
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigher and lower supramolecular orders for the design of self-assembled heterochiral tripeptide hydrogel biomaterials†
S.
Marchesan
*ab,
K. E.
Styan
b,
C. D.
Easton
b,
L.
Waddington
b and
A. V.
Vargiu
c
aCenter of Excellence for Nanostructured Materials, Chemical and Pharmaceutical Sciences Department, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: smarchesan@units.it
bCSIRO Manufacturing Flagship, Clayton, VIC 3168, Australia
cDepartment of Physics, University of Cagliari, Cittadella Universitaria, S.P. Monserrato-Sestu Km. 0.700, 09042 Monserrato (CA), Italy
First published on 31st August 2015
Abstract
The self-assembly behaviour of the eight stereoisomers of Val–Phe–Phe tripeptides under physiological conditions is assessed by several spectroscopy and microscopy techniques. We report the first examples of self-organised hydrogels from tripeptides in the L–D–L or D–L–D configuration, besides the expected gels with the D–L–L or L–D–D configuration, thus widening the scope for using amino acid chirality as a tool to drive self-assembly. Importantly, the positions of D- and L-amino acids in the gelling tripeptides determine a higher or lower supramolecular order, which translates into macroscopic gels with different rheological properties and thermal behaviours. The more durable hydrogels perform well in cytotoxicity assays, and also as peptides in solution. An appropriate design of the chirality of self-assembling sequences thus allows for the fine-tuning of the properties of the gel biomaterials. In conclusion, this study adds key details of supramolecular organization that will assist in the ex novo design of assembling chiral small molecules for their use as biomaterials.
Introduction
Nanomaterials are revolutionising the medicinal field through the addition of novel properties and the introduction of new therapeutic and diagnostic concepts.1,2 Self-assembling peptides have been rising as powerful tools in nanomedicine, not just through the formation of adaptive and biodegradable functional nanomaterials,3 but also through the creation of novel therapeutic paradigms.4 It is thus not surprising that the majority of self-assembling peptide systems have been patent-protected, with some constituting the core technology of spin-off companies.5Among the numerous peptide-based systems, low-molecular-weight gelators employing synthetic end-capping groups (e.g., naphthalene, fluorenylmethyloxycarbonyl, anthracene, etc.) play a prominent role thanks to their low-cost and ease of preparation, relative to longer peptides or more elaborate peptide derivatives.6–11 There is increasing interest in self-assembling unprotected tripeptides due to their use in medicine, since they are devoid of synthetic moieties that raise concerns over their fate and toxicity in vivo.12 However, the identification of novel self-assembling tripeptide motifs is still a holy grail in the field. In 2015, massive computational efforts towards the screening of all 8000 amino acid combinations has led to just a few hits.13 Clearly, there are unsolved complexities in the supramolecular behaviour of such simple and flexible small molecules that require further investigation.
We recently reported the use of amino acid chirality as a novel means to obtain hydrogels, under physiological conditions, from hydrophobic L-sequences that do not gel.14 In particular, we have shown that introduction of D-amino acids at the N-termini of tripeptide sequences bearing the Phe–Phe motif allows for the removal of steric clashes between amino acid side chains, leading to the interdigitation of tripeptide stacks into zippers.15 An analogous supramolecular behaviour is expected for enantiomers, i.e., displaying amino acids in the L–D–D configuration.
When we screened the first complete series of tripeptide stereoisomers for hydrogelation at neutral pH, indeed we identified D–L–L and L–D–D gelling peptides, thus confirming the special role played by the N-terminus.16 Other combinations of D- and L-amino acids in heterochiral tripeptides revealed some tendency towards self-assembly, although only isolated nanostructures were occasionally observed. By contrast, the homochiral analogues did not display self-organisation. While this study anticipated that other heterochiral tripeptides may self-assemble, it did not provide sufficient molecular insights into the supramolecular arrangements that make D–L–L and L–D–D tripeptides more successful in their self-organisation. Widening the scope of our methodology would be extremely useful within the supramolecular community, as other groups are adopting the approach to derive nanostructures and nanostructured hydrogels.17
In addition, the use of D-amino acids is particularly advantageous in medicine, not just to obtain biomaterials of higher enzymatic stability, but also for therapeutic effects.18 For instance, selected heterochiral self-assembling peptides display antimicrobial activity,19,20 and the introduction of D-amino acids has led to improved activity and selectivity of antimicrobial peptides.21 Short D-peptides bearing the Phe–Phe motif have also been successful binders of the amyloid beta peptide, thus reducing its cytotoxicity, and showing potential for the treatment of amyloidosis.22 Remarkably, covalent attachment of Phe–Phe D-peptides (but not L-peptides) to the anti-inflammatory drug naproxen has been shown to increase the COX-2 selectivity resulting in reduced side effects, thus providing a useful and innovative method for drug delivery.23 Therefore, we anticipate that gaining insights into the supramolecular behaviour of heterochiral peptides will be relevant not only for the biomaterials community, but also to understand the mechanisms of their unexpected therapeutic effects.
Experimental
Peptide synthesis and characterisation
Tripeptides were synthesized and purified using the standard Fmoc solid phase peptide synthesis with HBTU activation, following the same procedure described previously, employing reverse-phase HPLC (Agilent Technologies).14 The HPLC system was equipped with a preparative gradient pump (1100/1200), a preparative C-18 column (Luna, 10 μm, 100 Å, 150 × 21.20 mm, Phenomenex), an autosampler (G2260), and a diode array detector (G1365D). The gradient used consisted of acetonitrile (AcN)–water with 0.1% TFA using the following program (10 ml min−1 flow): t = 0–3 min, 25% AcN; t = 15 min, 55% AcN; t = 16–20 min, 95% AcN. The compounds were freeze-dried; their purity was verified by HPLC using the same equipment and gradient as above, but on an analytical C-18 column (Luna, 5 μm, 100 Å, 150 × 4.60 mm, Phenomenex; gradient 5–95% AcN over 15 min; 1 ml min−1 flow; tR = 9–11 min, see ESI†). The peptide identity was confirmed by ES-MS, 1H-NMR and 13C-NMR (see ESI†).Peptide gelation
Typically, 4.2 mg of peptide was added to 300 μl of 0.1 M sodium phosphate buffer (pH 11.8) and dissolved with the aid of sonication for 5 min in a water bath at room temperature, then diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with another 300 μl of 0.1 M sodium phosphate buffer (pH 5.6–5.7) to yield a final pH of 7.4. Self-assembling tripeptides formed hydrogels within minutes. All buffer solutions were filtered (0.2 μm) prior to use.
:1 with another 300 μl of 0.1 M sodium phosphate buffer (pH 5.6–5.7) to yield a final pH of 7.4. Self-assembling tripeptides formed hydrogels within minutes. All buffer solutions were filtered (0.2 μm) prior to use.
Rheometry
Dynamic time sweep rheological analysis was conducted on an Ares rheometer (TA Instruments, USA) with a 25 mm aluminium parallel plate geometry. A Peltier temperature controller was connected to the rheometer to maintain a temperature of 25 °C. The peptide samples were freshly prepared by mixing the two precursor solutions directly on the rheometer to register early stages of gelation. The samples were immediately analysed with a gap of 300 μm, a strain of 0.3% and a frequency of 10 rad s−1. After 60 min gelation, strain sweeps were recorded at a frequency of 10 rad s−1 and frequency sweeps at a strain of 0.3%. Data acquisition was repeated in triplicates.Differential scanning calorimetry (DSC)
DSC data were collected on a Q100 calorimeter (TA Instruments). The hydrogel samples were prepared directly in the DSC pans by dissolving the peptide in the alkaline buffer (1.3 mg in 100 μl), then transferring 10 μl of the peptide solution in the DSC pan, followed by another 10 μl of the second buffer. Pans were closed with their lids, and measurements started after 15 min at room temperature. DSC scans started with an isotherm at 20 °C for 5 min, followed by a 5 °C min−1 ramp up to 200 °C (i.e., below peptide decomposition temperature). Measurements were repeated at least in triplicates.Fourier-transformed infrared spectroscopy (FT-IR)
FT-IR spectra were collected on a Nicolet 6700 FT-IR spectrometer in ATR mode. A portion of the gel was transferred onto a clean piece of silicon wafer (1 cm × 1 cm), and then gently spread over the surface by pressing a coverslip on top; the coverslip was immediately removed and samples were dried under vacuum for 24 h. Dried samples on the silicon wafers were placed directly onto the ATR crystal. Scans were between 1800 and 1500 cm−1 with 80 accumulations at a resolution of 0.4 cm−1.Circular dichroism (CD) spectroscopy
The secondary structure of peptides was analysed using a 0.1 cm quartz cell on a Jasco J815 Spectropolarimeter, with 1 s integrations and a step size of 1 nm with a bandwidth of 1 nm over a range of wavelengths from 200 to 280 nm. Peptide samples were freshly prepared by mixing the two precursor solutions directly in the CD cell. Spectra were recorded after 15 min. Measurements were repeated at least 5 times, and to reduce the noise near 200 nm, their average was plotted.Thioflavin T (ThT) confocal fluorescence microscopy
Gel precursor solutions were prepared as indicated above and 12.5 μl of each were immediately placed on wells of a “μ-Slide Angiogenesis” uncoated (Ibidi, Germany, through DKSH Australia). 25 μl of a solution of ThT (200 μM in 50 mM glycine–NaOH, pH 7.5, 0.2 μm-filtered) were placed on top. After 15 min, the slides were imaged using a Leica SP5 microscope (63× water immersion objective, NA 1.2, ex. 458 nm, em. 468–600 nm). The samples treated and stained using an identical protocol but without the peptide were used as controls and did not reveal any of the fluorescent structures described in the text (data not shown).Cryo-transmission electron microscopy (cryo-TEM)
A locally built vitrification system was used to prepare the hydrogels for imaging in a thin layer of vitrified ice using cryo-TEM. The ambient temperature and humidity were 22 °C and 80%, respectively. 200-mesh copper grids coated with perforated carbon films (Lacey carbon films: ProSciTech, Qld, Australia) were glow discharged in nitrogen for 5 s immediately before use. Hydrogels were prepared as described above. At the time of analysis, hydrogels were disrupted by tapping against the glass vial. Approximately 4 μL aliquots of sample were pipetted onto each grid prior to plunging. In the case of samples which could not be liquefied adequately, the gel was smeared gently onto the grid. After 30 s adsorption the grid was blotted manually using Whatman 541 filter paper, for approximately 6–10 s. The blotting time was optimised for each sample. The grid was then plunged into liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid nitrogen until required. The samples were examined under low dose conditions using a Gatan 626 cryoholder (Gatan, Pleasanton, CA, USA) and Tecnai 12 TEM (FEI, Eindhoven, The Netherlands) at an operating voltage of 120 kV. Images were recorded using either a Megaview III CCD camera and AnalySIS camera control software (Olympus) using magnifications in the range 40000–110000×, or a FEI Eagle 4k × 4k CCD camera (FEI, Eindhoven, The Netherlands) at magnifications in the range 15000–30000×.
Atomic force microscopy (AFM)
An Asylum Research MFP-3D atomic force microscope (Santa Barbara, CA, USA) was used to measure surface topography in tapping mode employing ultrasharp silicon nitride tips (NSC15 noncontact silicon cantilevers, MikroMasch, Spain). The tips used in this study had a typical force constant of 40 N m−1 and a resonant frequency of 320 kHz. Typical scan settings involved the use of an applied piezo deflection voltage of 0.6–0.7 V at a scan rate of 0.7 Hz. All images were processed (1st order flattening algorithm). The samples were prepared in a glass vial as described above for the hydrogelation test and a small amount (∼30 μl) was spread onto a clean square of silicon wafer (1 cm × 1 cm) by gently pressing a glass coverslip on top. The samples were then dried at room temperature for 24 h.X-ray fiber diffraction (XRD)
A Bruker D8 Advance X-ray diffractometer with CuKα radiation (40 kV, 40 mA) equipped with a LynxEye silicon strip detector was employed to determine the X-ray diffraction (XRD) patterns. Each sample was scanned over the 2-theta range 1–60° with a step size of 0.02° and a count time of 1.6 s per step. An air scatter slit was used to reduce the beam intensity at low angles. The samples were mounted on zero background plates consisting of a silicon wafer located in a standard Bruker specimen holder, and were dried overnight in air.Molecular modelling
Model structures of zwitterionic tripeptides vFF and VfF were generated using MarvinSketch,24 and their antiparallel beta-sheet structures were built in an incremental fashion as follows. First, we manually assembled a three-strand antiparallel sheet using the software VMD 1.9.125 (see ESI,† Fig. SMAV1).Then, molecular optimization of atomic structures was performed using the AMBER1226 package both in vacuum and in the presence of model solvent (TIP3P model).27 The parm99SB-ILDN force field28 was used to model the peptides. First, up to 25000 cycles of restrained optimisation were performed applying smooth composite restraints (left-hand and right-hand parabolic restraints, respectively, centered at dl = 1.5 Å and dr = 2.9 Å; both force constants were set to 10 kcal mol−1 Å−2, and no restraints were applied between dl and dr) on the four H-bond distances between the following pairs of atoms (larger spheres in Fig. SMAV1, ESI†): OV1–HF6, OV4–HF3, OF5–HF8, and OF8–HF5. Then, the system was solvated within a truncated octahedral box with a 20 Å buffer, and a second restrained optimisation was performed. Finally, up to 25000 cycles of unrestrained optimisation were simulated. From Fig. SMAV2 (ESI†), it can be seen that the central phenylalanine residues get closer and tend to form a “hydrophobic spine” running along the direction of the beta-sheet.
In order to confirm this observation, we also built a five-strand sheet and optimised its structure using the same protocol described above. As seen from Fig. SMAV3 (ESI†), the formation of the hydrophobic spine was better seen in this structure. Finally, starting from the five-strand-optimised sheet, we manually built an antiparallel 12-strand sheet and we performed another set of optimisations.
For the VfF peptide, we started from the five-strand beta-sheet and then we built the 12-strand one, using the same protocol described above for the vFF peptide.
Structural analyses were performed using the VMD software through ad hoc scripts using the tcl language.
Cell assays
Tripeptides in solution were assessed for cytotoxicity in accordance with ISO 10993, and tripeptide gels were also probed for their ability to support cell viability and proliferation. For cytotoxicity studies, L929 mouse fibroblast cells were seeded at 10000 cells per well of a 96-well tissue culture plate in 100 μl of media (MEM + GlutaMAX™-I (GIBCO)), supplemented with 1 v% NEAA (non-essential amino acids, GIBCO), 2 v% anti–anti (antimycotic–antibiotic, GIBCO), and 10 v% FBS (fetal bovine serum, SAFC Biosciences) and cultured overnight at 37 °C, 5% CO2. Tripeptides were dissolved in the media at the highest concentration possible without occurrence of precipitation or gelation (i.e., 1 mg ml−1) and 1:1 serial dilutions down to <8 μg ml−1 were prepared. The tripeptide solutions were sterile-filtered and 100 μl was applied to monolayers that were then cultured further for 24 h. Cells were imaged using an inverted microscope (Olympus IX71) before quantitation by reduction of resazurin (120 μl of a 1:9 solution of PrestoBlue® in media for 1.5 h) and 100 μl assayed for fluorescence on a Pherastar fluorometer (ex. 540–20 nm, em. 590–20 nm). Media without tripeptides as a negative control and 5 v% dimethylsulphoxide (DMSO) in media as a positive control, were included. Monolayers remained subconfluent throughout. For gel studies, gel precursor solutions were prepared as indicated previously and 15 μl of each were mixed directly in triplicate wells of a “μ-Slide angiogenesis” uncoated (Ibidi, Germany, through DKSH Australia). Gels with lower peptide concentration were not tested to avoid premature gel dissolution during the assay. After 24 h, gels were pre-treated with 30 μl of media for 1 h. L929 cells were added to the gels (10000 cells per cm2 in 30 μl media), and cultured at 37 °C, 5% CO2 for up to 72 h, by handling the slides according to the manufacturers's instructions, including the addition of a few drops of deionised water in the empty space between the wells to minimise sample evaporation. Every 24 h, cells were either stained for viability or, if continuing to a later assay time point, had 30 μl of media exchanged for fresh media. Cell viability was investigated using the LIVE/DEAD® assay (Invitrogen), according to the manufacturers's instructions. Cells were imaged using an inverted microscope (Nikon Eclipse TE2000-U) for calcein (ex. 465–495 nm, em. 515–555 nm) and ethidium (ex. 510–560 nm, em. >590 nm).
Protease assay
Tripeptides (2.0 mg) were dissolved in 15 ml Falcon tubes with 5 ml of 50 mM sodium phosphate buffer (pH 7.5) containing a large excess of proteinase 6 from Aspergillus sp. (Fluka, 5.0 mg, ∼3 U mg−1). The tubes were incubated at 37 °C, 50 rpm, and 900 μl samples were analysed over time by HPLC (Abs @ 254 nm, acetonitrile as solvent A and water as solvent B, both containing 0.1% TFA, using the gradient program as follows: 2 min at 5%A, then ramp over 20 min up to 95%A, then 5 min at 95%A, then 3 min at 5%A). The assay on gels was performed in 15 ml Falcon tubes by dissolving 2.0 mg of each tripeptide in 150 μl of 0.1 M sodium phosphate buffer (pH 11.8) with 5 min ultrasonication, and then adding 150 μl of 0.1 M sodium phosphate buffer (pH 5.8) on top. The gels were left to self-assemble overnight. The following morning, 4.7 ml of 50 mM sodium phosphate buffer (pH 7.5) containing a large excess of proteinase 6 from Aspergillus sp. (Fluka, 5.0 mg, ∼3 U mg−1) was added on top. The tubes were incubated at 37 °C, 50 rpm, and after 1 h, 2 ml of 1 M NaOH was added on top to dissolve the gels and slow down the protease activity prior to HPLC analysis as above. The experiments were repeated three times.Results and discussion
Hydrogelation and inversion tests
Hydrogelation tests at physiological pH were performed for each Val–Phe–Phe stereoisomer, according to our established pH trigger method.16 Increasing peptide concentrations were tested until exceeding the peptide solubility limit; however, only four out of the eight stereoisomers formed homogeneous gels. In the other cases, we obtained either solutions (at low concentrations) or unstable heterogeneous systems that eventually precipitated (at high concentrations). In line with our previous results, homochiral tripeptides did not gel, whilst we observed the formation of self-supportive, homogeneous hydrogels for vFF (i.e., D–L–L configuration), and its enantiomer Vff (i.e., L–D–D configuration) (Fig. 1A). To our surprise, tripeptides VfF and vFf (i.e., L–D–L and D–L–D configurations, respectively) also formed hydrogels, although they were not self-supportive as observed in the inversion test (Fig. 1B). Clearly, VfF and vFf tripeptides have self-assembling and gelation abilities, but, in contrast with their stereoisomers vFF and Vff, they form supramolecular structures leading to gel matrices that are not capable of self-supporting their own weight, and in this work we investigated the reasons at the molecular level to understand such discrepancy. | ||
| Fig. 1 (A) vFF and Vff self-supportive hydrogels (top) displayed the highest moduli (bottom) among Val–Phe–Phe peptide stereoisomers. (B) VfF and vFf hydrogels were not self-supportive (right, see white arrows), and their moduli were lower (bottom). Note: rheometric data shown are for vFF (left) and VfF (right). Further data are in the ESI.† | ||
Rheometric analysis (Fig. 1 and ESI†) confirmed the gelling abilities of these four Val–Phe–Phe stereoisomers, with viscous and elastic moduli that showed independence from the applied frequency. All four gels showed similar rheological behaviours over time, with self-assembly proceeding very rapidly during the first five minutes, and moduli reaching a plateau within an hour (Fig. 1, bottom). In particular, vFF and Vff hydrogels displayed moduli that were significantly higher than those of VfF and vFf hydrogels, confirming the marked difference observed in the inversion test. The minimum gelation concentration was similar for all low-molecular-weight gelators, i.e., 10 mM for vFF or Vff, and 9 mM for VfF or vFf.
Differential scanning calorimetry (DSC) analysis was thus performed on the four gels to further investigate the soft materials (see ESI†). Thermal behaviour was analogous for enantiomers and different between vFF (or Vff) gels and VfF (or vFf) gels. All four gels displayed two distinct endotherm curves, of which the main one can be ascribed to the gel-to-sol transition, as verified in a separate test by visual observation of the gel melting upon heating to the desired temperature. The other, minor, DSC transition that follows can be ascribed to the melting of aggregates that are formed during heating.29
The first endotherm for the gel-to-sol transition was registered with a Tm ∼ 80 °C for all samples, as typically observed for amyloids.30 The gel-to-sol transition implies the loss of the 3D network that forms the gel matrix, and not necessarily the loss of the fibril nanostructure. However, the observed transition is compatible with amyloid fibril melting and the loss of the amyloid nanostructure, as reported by several others and recently reviewed.31
Remarkably, the onset temperature and the shape of the endotherms were very different. In particular, the self-supportive gels displayed high thermal stability and high homogeneity, as their transition started well over 60 °C (i.e., 65 ± 3 °C for vFF and 67 ± 3 °C for Vff), with a narrow endotherm halfwidth of ∼10 °C indicative of a highly cooperative process. By contrast, melting of the other two gels had an onset near human body temperature (i.e., 33 ± 1 °C for VfF and 31 ± 3 °C for vFf), thus resulting in a much wider and asymmetric endothermic curve. The width of the DSC transition can be ascribed to heterogeneity in thermal stability, thus to the presence of differently structured populations, each characterised by a specific Tm, as also observed for other fibrillar proteins that form gels.32 The data suggested a lower supramolecular order for the latter samples (i.e., VfF and vFf), with more heterogeneous and disordered gel states. Indeed, asymmetric melting transitions are uncommon for peptides and proteins, but have been observed for beta-sheet peptides able to self-assemble through a nucleation-growth process33 and for gel-forming and amyloid fibrillar proteins.31,32
Peptide secondary structure
Investigation of each stereoisomer secondary structure was performed to gain insights into the supramolecular behaviour. The tripeptide secondary structure was analysed by means of FT-IR and circular dichroism (CD) spectroscopy, and thioflavin T-fluorescence imaging. The highly sensitive ATR-IR technique provided spectra of the four self-assembling tripeptides that were quite similar, and all indicated the presence of anti-parallel beta-sheets (Fig. 2A). The amide II region was characterised by a maximum centered at 1640 cm−1 with a shoulder at ∼1680–1690 cm−1 for vFF and Vff. These beta-sheet signatures are in agreement with previous studies on vFF,14 as well as other self-assembling Phe–Phe derivatives15,34 and short hydrophobic sequences derived from the amyloid beta peptide fragments.35 The maximum was broader and shifted to 1620 cm−1, for VfF and vFf, suggesting prevalence of anti-parallel beta-sheets, but co-existence of other forms of less ordered beta-aggregates.36 The amide I region was dominated by a very broad signal indicating the formation of extended salt bridges between the peptide termini, as previously suggested for extended supramolecular assemblies of short hydrophobic peptides.37 By contrast, the FT-IR spectra of non-assembling stereoisomers had many different maxima in the amide I and II regions, indicating co-existence of different secondary conformations (see ESI†).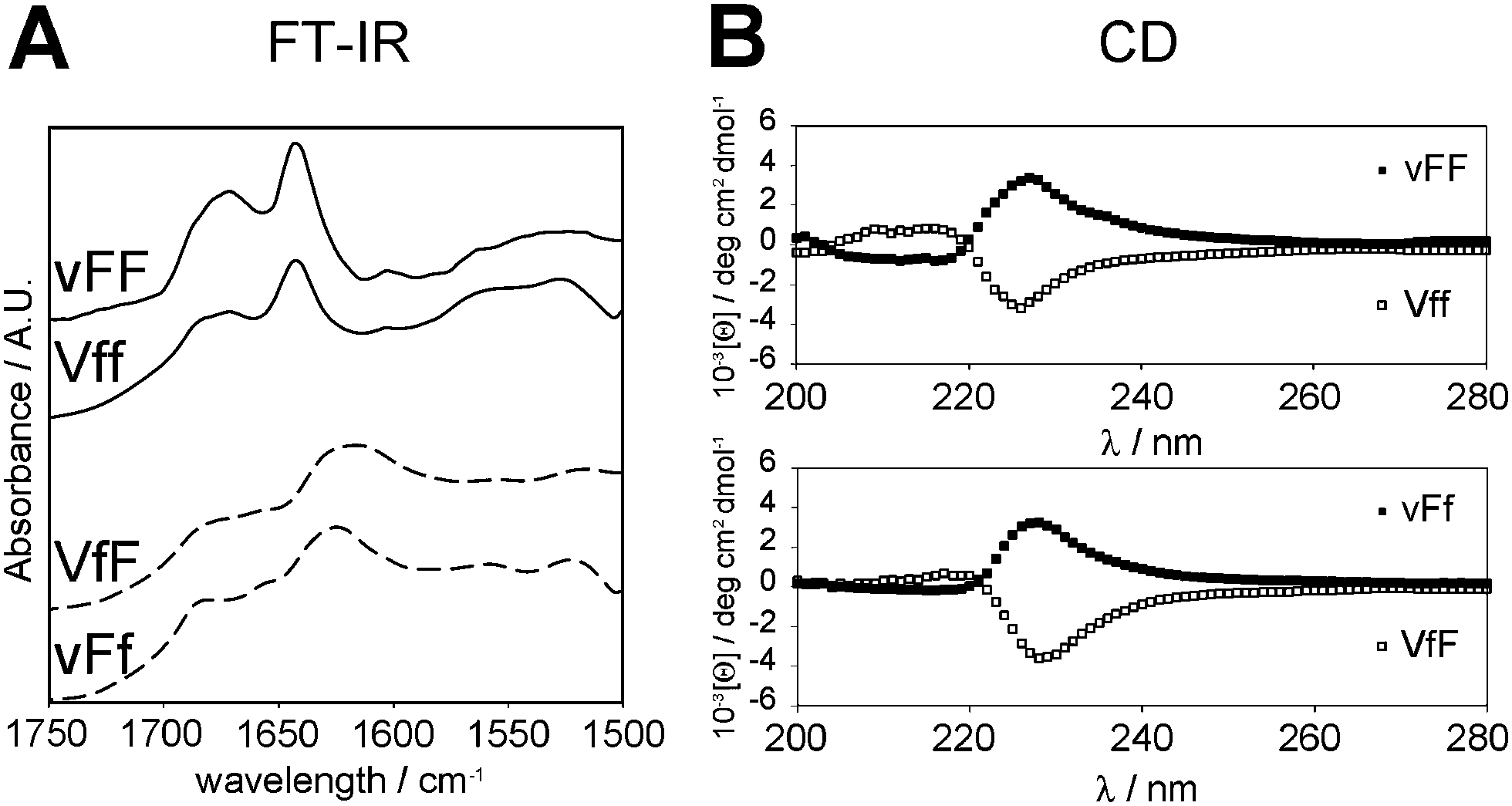 | ||
| Fig. 2 FT-IR (A) and CD (B) spectra of the four gelling tripeptides. | ||
Beta-sheets were observed for the four self-assembling peptides also by means of CD analysis in the region 200–220 nm (Fig. 2B). Such a beta-sheet signature is characteristic of similar, hydrophobic, self-assembling short fragments derived from the amyloid beta peptide.15,16,38 In particular, the signal was more intense for vFF and Vff, relative to VfF and vFf, in agreement with FT-IR data. In addition, the dominant signal centered at 227–230 nm was indicative of π–π stacking.
Interestingly, the signal for the four gelling tripeptides was of comparable intensity, even though the two phenylalanine residues of vFF and Vff have the same configuration, while those of VfF and vFf have opposite configuration. This fact can be ascribed to the ordered assembly of the tripeptides, since the non-gelling tripeptides VFf and vfF (which also have the phenylalanine residues in opposite configurations) have a very weak signal in the π–π stacking region (see ESI†). Importantly, both signatures of the beta-sheets and the aromatic interactions had a chirality (i.e., D- or L-) that was dictated by the central amino acid, even when it was only one out of the three residues with such a configuration (Table 1, highlighted in bold). This fact underlines the importance of chiral effects on the supramolecular behaviour, which can be directed by an appropriate molecular design.
| Peptide sequence | No. D-amino acids | No. L-amino acids | CD chirality |
|---|---|---|---|
| vFF | 1 | 2 | L |
| VfF | 1 | 2 | D |
| VFf | 1 | 2 | L |
| vfF | 2 | 1 | D |
| vFf | 2 | 1 | L |
| Vff | 2 | 1 | D |
Thioflavin T fluorescence imaging is another technique that reveals the presence of peptide extended beta-sheets, as established through various studies.39–41 Although the dye can also bind to structures other than proteins (e.g., DNA), the fluorescence arising from the dye binding to beta-structured peptide fibrils has made it a widely recognised and well-established amyloid marker.42 Upon dye staining, a continuous network of fluorescent amyloid fibers was revealed for vFF and Vff gels, while only a few, dim and isolated amyloid structures were seen for VfF and vFf (Fig. 3). We can rationalise this fact with a higher supramolecular order for vFF and Vff samples, and a more disordered assembly for VfF and vFf, where supramolecular arrangements are predominantly, but not exclusively, composed of beta-sheets, as suggested by IR and CD analyses. By contrast, no predominant nanostructure of interest was observed for the other stereoisomers (see ESI†). The homochiral tripeptide vff formed only occasional, dim, plate microcrystals similar to the non-assembling homochiral tripeptide Leu–Phe–Phe, with micelle-like nucleation points on the surface of the crystals.15 The other two stereoisomers, VFf and vfF, had only rare instances of dim structures, indicating hydrophobicity-driven aggregation, but lack of extended beta-sheets with supramolecular order forming a matrix as required for gelation of these peptides.
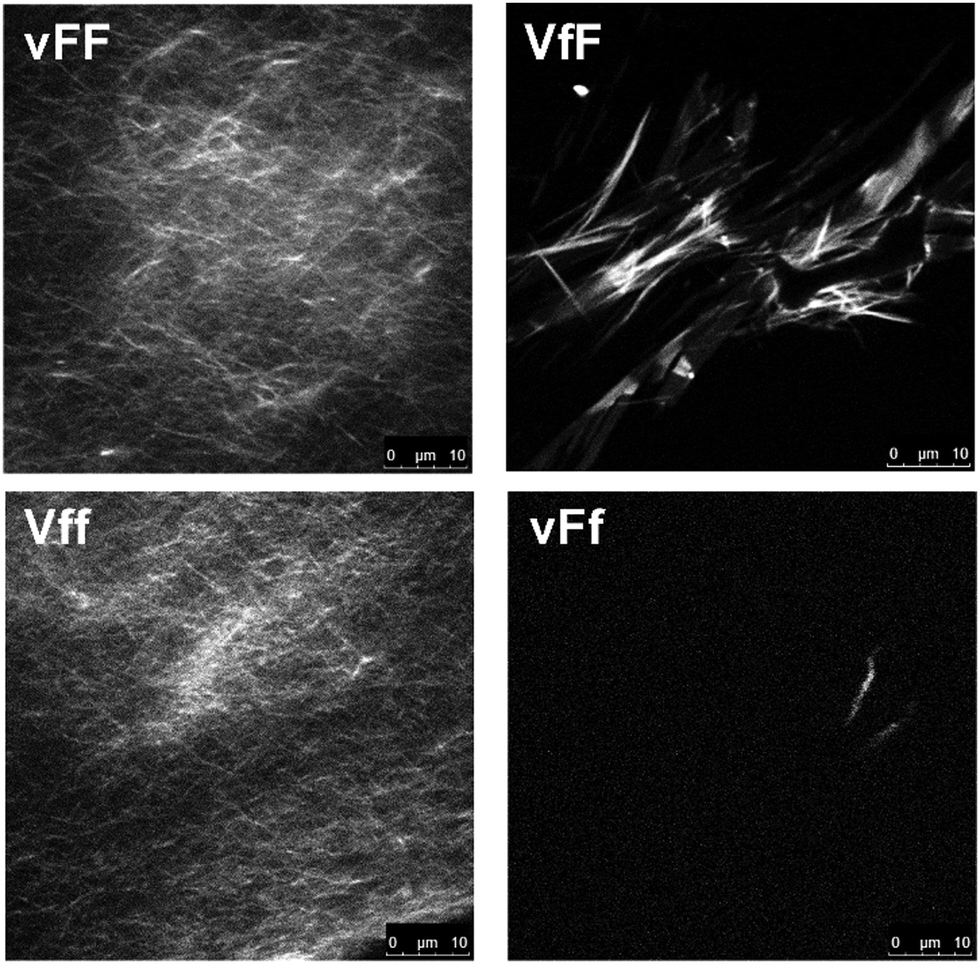 | ||
| Fig. 3 Thioflavin T fluorescence microscopy images of the four gelling tripeptides. | ||
Nanostructures
Cryo-TEM and AFM imaging were employed to investigate the nanostructures formed by the tripeptides, especially since thioflavin T imaging allows us to see only those arising from extended beta-sheets. Assembling heterochiral Val–Phe–Phe tripeptides formed nanotapes arising from the alignment of parallel fibrils as seen by cryo-TEM (Fig. 4, top). However, the individual fibrils formed by the assembling tripeptides were significantly thicker for the peptides forming self-supporting hydrogels (i.e., 17.1 ± 2.9 nm for vFF and 17.9 ± 2.4 for Vff, Fig. 4 on the left), relative to those forming weak hydrogels (i.e., 11.4 ± 1.2 nm for VfF and 11.3 ± 1.7 for vFf, Fig. 4 on the right). These data confirmed a lower supramolecular hierarchical order for the latter. No nanostructures were observed for the homochiral tripeptides, and only isolated instances of nanotapes were seen for the heterochiral VFf and vfF (see ESI†). AFM imaging qualitatively confirmed these observations, with large areas covered by nanotapes arising from aligned structures for vFF and Vff (Fig. 5, left) and only thinner structures of a highly homogeneous height seen for VfF and vFf (Fig. 5, right).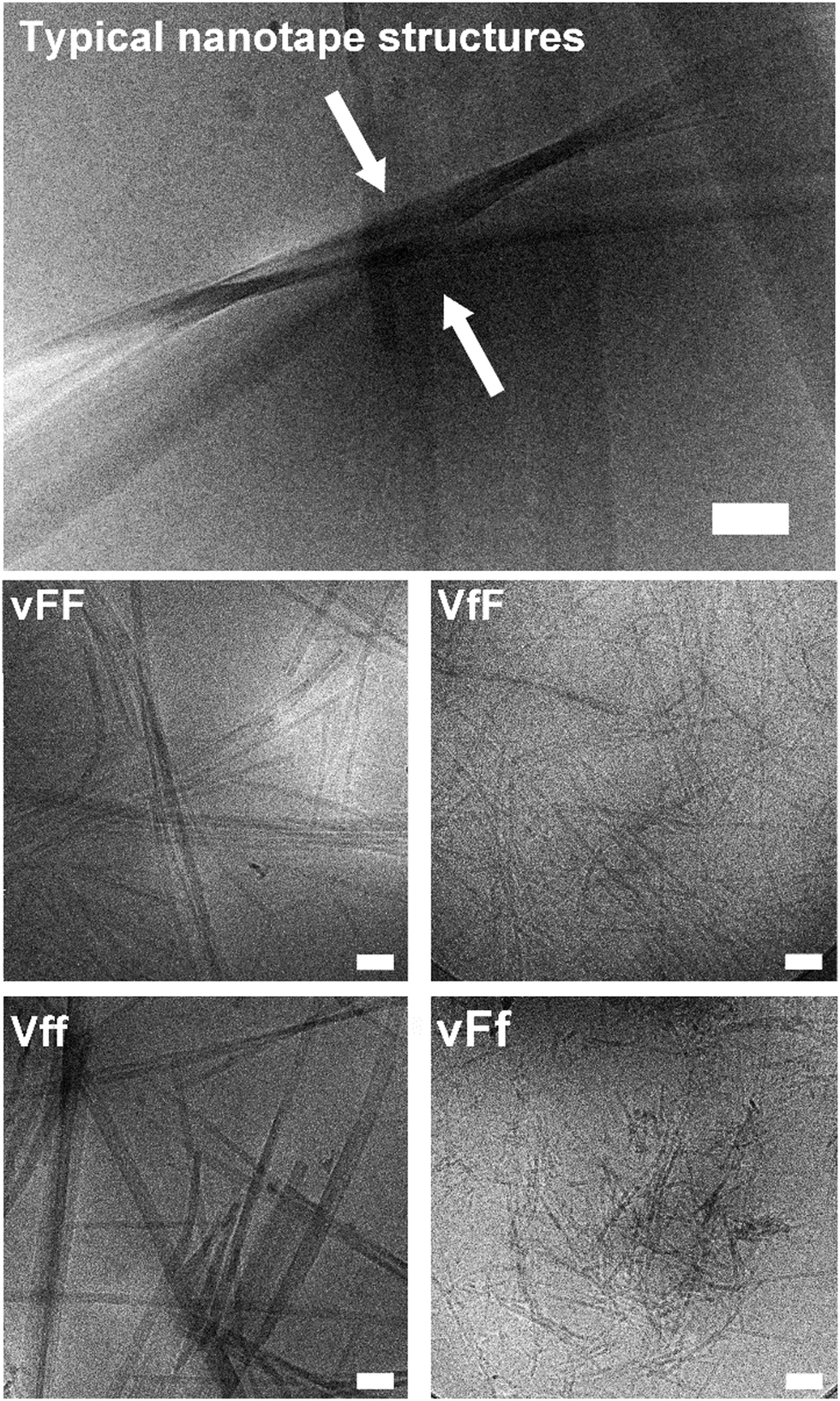 | ||
| Fig. 4 Cryo-TEM images of gelling peptides. Scale bar = 100 nm. | ||
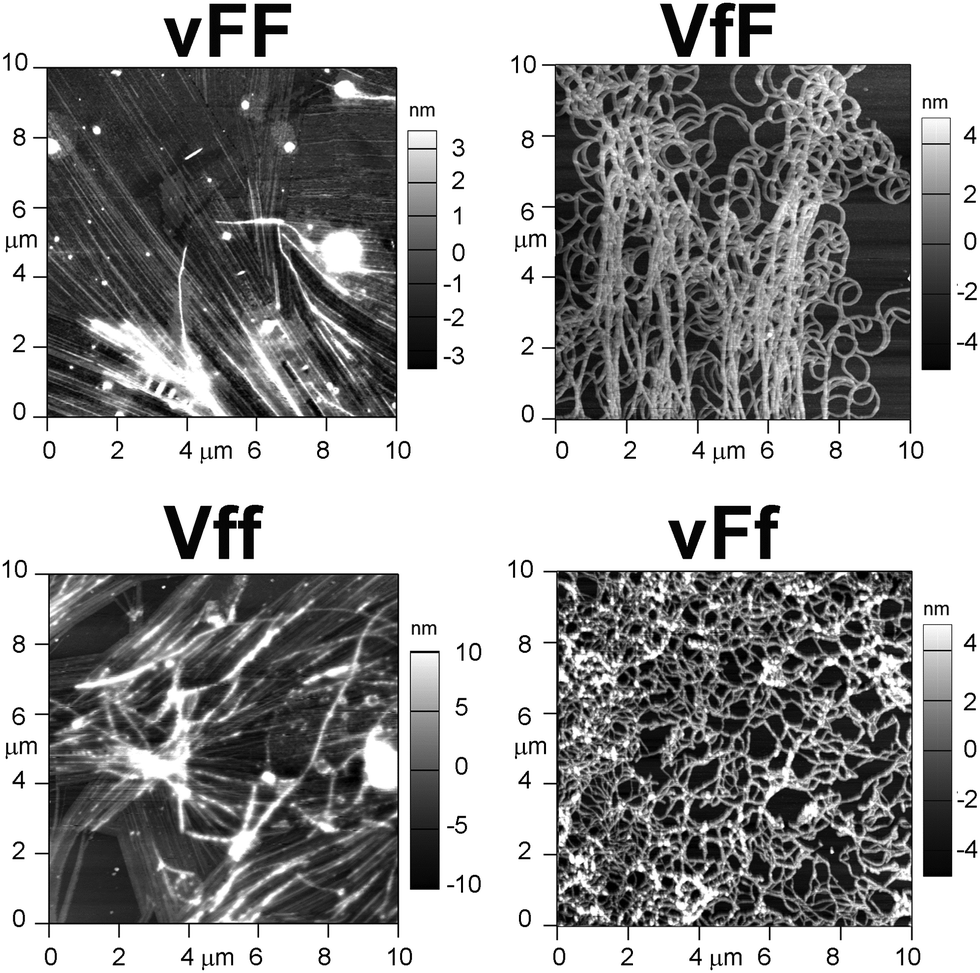 | ||
| Fig. 5 AFM images of the four gelling tripeptides highlight nanotapes arising from aligned fibrils only for vFF and Vff (left). | ||
Supramolecular order
XRD analysis was also employed to analyse the supramolecular order of gelling tripeptides. Unfortunately, all the tripeptides have a signal that is too weak for 2D XRD analysis, thus we had to rely on 1D data. As expected, self-assembled enantiomers displayed analogous XRD spectra (see ESI†), thus only one for each enantiomer pair is shown in Fig. 6. In line with similar compounds, typical amyloid signals were observed for all gelling peptides. The signals at 4.0 Å and 3.8 Å are characteristic of phenylalanine residues that interact via π–π stacking. Signals at 4.9 Å and 4.6 Å are compatible with distances between beta-strands; 16.7 Å and 18.8 Å could be attributed to peptide lengths, as confirmed by modelling (see the section below). | ||
| Fig. 6 XRD analysis of vFF and VfF hydrogels. | ||
A key difference between the XRD spectra of the self-supporting and the weak hydrogels composed of vFF (or Vff) and VfF (or vFf), respectively, is the presence of an intense signal at 9.4 Å for the former, and only a very weak signal at 10.8 Å for the latter. These are typical distances between every other beta-strand in an anti-parallel configuration.43 Therefore, it is likely that vFF (and Vff) antiparallel beta-sheets are packed more tightly with a higher supramolecular order, relative to VfF (and vFf), leading to a more intense signal. VfF (and vFf) anti-parallel beta-sheets display a less intense XRD signature owing to the peptide inherent disorder at the secondary conformation level. This translates not only in disordered beta-sheets, but also in the peptide folding with other secondary conformations, as suggested by the very weak beta-sheet signature seen for VfF (and its enantiomer vFf) in the FT-IR and CD spectra (Fig. 2) as well as in the thioflavin-T stained microscopy images (Fig. 3).
Molecular modelling with explicit water was thus used to shed light on the structural differences between vFF (or Vff) and VfF (or vFf) tripeptide gels. In vFF (Fig. 7A–C, top) we see a harmonious assembly of the peptides, with a helical turn of ∼36° between adjacent strands. The central phenylalanine residues are highlighted in colour. It can be seen how each peptide, in this system, is slightly curved (yellow ribbons in Fig. 7A). There is an almost perfect alternation between 3 and 2 H-bonds between adjacent beta-strands (Fig. 7C). Zwitterionic termini are solvated by water, while the central phenylalanineresidues are almost free of structural solvation, and form a regular and helical pile (Fig. 7B), thus confirming a tightly packed supramolecular structure, in agreement with XRD and rheological data. Modelling of a similar assembling tripeptide Leu–Phe–Phe in D–L–L configuration revealed how phenylalanine residues could engage in aromatic zippers holding together adjacent beta-sheets.15 Analogously, this model shows the tightly packed stack of phenylalanine residues in each beta-sheet. In addition, this model with explicit water molecules (Fig. 7C) shows that structural water is effectively excluded from such dry regions, similar to what has been observed for amyloid steric zippers,44 confirming their likely interdigitation with the corresponding phenylalanine stack of another beta-sheet, thus giving rise to the nanotapes.
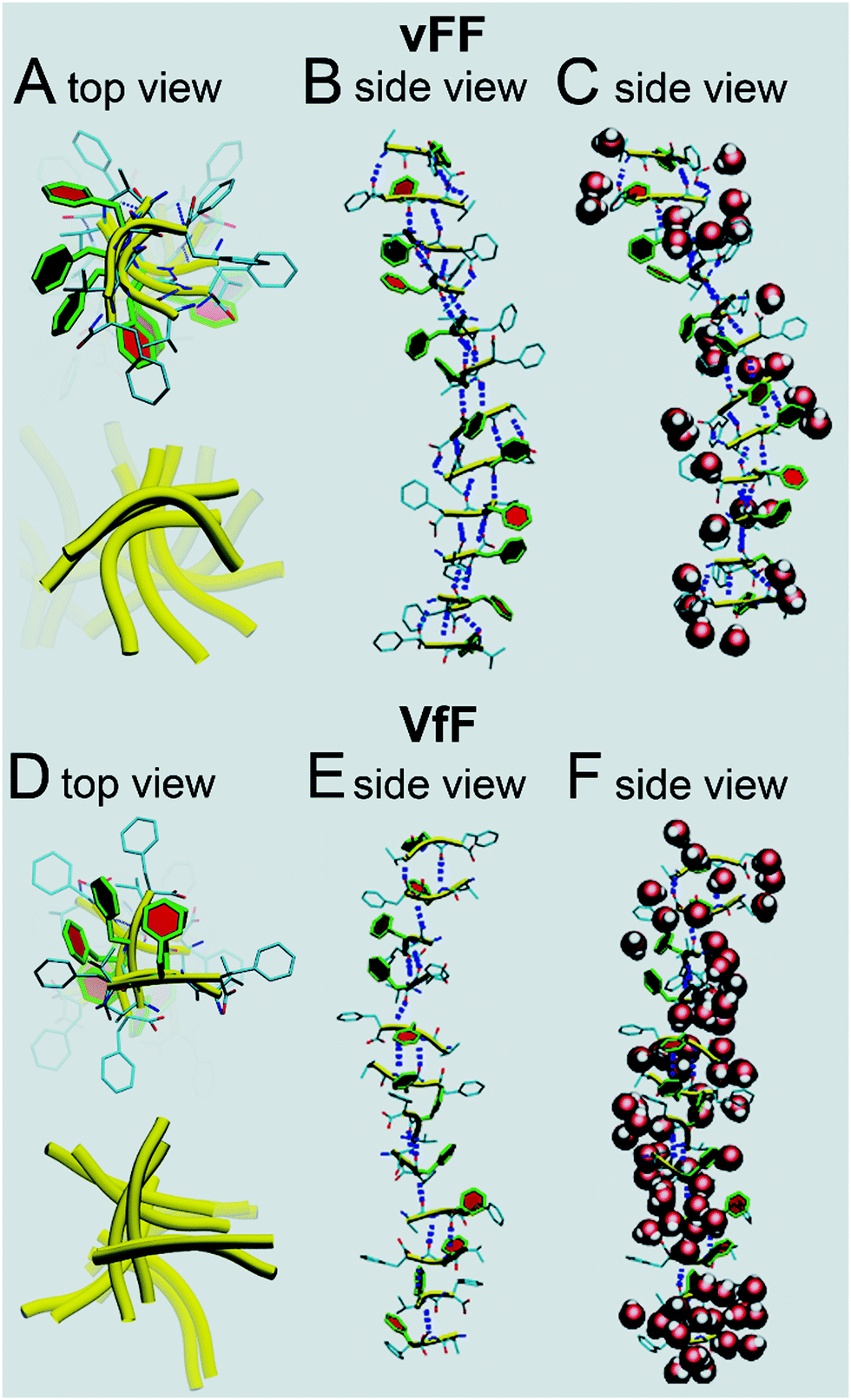 | ||
| Fig. 7 Molecular modelling of dodecamer stacks of gelling tripeptides vFF (A–C) and VfF (D–F). Peptide backbones are highlighted in yellow, hydrogen bonds in blue, central phenylalanine residues in color, and water molecules are depicted as red spheres (C and F). | ||
In VfF (Fig. 7D–F) several things are different. Peptides tend to assemble forming a pseudo helix, but this appears as an assembly of 2-strands of antiparallel beta-sheets forming an angle of approximately ∼90° between them (Fig. 7D) and linked by hydrophobic interactions. This is confirmed by the alternation of 2/3 H-bonds between strands belonging to a pair, and 1 or even 0 H-bonds between strands forming an angle of ∼90° (Fig. 7E). In accordance with this, the degree of hydration of this assembly is higher than in vFF, as structural water is not effectively excluded from the peptide stack (Fig. 7F). Therefore, this model suggests that hydrogels made of VfF (or vFf) are not durable in aqueous environments, as water readily penetrates in their structure, leading to peptide disassembly. Indeed, cell culture experiments (see the section below) confirmed this hypothesis.
We have also analysed the atomic distances in the modelled peptide stacks to find remarkable agreement with XRD data. The average peptide lengths correspond to 19.0 Å and 18.7 Å for vFF and VfF, respectively. When we analysed the distances between the α-carbons of the central phenylalanine residues we found interesting differences between vFF and VfF. In only vFF, there was a clear alternation of two very close values, indicating that beta-strand pairs would be repeatedly positioned at average distances of 4.3 Å and 5.0 Å, in agreement with the XRD signals at 4.6 Å and 4.9 Å, respectively. Instead, the beta-strands of VfF were more disordered, with various distances in the range 5–7 Å, thus explaining only a weak XRD signal at 4.9 Å for VfF. The distances between every other strand in our models corresponded to 9.3 Å and 10.4 Å for vFF and VfF, respectively, consistent with the values of 9.4 Å and 10.8 Å as seen by XRD. By contrast, the distances between central Phe residues corresponded to 5.4 Å and 5.8 Å for the two peptides, respectively. This is not too surprising, since the XRD values of 3.8 Å and 4.0 Å likely correspond to the π–π stacking between phenylalanine residues belonging to separate peptide stacks that engage in interdigitated zippers.
Overall, molecular modelling confirmed the XRD analysis and provided us with a realistic picture of the peptide stacks, well in agreement with our experimental data. Importantly, the models described here well illustrate the supramolecular features that allow vFF to achieve a higher order of peptide stacks to form a self-supporting hydrogel, while the inherent disorder of VfF and impossibility to effectively exclude water from the assemblies lead to a weak hydrogel that readily disassembles in aqueous solutions.
Cell culture experiments
The designed uncapped heterochiral tripeptides are ideal candidates for soft biomaterials. They are devoid of synthetic capping groups that may lead to cytotoxicity, do not require any use of organic solvents, and readily gel in phosphate buffer at physiological pH. There are also other grounds for application in nanomedicine. We have shown in separate studies that they can co-assemble with aromatic small molecules (e.g., drugs or dyes) for sustained delivery and/or imaging.12,20 Besides, similar sequences (i.e., DLeu–Phe–Phe, DPhe–Phe–Val, and Phe–DPhe–DVal) have performed well in fibroblast cell culture and cytotoxicity assays.16,20 Similar heterochiral tripeptides display mild antibacterial activity (i.e., DLeu–Phe–Phe) with lack of haemotoxicity, which is a common side effect of antimicrobial peptides.20 Due to all these reasons, we were keen to assess the performance of the assembling Val–Phe–Phe stereoisomers in cell culture.The hydrogels composed of VfF or vFf readily dissolved at 37 °C, while those composed of vFF and Vff were still present after 24 h of cell culture. This was anticipated by the DSC data, where the former had an endotherm onset temperature just above 30 °C, while the latter had one well above 60 °C. Modelling studies discussed above also supported a higher stability of the vFf due to a higher supramolecular order. However, after 72 h, vFF and Vff hydrogels also appeared to have partially dissolved (Fig. 8A), whether cells were present or not, suggesting a major role of prolonged heating in disruption of the self-assembled structures. To explore the potential contribution of protease degradation, peptides were tested for protease stability with an enzymatic amount that is far greater than might be present under cell culture conditions. While L-homochiral VFF was completely hydrolysed within 1 h, all four gelling tripeptides persisted in solution over the course of 24 h to a similar extent (∼25–30%, see ESI†). These data supported the hypothesis that the significant difference in gel stability observed between peptide stereoisomers under cell culture conditions is mainly ascribed to thermal dissolution. Besides the amino acid D-stereoconfiguration, self-assembly is another factor that provides protection against enzymatic degradation by rendering the tripeptides less accessible to the active site. Indeed, when the protease assay was repeated on self-assembled peptides to take into account the factor of supramolecular order as well, all peptides were preserved at significantly higher levels relative to the assay in solution (see ESI†). In fact, for certain applications it may be valuable to further increase hydrogel stability. To this end, peptide derivatisation with unnatural functional groups, cross-linking, or incorporation in the gel of other durable nanostructures promoting π–π stacking (e.g., nanocarbons45) may offer valuable options.
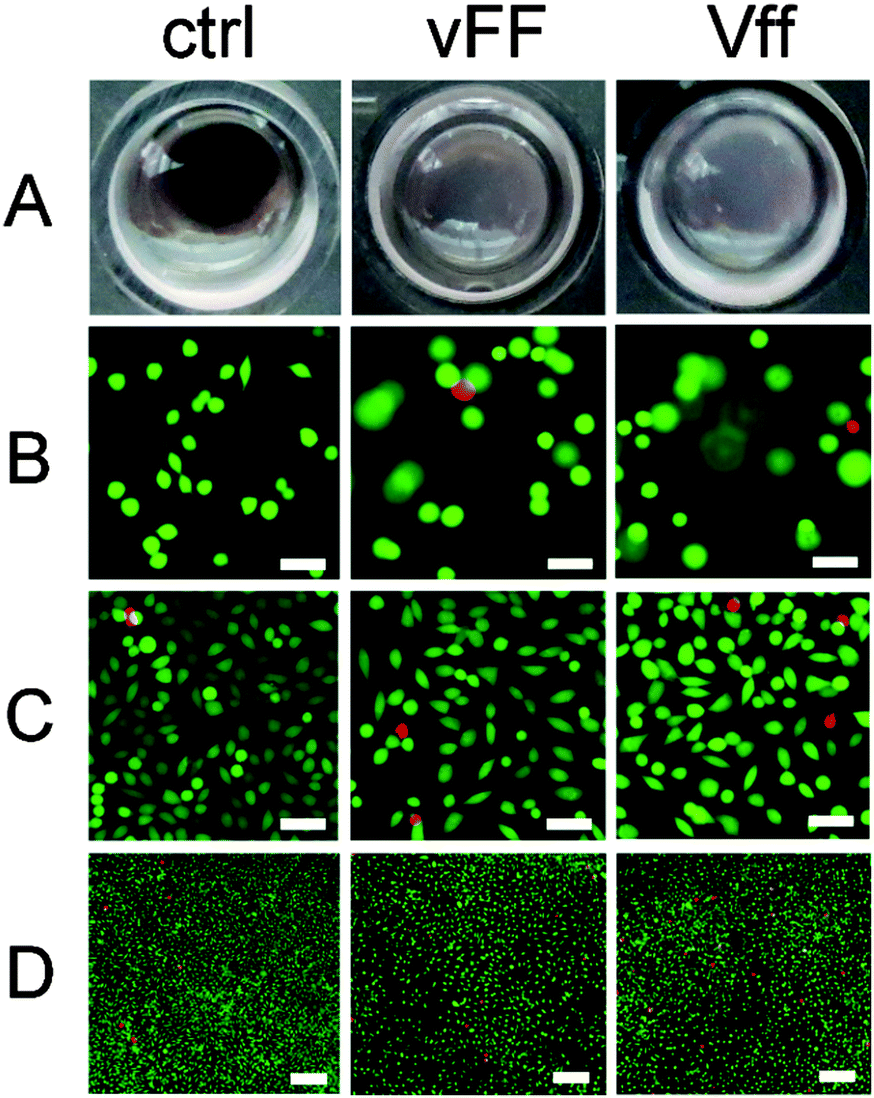 | ||
| Fig. 8 Images of heterochiral peptide gels over 3 days of fibroblast cell culture. (A) Photographs of the culture wells at 72 h. (B) Live/dead staining at 24 h show cells on different focal planes due to gel penetration (scale bar = 50 μm). (C and D) Live/dead staining at 72 h at high (C, scale bar = 50 μm) or low (D, scale bar = 200 μm) magnification. | ||
Fibroblasts penetrated to some extent in both vFF and Vff peptide hydrogels as imaging revealed cells on different focal planes throughout the gel depth during the first 24 h, when the gels were still present (Fig. 8B and ESI†). By contrast, at 72 h gels had significantly dissolved and cells were on the bottom of the wells as in the plastic controls (Fig. 8C and D), in contrast to what was observed for the more durable Phe–Phe–Val16 peptide gels. Live/dead staining at 72 h did not highlight any major toxic effect, and cell spreading occurred on both hydrogels (Fig. 8C). Although it appeared that cell density and viability were slightly lower relative to the control (Fig. 8D), the difference was within the experimental error. Interestingly, no substantial difference was observed between the enantiomeric assembled peptides, despite the fact that Vff has an unnatural supramolecular chirality (i.e., as expected for D-peptides).
Tests performed on the heterochiral peptides in solution indicated no cytotoxicity (see ESI†). Considering that the Val–Phe–Phe sequence is one of the core motifs of the amyloid beta peptide responsible for its propensity towards self-aggregation,46 this outcome is slightly surprising, although in agreement with the lack of fibroblast cytotoxicity observed in vitro for the self-assembling Phe–Phe–Val regioisomers.16 The data hold promise for the application of these self-assembling tripeptides as biomaterials, although further studies will be required to investigate their effects in a biological setting.
Conclusions
This is the first report of hydrogels formed under physiological conditions from uncapped, heterochiral tripeptides with the central amino acid in an opposite configuration relative to the other two (i.e., L–D–L or D–L–D stereochemistry). This study therefore highlights that our previous reports on hydrogels formed by heterochiral, uncapped tripeptides (in D–L–L or L–D–D configuration) have a larger scope than anticipated.14,15 Importantly, it confirms that amino acid chirality is a key tool to drive peptide self-assembly and elucidates relevant mechanisms of supramolecular order that will assist the design ex novo of self-assembling chiral small molecules.For the first time, we unveiled key details of heterochiral tripeptide stacked assemblies that give rise to a higher (for vFF and Vff) or lower (for VfF and vFf) supramolecular order. As a result, hydrogels manifest different properties and stability at 37 °C, giving a scope for the fine-tuning of their dissolution times, depending on the desired application.
Besides, we observed that while all heterochiral Val–Phe–Phe peptides have some tendency towards self-organization into nanotapes, only selected peptides (in the D–L–L, L–D–D, L–D–L or D–L–D configuration) gel under physiological conditions. Importantly, we observed the formation of dry hydrophobic channels from the ordered vFF (or Vff) stacks that effectively exclude water molecules and are essential for the gel durability. By contrast, VfF and vFf can form only assemblies of a lower supramolecular order that cannot efficiently exclude water molecules, resulting in gels with lower moduli and thermo-stability.
Finally, our cytotoxicity tests reveal interesting results for the future development of cost-effective biomaterial gels from heterochiral assembling tripeptides. While no cytotoxicity is observed in solution and fibroblast cell viability and proliferation in the supramolecular assemblies appear to be comparable to controls over 3 days, further studies will be needed to fully assess the impact of self-assembling peptides in a biological context. Importantly, both vFF and Vff gelling peptides performed well in cell culture experiments, and with no difference observed between enantiomers, raising interesting questions as it regards cell interactions with supramolecular chirality.
In conclusion, we believe this study is a relevant addition for the exploitation of chirality at the molecular and supramolecular levels towards the design of innovative and self-organised soft biomaterials.
Acknowledgements
The authors would like to acknowledge Firdawosia Kushkaki (LaTrobe University, Bundoora, Victoria, Australia) for the synthesis of selected peptides and Liz Goodall (CSIRO, Clayton, Victoria, Australia) for XRD data acquisition. SM is grateful to the Italian Ministry of Education, University and Research (MIUR) SIR program for funding under grant agreement no. RBSI14A7PL, HOT-SPOT project.References
- H. Cabral, K. Miyata and A. Kishimura, Adv. Drug Delivery Rev., 2014, 74, 35–52 CrossRef CAS PubMed.
- A. Kaushik, R. D. Jayant, V. Sagar and M. Nair, Expert Opin. Drug Delivery, 2014, 11, 1635–1646 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- Y. Kuang, J. Shi, J. Li, D. Yuan, K. A. Alberti, Q. Xu and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 8104–8107 CrossRef CAS PubMed.
- S. Marchesan, Recent Pat. Nanomed., 2014, 4, 77–81 CrossRef CAS.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160–173 CrossRef CAS PubMed.
- S. Awhida, E. R. Draper, T. O. McDonald and D. J. Adams, J. Colloid Interface Sci., 2015, 455, 24–31 CrossRef CAS PubMed.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 6595–6597 RSC.
- C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS PubMed.
- Y. Shi, J. Wang, H. Wang, Y. Hu, X. Chen and Z. Yang, PLoS One, 2014, 9, e106968 Search PubMed.
- J. Zhang, C. Ou, Y. Shi, L. Wang, M. Chen and Z. Yang, Chem. Commun., 2014, 50, 12873–12876 RSC.
- S. Marchesan, L. Waddington, C. D. Easton, F. Kushkaki, K. M. McLean, J. S. Forsythe and P. G. Hartley, BioNanoScience, 2013, 3, 21–29 CrossRef.
- W. J. M. FrederixPim, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- S. Marchesan, C. D. Easton, F. Kushkaki, L. Waddington and P. G. Hartley, Chem. Commun., 2012, 48, 2195–2197 RSC.
- S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752–6760 RSC.
- S. Marchesan, C. D. Easton, K. E. Styan, L. J. Waddington, F. Kushkaki, L. Goodall, K. M. McLean, J. S. Forsythe and P. G. Hartley, Nanoscale, 2014, 6, 5172–5180 RSC.
- C. G. Pappas, P. W. J. M. Frederix, T. Mutasa, S. Fleming, Y. M. Abul-Haija, S. M. Kelly, A. Gachagan, D. Kalafatovic, J. Trevino, R. V. Ulijn and S. Bai, Chem. Commun., 2015, 51, 8465–8468 RSC.
- M. Melchionna, K. E. Styan and S. Marchesan, Curr. Top. Med. Chem., 2015 Search PubMed , in press.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
- S. Marchesan, Y. Qu, L. J. Waddington, C. D. Easton, V. Glattauer, T. J. Lithgow, K. M. McLean, J. S. Forsythe and P. G. Hartley, Biomaterials, 2013, 34, 3678–3687 CrossRef CAS PubMed.
- H.-W. Liu, S.-H. Hu, Y.-W. Chen and S.-Y. Chen, J. Mater. Chem., 2012, 22, 17311–17320 RSC.
- V. Castelletto, G. Cheng and I. W. Hamley, Chem. Commun., 2011, 47, 12470–12472 RSC.
- J. Li, Y. Kuang, Y. Gao, X. Du, J. Shi and B. Xu, J. Am. Chem. Soc., 2012, 135, 542–545 CrossRef PubMed.
- Marvin 2.5.9, 2012, ChemAxon (http://www.chemaxon.com).
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS , 27–38.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvai, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 2012, University of California, San Francisco, 2012 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- K. Kar, Y.-H. Wang and B. Brodsky, Protein Sci., 2008, 17, 1086–1095 CrossRef CAS PubMed.
- B. Morel, L. Varela and F. Conejero-Lara, J. Phys. Chem. B, 2010, 114, 4010–4019 CrossRef CAS PubMed.
- K. Sasahara and Y. Goto, Biophys. Rev., 2013, 5, 259–269 CrossRef CAS.
- E. Badea, G. Della Gatta and T. Usacheva, Polym. Degrad. Stab., 2012, 97, 346–353 CrossRef CAS PubMed.
- W. C. Wimley and S. H. White, J. Mol. Biol., 2004, 342, 703–711 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10–S19 CrossRef CAS PubMed.
- R. S. Jacob, D. Ghosh, P. K. Singh, S. K. Basu, N. N. Jha, S. Das, P. K. Sukul, S. Patil, S. Sathaye, A. Kumar, A. Chowdhury, S. Malik, S. Sen and S. K. Maji, Biomaterials, 2015, 54, 97–105 CrossRef CAS PubMed.
- B. Shivu, S. Seshadri, J. Li, K. A. Oberg, V. N. Uversky and A. L. Fink, Biochemistry, 2013, 52, 5176–5183 CrossRef CAS PubMed.
- N. S. de Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732–1741 CrossRef PubMed.
- V. Castelletto, I. W. Hamley, P. J. Harris, U. Olsson and N. Spencer, J. Phys. Chem. B, 2009, 113, 9978–9987 CrossRef CAS PubMed.
- C. Wu, M. Biancalana, S. Koide and J. E. Shea, J. Mol. Biol., 2009, 394, 627–633 CrossRef CAS PubMed.
- R. Sabate, L. Rodriguez-Santiago, M. Sodupe, S. J. Saupe and S. Ventura, Chem. Commun., 2013, 49, 5745–5747 RSC.
- I. M. Kuznetsova, A. I. Sulatskaya, V. N. Uversky and K. K. Turoverov, PLoS One, 2012, 7, e30724 CAS.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- K. Morris and L. Serpell, in Amyloid Proteins, ed. E. M. Sigurdsson, M. Calero and M. Gasset, Humana Press, 2012, pp. 121–135 Search PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- D. Iglesias, S. Bosi, M. Melchionna, T. D. Ros and S. Marchesan, Curr. Top. Med. Chem., 2015 Search PubMed , in press.
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic peptide characterisation data (1H and 13C NMR, ESI-MS, HPLC), additional rheometric data, microscopy images, and cytotoxicity assay and protease test data. See DOI: 10.1039/c5tb00858a |
| This journal is © The Royal Society of Chemistry 2015 |