distributed polymer networks are formed after second cross-linking.35–37
 | ||
| Fig. 4 (a) Images of PEG20 and DMMN-2-20 gels after compression failure. (b) 〈I〉E of HA2.0, PEG20, and DMMN-2-20 gels; schematic diagrams of PEG20 and DMMN-2-20 gel networks. Statistical significance is indicated with **(p ≤ 0.01). | ||
 | ||
| Fig. 5 Schematic fabrication process of the PEG gel (Route 1) and DMMN gel (Route 2). SIPN stands for semi-interpenetrating networks. | ||
The obtained DMMN gels showed excellent mechanical properties, especially for the DMMN-2-20 gel. As shown in Fig. 6a–c, the DMMN-2-20 gel possesses excellent anti-compression ability in spite of its high water content (95.9 ± 0.1%, Table 1). It achieves an outstanding compressive fracture stress of 50.1 ± 4.4 MPa, which is about 2500 times and 44 times higher than that of the HA2.0 gel (0.02 ± 0.01 MPa) and PEG20 gel (1.14 ± 0.37 MPa), respectively. The fracture energy of the DMMN-2-20 gel (2374 ± 372 kJ m−3) is also obviously much higher than those of the HA2.0 gel (1.6 ± 0.4 kJ m−3) and PEG20 gel (101 ± 30 kJ m−3). The superior mechanical strength and large recoverable deformation are vividly demonstrated by loading a person's body weight (approx. 74 kg, i.e., 163 pounds) on the DMMN-2-20 gel (diameter 12.5 mm, height 10.8 mm) (Video, ESI†). The gel can fully recover from the compressed state upon removal of load and retain its intactness and resilience, exhibiting rubber-like behaviors. The cyclic loading experiments are carried out to explore energy dissipation of DMMN gels. As shown in Fig. 6d, the DMMN-2-20 gel dissipates energy effectively, as demonstrated by the large hysteresis loop in the first loading–unloading curve. The pronounced hysteresis is also observed in the first loading–unloading cycle of the PEG20 gel (Fig. 6e), suggesting that the second PEG network in DMMN gels can effectively dissipate energy under stress. This result indicates that the PEG network as the second network is significantly different from the widely used polyacrylamide (PAAm) network that shows negligible hysteresis in the loading–unloading cycle.6,20 Immediately second loading–unloading cycles of the PEG20 gel and DMMN-2-20 gel are conducted. The hysteresis loops for both gels become much smaller at the second loading–unloading cycles. These results demonstrate that the energy dissipation of DMMN gels under stress might mainly be attributed to the rearrangement of PEG20K chains of the second network, such as coil-stretch conformation transition.21,38
 | ||
| Fig. 6 (a) The PEG20 gel is easily sliced with a scalpel, while (b) the DMMN-2-20 gel can resist with a strain up to 60%. (c) Representative stress–strain curves of HA2.0, PEG20, and DMMN-2-20 gels. (d) Loading–unloading cycles of the DMMN-2-20 gel. (e) Loading–unloading cycles of the PEG20 gel. | ||
To further characterize the DMMN gel system, the influence of the molecular weight of the PEG-DA precursor and the concentration of the HA–MA solution on the mechanical properties of DMMN gels are evaluated, respectively (Table 1). The DMMN-2-4K gel is fabricated with the same 2% (g mL−1) HA–MA solution as the first network and 20% (g mL−1) PEG4K-DA solution as the second network. However, the fracture stress of the DMMN-2-4K gel (2.73 ± 0.77 MPa) are significantly lower than that of the DMMN-2-20 gel. Weng et al. report a series of HA/DAAm DN gels, using 2% (g mL−1) HA gel as the first network and the small monomer N,N-dimethylacrylamide (DAAm) as the second network, of which the maximum fracture stress is 5.2 MPa.39 Compared with the DMMN-2-4K gel and HA/DAAm DN gel, we can conclude that the high molecular weight of the PEG-DA precursor plays an important role in generating high-strength double-network gels. The PEG20K networks in the DMMN-2-20 gel have lower crosslink density than PEG4K networks in the DMMN-2-4K gel due to the higher molecular weight;21 the lower crosslink density is beneficial to the extension of PEG20K chains under stress.21 Besides, the chain length of PEG20K networks in the DMMN-2-20 gel is higher than that of PEG4K networks in the DMMN-2-4K gel. Therefore, the DMMN-2-20 gel can achieve a higher strain and absorb more fracture energy than the DMMN-2-4K gel. When keeping the concentration of PEG20K-DA constant at 20% (g mL−1), the increased concentration of the HA–MA solution from 2% to 2.8% and 3.5% (g mL−1) makes the fracture stress of the DMMN gels drop greatly from 50.1 ± 4.4 to 8.22 ± 3.7 and 1.03 ± 0.16 MPa. The above results suggest that higher molecular weight of PEG-DA precursors (for the second network) and lower initial concentration of the HA–MA precursor solution (for the first network) are beneficial for producing high-strength DMMN gels.
The mass ratio of the second network to the first network is regarded as the crucial structure parameter for preparing robust DN gels.10 As shown in Fig. 7, similar to previously reported DN gels,6 the fracture stress of the DMMN gels increases with the increase of the mass ratio of the second to the first network, except for those of DMMN-2-10 and DMMN-2.8-20 gels. They exhibit a similar mass ratio of PEG to HA network (Table 1 and Fig. 7), however, the fracture stress and strain of the DMMN-2–10 gel (17.7 ± 3.9 MPa and 95.8 ± 1.2%) is significantly higher than those of the DMMN-2.8-20 gel (8.22 ± 3.7 MPa and 88.4 ± 1.6%). Furthermore, though the mass ratio of DMMN-2-4K is up to 183.9 ± 11.9, its fracture stress and fracture strain is only 2.73 ± 0.77 and 66.6 ± 0.9%, respectively. This result further demonstrates that the relative lower concentration of the HA–MA precursor solution as well as the higher molecular weight of the PEG-DA precursor is crucial for preparing high-strength DMMN gels. Cytotoxicity of DN gels is another major concern for biomedical applications,10 and the DMMN gels exhibit excellent cytocompatibility due to the widely recognized materials (namely PEG and HA) as well as the well established photo-crosslinking method (ESI,† Fig. S2).
 | ||
| Fig. 7 Mass ratios of the PEG20K network to HA network in DMMN gels. The statistical significance is indicated with **(p ≤ 0.01). | ||
4 Conclusions
In this study, strong and resilient DMMN gels that possess DN and a more evenly distributed polymer network structure have been developed with biocompatible HA–MA and PEG20K-DA precursors for the first network and second network, respectively. A relatively loose HA network, synthesized from lower concentration of the HA–MA solution, and a relatively tight PEG20K network, synthesized from higher concentration of the PEG20K-DA solution, help enhance the mechanical properties of resultant DMMN gels. This novel DMMN gel system almost integrates the qualities of the homogeneous gel and DN gel, and thus it shows higher mechanical strength and resilience. This study represents a protocol to prepare robust and biocompatible gels that may expand the biomedical applications of hydrogels.Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (Grant No. 20904042) and Natural Science Foundation of Jiangsu Province, China (Grant No. BK20131187).References
- P. Calvert, Adv. Mater., 2009, 21, 743 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- L. Tang, W. Liu and G. Liu, Adv. Mater., 2010, 22, 2652 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS PubMed.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z. F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325 CrossRef CAS PubMed.
- H. Zhang, D. Zhai and Y. He, RSC Adv., 2014, 4, 44600 RSC.
- J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379 CrossRef CAS.
- T. Sakai, Y. Akagi, T. Matsunaga, M. Kurakazu, U. Chung and M. Shibayama, Macromol. Rapid Commun., 2010, 31, 1954 CrossRef CAS PubMed.
- Y. H. Na, T. Kurokawa, Y. Katsuyama, H. Tsukeshiba, J. P. Gong, Y. Osada, S. Okabe, T. Karino and M. Shibayama, Macromolecules, 2004, 37, 5370 CrossRef CAS.
- Y. Tanaka, R. Kuwabara, Y. H. Na, T. Kurokawa, J. P. Gong and Y. Osada, J. Phys. Chem. B, 2005, 109, 11559 CrossRef CAS PubMed.
- H. Tsukeshiba, M. Huang, Y. H. Na, T. Kurokawa, R. Kuwabara, Y. Tanaka, H. Furukawa, Y. Osada and J. P. Gong, J. Phys. Chem. B, 2005, 109, 16304 CrossRef CAS PubMed.
- M. Huang, H. Furukawa, Y. Tanaka, T. Nakajima, Y. Osada and J. P. Gong, Macromolecules, 2007, 40, 6658 CrossRef CAS.
- X. Wang, H. Wang and H. R. Brown, Soft Matter, 2011, 7, 211 RSC.
- W. Yang, H. Furukawa and J. P. Gong, Adv. Mater., 2008, 20, 4499 CrossRef CAS PubMed.
- M. A. Haque, G. Kamita, T. Kurokawa, K. Tsujii and J. P. Gong, Adv. Mater., 2010, 22, 5110 CrossRef CAS PubMed.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171 CrossRef CAS PubMed.
- C. J. Fan, C. Zhang, Y. Jing, L. Q. Liao and L. J. Liu, RSC Adv., 2013, 3, 157 RSC.
- C. J. Fan, L. Q. Liao, C. Zhang and L. J. Liu, J. Mater. Chem. B, 2013, 1, 4251 RSC.
- C. J. Fan, J. Tu, X. Yang, L. Q. Liao and L. J. Liu, Carbohydr. Polym., 2011, 86, 1484 CrossRef CAS PubMed.
- L. Z. Zhang, C. C. Zhao, J. P. Zhou and T. Kondo, J. Mater. Chem. C, 2013, 1, 5756 RSC.
- Y. Zhang, S. Li and L. Zhang, J. Phys. Chem. B, 2010, 114, 4945 CrossRef CAS PubMed.
- M. Y. Lee, W. H. Kong, H. S. Jung and S. K. Hahn, RSC Adv., 2014, 4, 19338 RSC.
- T. C. Lin, J. H. Chen, Y. H. Chen, T. M. Teng, C. H. Su and S. H. Hsu, J. Mater. Chem. B, 2013, 1, 5977 RSC.
- K. L. Linegar, A. E. Adeniran, A. F. Kostko and M. A. Anisimov, Colloid J., 2010, 72, 279 CrossRef CAS.
- H. Shin, B. D. Olsen and A. Khademhosseini, Biomaterials, 2012, 33, 3143 CrossRef CAS PubMed.
- G. J. Lake and A. G. Thomas, Proc. R. Soc. London, 1967, 300, 108 CrossRef CAS.
- M. Schmidt and W. Burchard, Macromolecules, 1981, 14, 210 CrossRef CAS.
- M. Shibayama, Macromol. Chem. Phys., 1998, 199, l CrossRef.
- V. Nesetova and E. Z. Lajtai, Int. J. Rock Mech. Min. Sci., 1973, 10, 265 CrossRef.
- T. V. Vliet and P. Walstra, Faraday Discuss., 1995, 101, 359 RSC.
- P. G. De Gennes, Macromolecules, 1976, 9, 587 CrossRef CAS; P. G. De Gennes, Macromolecules, 1976, 9, 594 CrossRef.
- W. Hess, Macromolecules, 1986, 19, 1395 CrossRef CAS.
- T. T. Perkins, D. E. Smith and S. Chu, Science, 1994, 264, 819 CAS.
- S. S. Jang, W. A. Goddard and M. Y. S. Kalani, J. Phys. Chem. B, 2007, 111, 1729 CrossRef CAS PubMed.
- L. Weng, A. Gouldstone, Y. Wu and W. Chen, Biomaterials, 2008, 29, 2153 CrossRef CAS PubMed.
- E. S. Matsuo, M. Orkisz, S. T. Sun, Y. Li and T. Tanaka, Macromolecules, 1994, 27, 6791 CrossRef CAS.
- S. L. Gibson, R. L. Jones, N. R. Washburn and F. Horkay, Macromolecules, 2005, 38, 2897 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00867k |
| This journal is © The Royal Society of Chemistry 2015 |