
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOctacalcium phosphate – a metastable mineral phase controls the evolution of scaffold forming proteins†
Wolfgang
Pompe
*a,
Hartmut
Worch
a,
Wouter J. E. M.
Habraken
b,
Paul
Simon
c,
Rüdiger
Kniep
c,
Hermann
Ehrlich
d and
Peter
Paufler
e
aInstitute of Materials Science, Technical University Dresden, D-01062 Dresden, Germany. E-mail: wolfgang.pompe@t-online.de
bMax Planck Institute of Colloids and Interfaces, Department of Biomaterials, Research Campus Golm, 14424 Potsdam, Germany
cMax Planck Institute of Chemical Physics of Solids, Nöthnitzer Str. 40, 01187 Dresden, Germany
dInstitute of Experimental Physics, TU Bergakademie Freiberg, Leipziger Str. 23, 09599 Freiberg, Germany
eInstitute of Structure Physics, Technical University Dresden, D-01062 Dresden, Germany
First published on 1st June 2015
Abstract
The molecular structure of collagen type 1 can be understood as the result of evolutionary selection in the process of formation of calcium phosphate based biocomposites acting as load bearing components in living organisms. The evolutionary selection fulfills the principle of ‘survival of the fittest’ in a particular biological environment. Disk-like post-nucleation complexes of Ca2(HPO4)32− organized in ribbon-like assemblies in the metastable octacalcium phosphate (OCP) phase, and Ca3 triangles in the stable HAP phase had formed the crystallographic motifs in this selection process. The rotational as well as the translational symmetry of the major tropocollagen (TC) helix agree nearly perfectly with the corresponding symmetries of the OCP structure. The sequence of (Gly–X–Y) motifs of the three α chains constituting the TC molecule enables an optimized structural fit for the nucleation of Ca3 triangles, the directed growth of nanostructured OCP, and the subsequent formation of hydroxyapatite (HAP) in collagen macrofibrils by a topotaxial transition. The known connection between genetic defects of collagen type 1 and Osteogenesis imperfecta should motivate the search for similar dependences of other bone diseases on a disturbed molecular structure of collagen on the genetic scale.
1. Introduction
For many years the study of biomineralization has attracted the interest of biologists, chemists, physicists, materials scientists, as well as more recently also scientists of various engineering disciplines. The discovery of a large number of specific mechanisms of biomineralization has already established innovations in chemical processing, ‘white’ biotechnology, medical techniques, as well as in materials engineering for electronic and optical applications. At the same time more general principles have been derived which can be regarded as the basis for a general theory of biomineralization.1–13 Strikingly, the large variety of biomineralization processes in nature is realized only with a limited number of biomolecular and inorganic constituents. As we would like to show by means of calcium phosphate–collagen based biocomposites as an example, this restriction is caused by the evolutionary selection of materials combinations fulfilling the principle of ‘survival of the fittest’ in a particular biological environment.Depending on the extent of the biological impact, biologically mediated, induced, and controlled mineralization are distinguished.6 In the case of distinct control of the mineralization process by the living organism, the mineralization products are characterized by high functionality precisely tailored for an optimized reaction on a specific environmental impact. Often the biological control yields a full hierarchy of mineralized structures ranging from the subnanometer scale up to some hundreds of micrometers. Load bearing structures as bones, teeth, spicules, and shells are examples of such hierarchically composed mineralized tissues. The tissues are the result of activities of cellular structures regulating the transfer of chemical and physical information on the nanoscale.14 Each of those structures is characterized by specific chemical or physical motifs governing the interactions on the nanoscale. Often, this kind of ‘nanotectonics’ is supported by the conservation or controlled transformation of internal symmetries of the interacting nanoscale structures. The limited number of such characteristic nanoscale structural motifs is an essential precondition for an effective evolution of biological structures with higher complexity. In protein evolution, for example, characteristic length increments of amino acid chains have been identified which are the structural units of closed loops (typically 25–35 residues) or domains (typically 100–200 residues).15
Under the aspect of ‘nanotectonics’, biomineralization is of particular interest as far as biomolecular motifs as structured proteins, polysaccharides, or lipids meet inorganic nanostructures as ionic pre-clusters of amorphous phases (e.g. biosilica) or crystalline phases (e.g. calcium phosphate, calcium carbonate, iron oxide). As pointed out by Stephen Mann already in 1988, molecular complementarity at the inorganic–organic interfaces governs the processes of biomineralization.13 Electrostatic accumulation of inorganic species on an organic substrate, structural correspondence (or epitaxy) between the macromolecular surface and crystal nuclei, and stereochemical requirements govern the molecular recognition at the interfaces. The molecular complementary is reflected in the chemical and physical motifs observed in biomineralization. For example, the recently discovered hydroxylated fibrillar collagen of glass sponge origin that contains an unusual chemical [Gly–3Hyp–4Hyp] motif is predisposed for silica precipitation and provides a template for biosilicification in nature.16 Otherwise, in the case of biomineralization of fluorapatite in the presence of collagen, the chemical interaction of the [Gly–X–Y] motifs with calcium and fluoride ions yields Ca3F motifs.17 The Ca3F motifs staggered along the tropocollagen fiber are the nuclei for the preferential orientation of the fluorapatite crystals along the collagen molecules.
However, there is not only a simple ‘one-way transfer’ of the structural information from the biomolecular motifs to the nucleating inorganic structures. As we will show in the following for calcium phosphate biominerals, the evolutionary optimization of their structures has led to optimized biomolecular structures controlled by inorganic structural motifs. Calcium phosphate fulfills a number of preconditions for biomineralization because of the high availability of soluble reaction components in the aqueous environment, biocompatibility, crystallization at room temperature, low solubility of the equilibrium phase, and last but not least the existence of metastable phases. The metastable phases can be formed by kinetically controlled growth (Ostwald stage rule18). For high supersaturation of the reactive components in the aqueous solution, often the initial stage is an amorphous phase.19 For nanosize structures, low interface energy (low in comparison to that of the equilibrium phase) can favor reaction paths leading to metastable phases.20,21 Furthermore, their selective stabilization can be caused by adsorbing biopolymers.9 The need for structural adaption and self-healing mechanisms of the final functional structure explains that metastable phases often can be observed in the process of biomineralization. It can be assumed that structural motifs of the favored metastable inorganic phases control the evolutionary selection of optimized organic components of the biomineral.
2. Molecular complementarity in biomineralization of calcium phosphate
2.1 Structural motifs of calcium phosphate phases
Hydroxyapatite (HAP) Ca10(PO4)6(OH)2 (in vivo commonly defect HAP Ca10−yHy(PO4)6(OH)2−y) is the thermodynamically stable phase for pH > 4 in the calcium phosphate system, whereas dicalcium phosphate dihydrate (DCPD) CaHPO4·2H2O is more stable for pH < 4. The HAP phase precipitates under in vivo conditions in hexagonal P63/m geometry with the lattice parameters aHAP = 0.9418 nm, cHAP = 0.6884 nm22 (Fig. 1a). Owing to kinetic barriers as well as depending on the supersaturation and on the presence of growth activating or inhibiting additional molecules, various transient metastable calcium phosphate phases can be observed under in vitro and in vivo conditions.23,24 In the range from pH ≅ 4 to pH ≅ 6.5 octacalcium phosphate (OCP) Ca8(HPO4)2(PO4)4·5H2O is the favored metastable phase. However, near neutral pH and high supersaturation σ (for example, σHAP = 3.36, σACP = 0.04) ACP is the first insoluble phase that precipitates on collagen, as was shown in a study by Habraken et al.25 Therefore, ACP should not be neglected as a precursor when the OCP or hydroxyapatite (HAP) formation on collagen is discussed. By a combination of in situ investigation and theoretical modeling25 it has been shown that the whole process of crystallization of calcium phosphate in solution starts with the aggregation and subsequent transformation of pre-nucleation complexes Ca(HPO4)34−. There are also other types of calcium phosphate complexes described in the literature, i.e. CaHPO4, CaPO4− and CaH2PO4+ ion pairs, as noted by the papers of Chughtai et al.,26 Lu and Leng,27 and Ohtsuki et al.28 In the study of Habraken et al.25 the presence of these ions pairs was also taken into account, representing 14% of all bound calcium in the pre-nucleation stage (mostly CaHPO4), whereas the Ca(HPO4)34−–Ca(HPO4)2(H2PO4)3− complex represented 86% of the amount of bound Ca2+ in the pre-nucleation stage. When the calcium triphosphate complex is taken into account in the speciation calculations, the amount of ion pairs logically decreases, which explains the difference in abundance of these ions pairs between the studies of Habraken et al.25 and Lu and Leng27 or Ohtsuki et al.28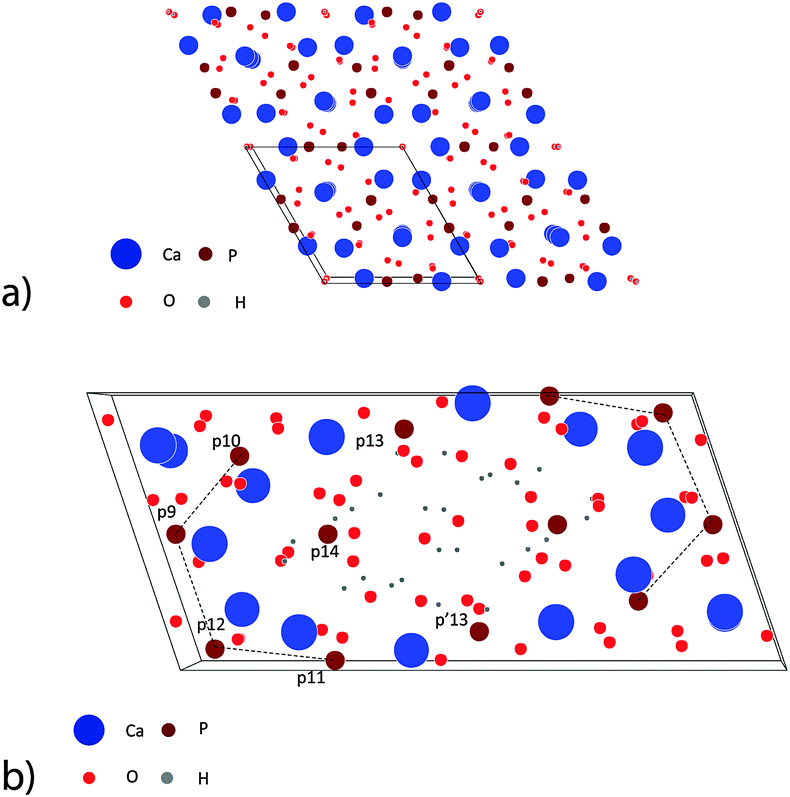 | ||
| Fig. 1 Simulation of the atomistic structure of HAP and OCP unit cells.67 (a) View on four unit cells of HAP in the [001] direction. (b) View on one unit cell of OCP in the [001] direction. The phosphate ions in the first half-cell are numbered using a notion introduced by Brown.30 p′13 denotes the phosphate ion situated mirror-symmetrically to p13 in the second half-cell. | ||
Ab initio calculations of the structure of the pre-nucleation complex result in a triangular arrangement of the phosphate ions around the Ca2+ ion. The disc-like complex has a diameter of about 1.1 nm and a height of about 0.5 nm. In a following reaction step the cluster takes up one more Ca2+ ion in the solution which leads to the precipitation of ACP that is composed of post-nucleation complexes (Ca2(HPO4)32−) with a diameter of about 1.2 nm25 (Fig. 2a). During this process, first hydrated aggregates of complexes with pre-nucleation chemistry are precipitated as a fractal polymer-like network. This structure is thermodynamically stabilized by its low interface energy in aqueous solution. The uptake of Ca2+, corresponding to the complete chemical transformation from the pre- to post-nucleation complex, leads to a densification of the amorphous network, and the appearance of the more typical spherical ACP morphology. When the Ca/P ratio increases up to Ca/P = 1, ribbons are observed which are composed of oriented post-nucleation complexes arranged in parallel fibers. When the Ca/P ratio reaches the value Ca/P = 1.33, the ribbons transform to crystalline octacalcium phosphate (OCP) Ca8(HPO4)2(PO4)4·5H2O plates. In all the phases the negatively charged post-nucleation complexes are present as characteristic structural motifs stabilized by Ca2+ ions.
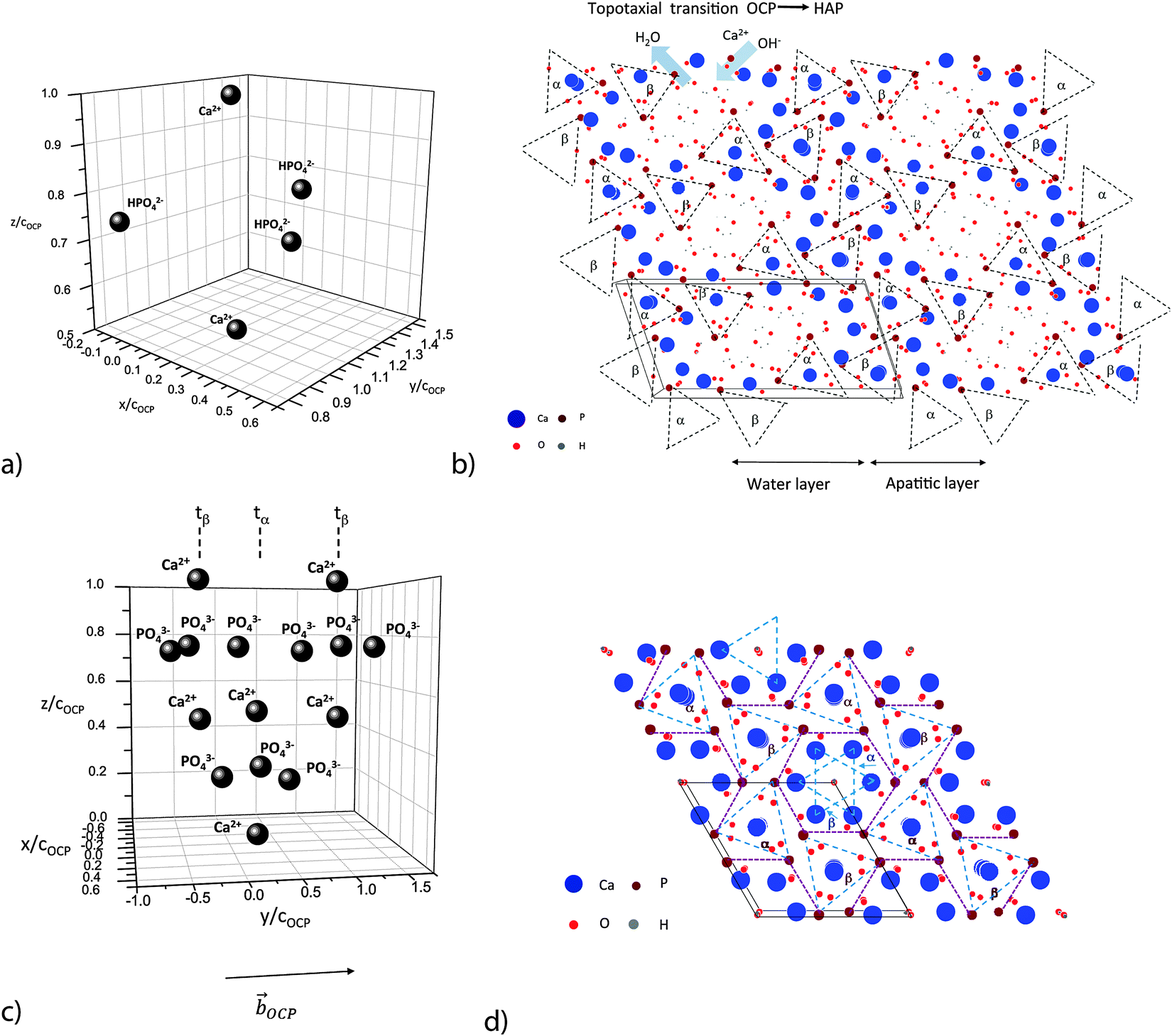 | ||
| Fig. 2 Ribbon-like assembly of Ca2(HPO4)32− motifs: (a) post-nucleation complex Ca2(HPO4)32−; cOCP = 0.687 nm, OCP lattice distance in the [001] direction. (b) View on the (001) face of 6 OCP unit cells.67 Note the ribbon-like patterns of the two families α and β of post-nucleation complexes. α and β denote triangular phosphate ion groups, placed at the (nearly) same height at the (001) plane (α := about 0.25cOCP; β := about 0.75cOCP) and sandwiched between two calcium ions situated axially at a distance of ±0.25cOCP. (c) Distribution of three deprotonated post-nucleation complexes along the [010] axis, one in a phosphate ion triangle configuration tα and two in a tβ configuration. (d) View on the (001) face of 4 HAP unit cells.67 Note the stabilization of the ribbon-like assembly of the phosphate ion triangles by the rod-like assembly of Ca2+ ion triangles. | ||
OCP grows in the triclinic space group P1, characterized by the following lattice parameters: aOCP = 1.987 nm, bOCP = 0.963 nm, cOCP = 0.687 nm, α = 90.13°, β = 92.13°, γ = 108.36°.29 The OCP shows a layered structure along the [100] direction (Fig. 1b). The unit cell of OCP is composed of two ribbon-like assemblies of post-nucleation complexes, the so-called ‘apatitic layers’, separated by a highly hydrated ‘water layer’30 (Fig. 2b). The ribbons are constituted of two periodically distributed families α and β of deprotonated post-nucleation complexes (NC), where the phosphate ion triangles tα and tβ are (nearly) placed at the (001) plane. The neighboring phosphate ion triangles tα and tβ are shifted by 0.5cOCP along the c axis as shown in Fig. 2c. The negatively charged ribbons are stabilized by two positively charged layers of Ca ions. The calcium triphosphate post-nucleation complex is also the main structural motif constituting the HAP phase25 (Fig. 2d). In the HAP crystal the highly ordered two-dimensional network of NC ribbons is stabilized by rod-like stacks of Ca2+ triangles oriented along the [001] axis. Each stack consists of a periodic assembly of pairs of Ca3(OH)5+ motifs twisted by 60° around the [001] axis. The periodicity length equals the lattice parameter cHAP.
In the OCP unit cell the phosphate and calcium ions occupy nearly the same positions as in the HAP structure. Only at two of the ‘hexagonal’ positions in the HAP unit cell, phosphate and calcium ions are missing in the OCP structure. The ‘water layer’ is a channel filled with water of hydration and ‘non-apatitic’ phosphate ions (p13 and p14 in Fig. 1b). Whereas the p14 ion occupies a region where in the HAP structure two calcium ions are placed, the p13 ions tie the apatite layers together over calcium ion bridges. Recently Wang et al. have identified a similar ‘water layer’ covering HAP crystals grown in bone as well as by biomimetic processing. By solid-state nuclear magnetic resonance, wide-angle X-ray scattering, and transmission electron microscopy a distinct thin amorphous mineral domain has been detected for Ca2+, HPO43−, and CO32−, and a rigid water network as the main components.31 It seems that there is a close correspondence to the polymer-like network of hydrated aggregates of the post-nucleation complexes observed as precursors in the experiments of Habraken et al.25
The sequential transformation of OCP to the HAP phase can proceed either by a dissolution–precipitation reaction, or by a direct solid-transformation.23,30,32 The structural similarity of the OCP and HAP structures enables HAP to grow epitaxially on the (100) surface of OCP. There are two alternative growth modes with the c-axes of HAP and OCP being either antiparallel29 or parallel,30 respectively (see also Fig. S2 in Supplementary Material 2, ESI†). Brown (1962)30 has identified a configuration where the direction [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20]HAP of the unit cell of HAP is ‘glued’ with one unit cell of OCP along the direction [010]OCP, and [0001]HAP is parallel to [001]OCP. A second model has been proposed by Fernandez et al.,29 in which [1
20]HAP of the unit cell of HAP is ‘glued’ with one unit cell of OCP along the direction [010]OCP, and [0001]HAP is parallel to [001]OCP. A second model has been proposed by Fernandez et al.,29 in which [1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 10]HAP is ‘glued’ with [010]OCP, and the c-axes of HAP and OCP are antiparallel. Using a molecular simulation model of the minimum free energy of the two growth modes, Fernandez et al. have shown that the interface energy of the mode with antiparallel c-axes is by about 5% lower than that of the mode with a parallel c-axis.29 Therefore, this transition should be thermodynamically favored. As shown by Nelson et al.,33 the lattice mismatch between HAP and OCP leads to the formation of a semi-coherent interface with a network of misfit dislocations. The solid-transformation is constrained by the motion of HPO42−, Ca2+ and OH− ions and the loss of crystal water along the (100) planes. In conclusion, the elastic misfit strains, the necessary chemical reaction, and the limited loss of crystal water can hinder the solid-transformation when the lateral width of the (100) crystal faces in the OCP crystal becomes too large.
10]HAP is ‘glued’ with [010]OCP, and the c-axes of HAP and OCP are antiparallel. Using a molecular simulation model of the minimum free energy of the two growth modes, Fernandez et al. have shown that the interface energy of the mode with antiparallel c-axes is by about 5% lower than that of the mode with a parallel c-axis.29 Therefore, this transition should be thermodynamically favored. As shown by Nelson et al.,33 the lattice mismatch between HAP and OCP leads to the formation of a semi-coherent interface with a network of misfit dislocations. The solid-transformation is constrained by the motion of HPO42−, Ca2+ and OH− ions and the loss of crystal water along the (100) planes. In conclusion, the elastic misfit strains, the necessary chemical reaction, and the limited loss of crystal water can hinder the solid-transformation when the lateral width of the (100) crystal faces in the OCP crystal becomes too large.
2.2 Structural motifs of collagen
In biomineralization, the reaction path of calcium phosphate formation is strongly influenced by the biopolymers present. Hard tissues in vertebrates, such as bone or dentin, contain collagen as the common structural protein. The assembly of the insoluble collagen macromolecules determines the final structure of the nanocomposite composed of the interpenetrating organic and inorganic phases. The three-dimensional suprafibrillar organization of the collagen governs the size, shape and arrangement of the nanocrystalline mineral phase, which results in an optimal relation between stiffness, strength, and fracture toughness of the hard tissue. Furthermore, soluble non-collagenous proteins (NCPs) as phosphoproteins, glycoproteins, and proteogylcans play a major role in nucleation and inhibition of mineral growth.6 There is still an ongoing debate on what could be the contribution of the various biopolymers to the finally formed structure.10,34 Particularly, they can stabilize amorphous precursors of the mineral phase. These so-called polymer-induced precursor phases (PILP) can initiate the intrafibrillar heterogeneous mineralization in the hole zones of collagen fibers.4 It is obvious that collagen alone can never be responsible for the mineral structure grown under in vivo conditions. However, there is clear evidence that collagen offers nucleation sites for calcium phosphate precipitation and guides the growing mineral structure.35 Recently, Wang et al. demonstrated that under in vitro conditions a highly organized bone-like HAP structure can be precipitated in a dense collagen matrix without the presence of any other biopolymers.36 This structure has been realized by mimicking an extracellular fluid which allows the co-precipitation of monodisperse collagen fibrils and carbonated hydroxyapatite by continuous injection of the carbonated apatite ion precursors with the collagen molecules.The questions arise, whether there is a straight structural relation between the molecular structure of collagen and the structural motifs of the related calcium phosphate phases enabling the biological control of mineralization, and which calcium phosphate phase has played the decisive role in the evolutionary selection of the collagen structure optimizing the mineralization process. To find an answer to these questions, we have to come back to the possible scenarios of calcium phosphate formation in a supersaturated aqueous solution. At near neutral pH, ACP formed in earlier stages transforms into the transient OCP phase which can then hydrolyze under basic conditions into HAP. Therefore, it could be hypothesized that the collagen-controlled nucleation of metastable OCP and the subsequent OCP–HAP transformation (or epitaxy mediated growth) are the critical mechanisms governing the final structure of the biocomposite. There should be molecular complementarity between the structure of the two inorganic motifs, the post-nucleation complexes Ca2(HPO4)32− in the OCP structure and the Ca3(OH)5+ motif, and the molecular structure of collagen.
The term ‘collagen’ comprises a large family of insoluble proteins acting as structural components in various connective tissues of organisms.37 The fibrous protein can form long ropes in tendon, two-dimensional sheets in skin, or cross-linked three-dimensional mineralized fiber networks in bone, for instance. In vertebrates, at least 27 collagen types with 42 distinct polypeptide chains have been identified. The fibrous collagen is composed of three polypeptide chains. These so-called α chains α1, α2, and α3 consist of a repeat of amino acid triplets (Gly–X–Y), where X and Y are often proline (Pro) or hydroxyproline (Hyp). There are homo- as well as heterotrimers. For instance, in collagen type 1 the fiber-forming molecule, the tropocollagen, is composed of two α1 chains and one α2 chain. Nowadays, collagen is considered an ancient protein with a history of more than 600 millions years as it has been identified in sponges, one of the first multicellular organisms arisen in the Precambrium.16 Probably collagen is even older because of the identification of so-called ‘fungal collagens’ within fimbriae of some fungi.38 The unexpected find of collagen in Mycota species (arisen about 1 billion years ago) suggests that it may have evolved from a common ancestor that existed before the divergence of fungi and animals. Furthermore, it was hypothesized that native fungal fimbriae can function as a mammalian extracellular matrix component. They can act as a substrate which permits corresponding animal cells to adhere, spread, and proliferate in a manner similar to animal collagens.39 The gene coding α chains of fibrillar collagen derived from the common ancestor are formed from 45 or 54 nucleotides coding 15 or 18 amino acids which correspond to five or six amino acid triplets (Gly–X–Y).40 This ancient gene has grown by duplication and mutation resulting in a family of larger genes. These genes codify for a collagen transcript in the range of 1200–1300 residues. In these molecules the (Gly–X–Y) unit is always conserved. It means that all α chains of fibrillar collagen are composed of an uninterrupted sequence of about 300 (Gly–X–Y) triplets and two shorter terminal domains (so-called telopeptides) of different structure. The primary structure of the molecules shows periodicity of 18 residues of some amino acids (lysine, glutamine and arginine) corresponding to the structure of the above-mentioned ancestor peptide, whereas polar and hydrophobic residues are arranged with a periodicity of every ∼234 residues.40 Collagen-related structural motifs (CSMs) containing (Gly–X–Y) repeats are also found in numerous proteins in procaryotic and eucaryotic organisms. There are bacterial proteins containing a CSM that could be functionally useful. They are either surface structures or spore components. Furthermore, CSMs have also been detected in viral proteins which act as structural components of the viral particle.41 The limited occurrence of CSMs in eubacteria and lower eukaryotes, and the absence of CSMs in archaebacteria, suggests that the DNA encoding CSMs has been transferred horizontally, possibly from multi-cellular organisms to bacteria.
In collagen type 1, each of the α chains forms a left-handed minor helix with 10 residues per 3 turns and a rise per turn of about 0.3 nm. The steric repulsion of the amino acids in the X-position and Y-position determines the folding of the α chains into a right-handed major helix, about 1.5 nm wide and over 300 nm long. It accommodates 30 residues per turn and has a pitch of about 8.58 nm. The neighboring helices have an angular separation of 108°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 (Fig. 3a). The tropocollagen (TC) molecule is a very reactive molecule after cleaving most of the terminal globular domains by proteases. TC undergoes spontaneously fibrillogenesis by an entropy-driven process by loss of surface water from the monomer. The supramolecular structures are initially stabilized by non-covalent interactions (e.g. polar and hydrophobic interactions). In a later stage covalent cross-linking can also occur.40
42 (Fig. 3a). The tropocollagen (TC) molecule is a very reactive molecule after cleaving most of the terminal globular domains by proteases. TC undergoes spontaneously fibrillogenesis by an entropy-driven process by loss of surface water from the monomer. The supramolecular structures are initially stabilized by non-covalent interactions (e.g. polar and hydrophobic interactions). In a later stage covalent cross-linking can also occur.40
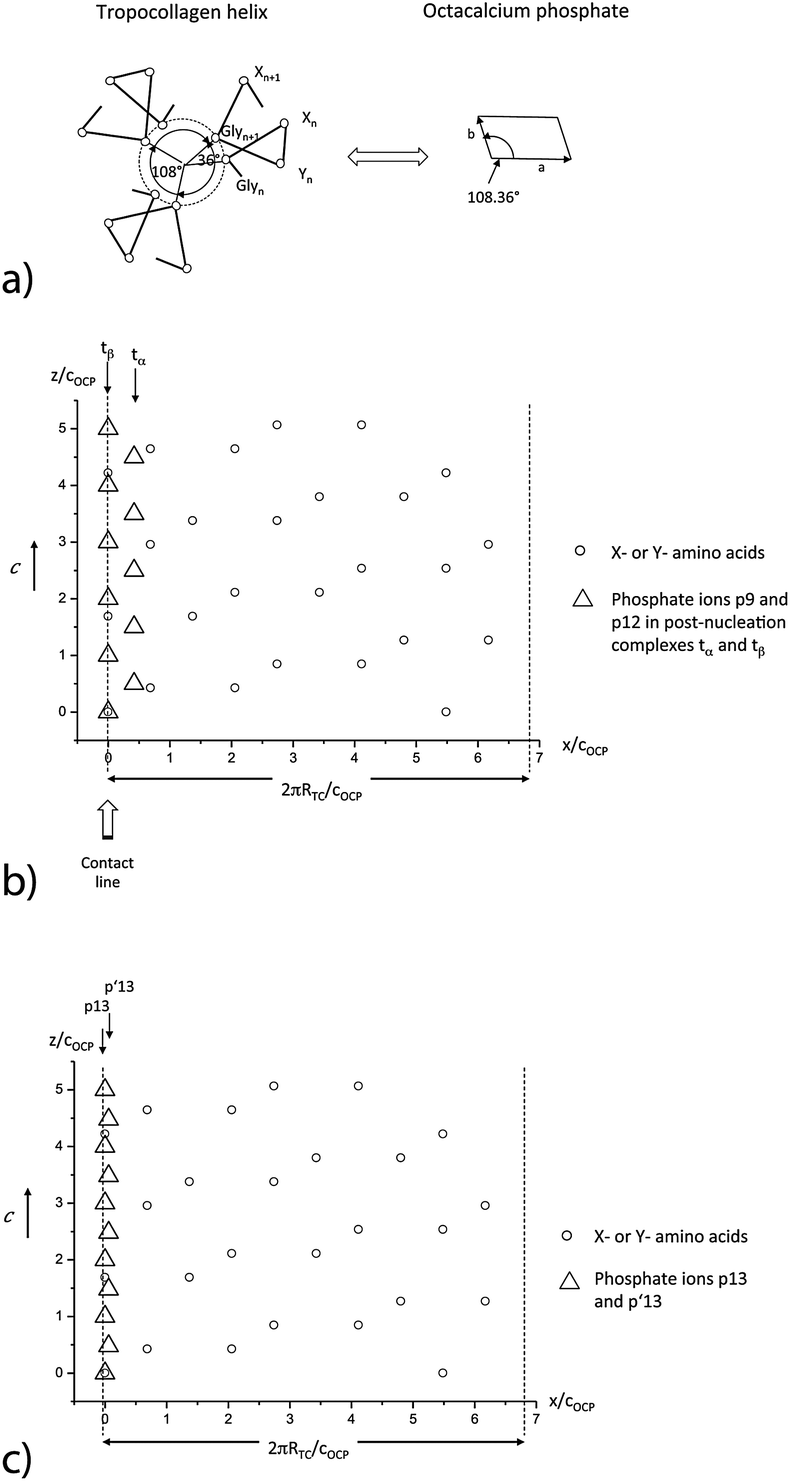 | ||
| Fig. 3 (a) Basic symmetry relationship between tropocollagen and the (001) face of octacalcium phosphate. (b) Superposition of the positions of the ‘apatitic’ phosphate ions p9 and p12 of the triangular phosphate complexes tα and tβ and of X- and Y-amino acids at the surface of a TC molecule aligned along the c axis of OCP (RTC = radius of the TC molecule); ‘apatitic’ phosphate ions p9 (tα: relative positions x/cOCP = 0.42, y/cOCP = 0.626) and p12 (tβ: relative positions x/cOCP = 0, y/cOCP = 0.099) at the (100) face. (c) Superposition of the positions the ‘non-apatitic’ phosphate ions p13 (relative positions x/cOCP = 0, y/cOCP = 1.210), and p′13 (relative positions x/cOCP = 0.058, y/cOCP = 0.190) at the (010) in the ‘water layer’ of OCP with positions of X- or Y-amino acids. | ||
The periodicity in the molecular structure of TC causes a characteristic stacking of neighboring TC molecules in the fibrils. In the stack, each TC can be divided into five segments where four of the segments have the same length of D = 67 nm but dissimilar charge profiles. The final fifth segment is only 0.4 D long. Each segment comprises positively as well as negatively charged motifs of amino acids. When TC molecules aggregate into fibrils, then adjacent molecules are staggered by 1 D. Longitudinal neighbors are separated by intervals of 0.6 D. Therefore, a periodic pattern of gaps is formed in a collagen fibril. As pointed out by Smith,43 the thinnest fiber reproducing such a structural feature is a microfibril (MF). The MF is formed by a monolayer of the five TC molecules which is close to a cylindrical shape, with a diameter of the wet molecule of about 4–5 nm (see also Fig. 5). The neighboring TC molecules have an angular separation of 72°. In the monolayer, each molecule is staggered by 1 D with respect to the adjacent molecules. Owing to the 4 successive peptide sequences of the same length and the final segment of length 0.4, five gaps of 0.6 D are disposed in a discontinuous spiral along the surface of the MF.
The high regularity of the TC molecule, showing the nanoscale periodic repeat of amino acid triplets (Gly–X–Y) with dominating proline (Pro) or hydroxyproline (Hyp) motifs as well as also the periodic arrangement of groups of positively charged amino acids (near neutral pH), indicates a possibility of a periodic deposition of inorganic structural motifs along the TC molecule. Furthermore, the simultaneous appearance of the characteristic angle of 108° in the structure of the TC molecule and of 72° in the MF as well as of the angle γ = 108.36° in the (001) plane of the OCP lattice does not seem to be incidental. These are strong indications for the evolutionary optimization of the tropocollagen molecule to provide an optimal structural fit in biomineralization of OCP as well as HAP as shown in the following section.
3. OCP-directed structure of collagen type 1
3.1 OCP adsorption sites
As the short range as well as long range order of OCP and HAP can be described by Ca2(HPO4)32− post-nucleation complexes, stabilized by additional Ca3(OH)5− motifs, this finding has been used as a starting point to analyze possible structural relationships between the metastable OCP phase (as well the HAP phase) and collagen.In order to incorporate OCP crystals into some tissue to increase stiffness and tensile strength, a highly ordered alignment of the nanosize crystals and collagen molecules along the maximum stress direction is the favored solution. In the presence of divalent ions (Ca2+), the TC molecule is positively charged near neutral pH owing to excess of basic residues in its sequence of amino acids. As shown by Freudenberg et al.,44 the isoelectric point shifts with increasing ionic strength in CaCl2 solutions from pH 7.5 to above pH 9. Therefore, Ca2(HPO4)32− post-nucleation complexes can be adsorbed at positively charged (X–Y–) motifs near neutral pH.25 At OCP crystals, the ribbon-like tαtβ-assemblies are favored adsorption sites for TC molecules on the (100) and (00) face as well as at the ‘apatitic layers’ on the (010) and (00) face of OCP. Furthermore, the ‘non-apatitic’ phosphate ions p13, p′13 situated in the ‘water layer’ (see Fig. 1b) act also as favored adsorption sites on the (010) and (00) face for the positively charged motifs of the TC molecule. Ca2+ ions can be coordinated by carbonyl oxygen (partial charges of −0.51 to −0.57) and by oxygen atoms from hydroxy groups of the Hyp residues (partial charge −0.61). In an atomistic simulation of the nucleation mechanism of fluorapatite-collagen, Kawska et al. have shown by means of a ‘model TC molecule’ built of three (Gly–Pro–Hyp)12 polypeptide chains17 that single calcium ions are incorporated between the strands of the triple helix. One to two hydrogen bonds connecting the triple helix are opened. They are replaced by electrostatic O⋯Ca⋯O interactions while keeping the overall triple helical structure without any essential change. Such structure causes a stiffening of the TC molecule. In later stages, Ca3F5+ motifs are formed lateral to the TC molecule. These triangle motifs are oriented preferentially along the TC molecule.
There is a remarkable correspondence of the molecular structure of the TC helix as well as Ca3 triangles to the periodic arrangement of the ‘apatitic’ phosphate ions which enables a perfect alignment of the TC molecule along the c-axis of OCP (Fig. 3b, for more details see Supplementary Material 1, ESI†). As the periodicity length of the OCP structure equals cOCP = 0.687 nm along the c-axis there is a fit of the charged (X–Y–) motifs of the TC molecule with the triangular arrays of phosphate ions in the tα,tβ-ribbons. The same correspondence can be observed for the p13 and p′13 phosphate ions (Fig. 3c). The periodicity of the OCP structure and the repeat length of the TC molecule lead to an ideal fit with a repeat of about 3 nm along the c-axis of OCP and the long axis of TC. Furthermore, the graphs show that the periodicity of available binding sites equals cOCP = 0.687 nm if also interaction partners with small deviations (less than 0.3 nm) in the c direction are included.
3.2 OCP mineralization of tropocollagen
The remarkable similarity of the symmetry of the (001) plane of OCP (angle γ = 108.36°) and the axial symmetry of TC molecules (characteristic angle of 108°) suggests the idea that the cross-section of the TC molecule, too, is geometrically optimized for its embedding into the calcium phosphate matrix. To substantiate this hypothesis we consider the various stages of the formation of OCP on collagen type 1 by means of a simplified phenomenological model. For this purpose we can start from:• experimental data on the initial stages of calcium phosphate precipitates in a supersaturated aqueous solution of Ca2+ ions and Ca(HPO4)34−–Ca2(HPO4)32− complexes recently published by Habraken et al.,25
• extensive studies of the solid-to-solid OCP–HAP transition.29–32
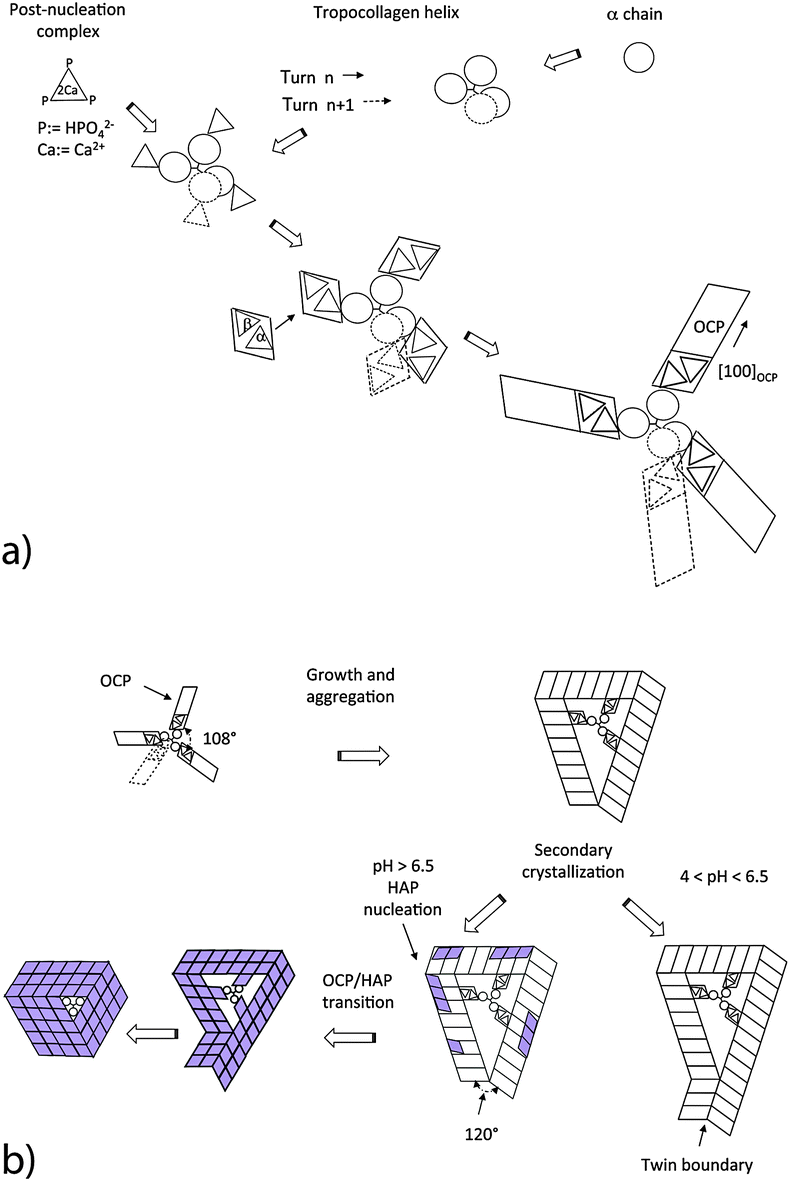
The mineralization is initiated by a random nucleation of oriented post-nucleation complexes along a single TC molecule functionalized with Ca3 triangles (Fig. 4a). The adsorbed Ca2(HPO4)32− complexes act as nucleation sites for the growth of crystalline OCP unit cells. The symmetry fit between individual OCP nuclei and the helical arrangement of the three α chains results in a helical arrangement of crystalline OCP units leading to a ‘replica’ of the major TC helix. Possible reaction steps of OCP nucleation are schematically shown in Fig. 4a. The known structure of OCP and the result of HRTEM studies on trabecular rat bone (P. Simon, private communication, 2015) have led us to the assumption that adsorbed post-nucleation complexes (NCs) together with additional Ca ions and ordered water molecules form nuclei at the individual α chains. The NCs organized in ribbon-like structures are oriented parallel to the fiber axis of the TC molecule. From these progenitor nuclei, complete OCP unit cells can grow oriented with their [100] axis radially to the TC molecule. With their circumferential distribution they follow the 108° pattern of the α chains of the TC molecule. The growth of the individual crystalline units is governed by the differences of the various interface energies. Analogous to the growth of OCP in a collagen-free solution, it can be assumed that thin OCP platelets are formed with the (100) face of minimum interface energy (Fig. 4b). The favored growth directions of the individual platelets are the [010] and [001] directions. The platelets are arranged along the TC molecule randomly. When the TC helix is completely covered with OCP crystal units, a so-called mesocrystal45 of initially independently grown crystalline platelets is formed by secondary crystallization. Usually the structure of the mesocrystal will not be highly ordered. However, proteins with their high structural regularity are promising candidates for higher biological control.
 | ||
| Fig. 4 Sequence of nucleation and growth steps of an OCP coating on an individual TC molecule, and the following HAP nanorod formation (view along the [001] axis). (a) Site specific nucleation. (b) OCP growth with the radially oriented [100] axis of the unit cells. | ||
The various α chains have almost identical charge motifs (at least the two α1 chains). Thus we assume that the OCP distribution of binding sites along the TC-fibril follows the right handed major helix. After a full turn of the major helix the shift of binding sites is about 0.9 nm along the TC axis. As the lattice parameter cOCP = 0.686 nm, the OCP crystal can form a helical structure with a screw dislocation in the center oriented along the c-axis when the TC template shrinks in the axial direction during the secondary crystallization of OCP. As shown in Fig. 4b, the OCP symmetry permits only three crystalline units to aggregate into an undisturbed coating. A fourth unit would not fit with the first one. However, three OCP units of equal length would cause a structural defect related to a gap of 34.92° per cycle in the (001) plane (because of the angle γ = 108.36° of the unit cell in the (001) plane). This gap can be closed by the nucleation of a twinned layer at the defect corner (Fig. 4b).
3.3 OCP mineralization of microfibrils
A similar correspondence of the crystallographic symmetries can be observed in the case of OCP mineralization of the collagen microfibril (Fig. 5). Depending on the particular stacking geometry of the individual TC molecules along the Smith five-stranded microfibril,43 it has to be distinguished between nucleation in the region of overlap of all five TC molecules and in the gap region missing one of the TC molecules in the cross section of the MF. Obviously, the five-fold symmetry of the microfibril enables a perfect fit of neighboring OCP crystals nucleated at single TC molecules as the angle of 360°/5 = 72° between neighboring TC molecules is approximately equal to the complementary angle 180°–108.36° = 71.64° of the (001) face of the OCP unit cell. Other than with mineralization of individual TCs, a defect-free complete OCP coating is formed in this case. It can be assumed that the contact interfaces between the touching OCP units are amorphous layers composed of Ca2(HPO4)32− post-nucleation complexes and a water-network. Thus the interface energy Egb per microfibril length LMF can be described phenomenologically as| Egb/LMF = 5Δgb·T, | (1) |
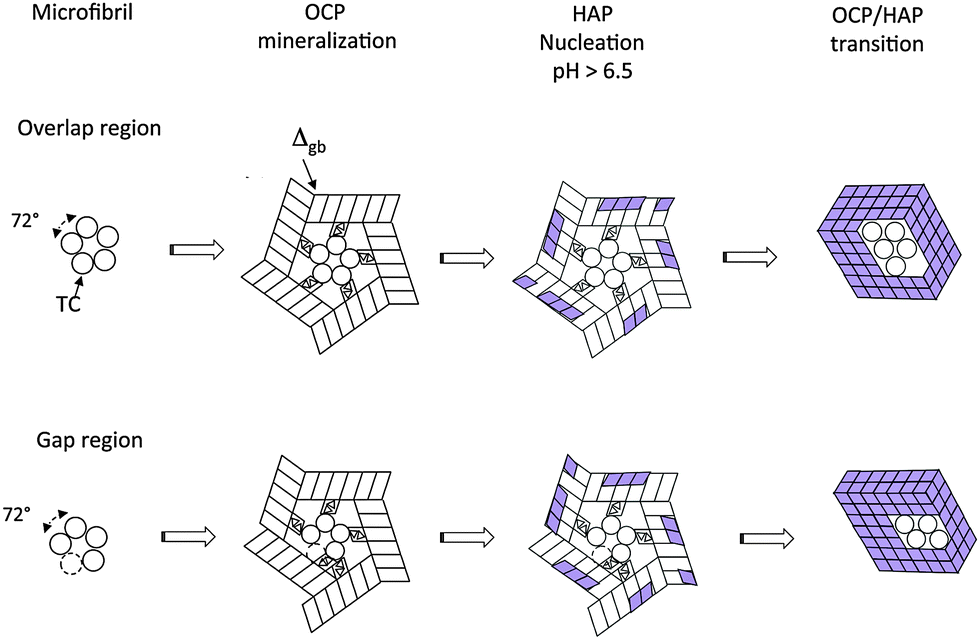 | ||
| Fig. 5 Mineralization of a microfibril at its overlap region and at its gap region. Details of the reaction path. | ||
4. Structure change by OCP–HAP transition
The structures derived in the last section can only exist metastably in the range from pH 4 to pH 6.5 or as a transient phase for pH > 6.5. There are numerous experimental studies analyzing the OCP–HAP transition in vitro.29,32,33,46 This transition has been observed by Raman spectroscopic experiments in vivo, too.47,48 As pointed out by Zhan et al.,32 the ‘apatitic layers’ of the OCP unit cell act as nucleation sites for the HAP phase. The OCP–HAP transition can be triggered by a change of the external process conditions increasing the pH from acidic to alkaline (see also the example for producing HAP nanorods, discussed in Section 5). As OCP is metastable near neutral pH, the transition can be initiated also by in-diffusion of Ca2+ and OH− ions and out-diffusion of H2O as schematically shown in Fig. 2b.Nuclear magnetic resonance (NMR) spectroscopy data reveal that the crystal transforms topotaxially.46 It means that the lattice transformation, which can be connected with a chemical reaction, results in a product phase showing one or more crystallographically equivalent, orientational relationships to the lattice of the original phase. The NMR studies revealed that the OCP–HAP transition mode with a parallel c-axis is favored. Phosphate ions, Ca2+, OH− ions and water molecules can move along the hydrated channels of the OCP ‘water layer’. Short displacements (about 0.5 nm) of the phosphate ions and their chemical reaction with additional Ca2+ and OH− ions diffusing into the ‘water layer’ are needed to yield the HAP structure. Starting from structures derived in the previous section, possible sequences of transition stages which lead to HAP mineralized individual TC molecules and microfibrils are shown in Fig. 4b and 5, respectively. First, HAP nuclei should be formed randomly at the surface of the OCP layer. The lattice misfit between OCP and HAP causes residual stresses during the phase transition. Stress relaxation due to microtwinning and surface diffusion leads to a remarkable change of the overall structure (see also Supplementary Material 2, ESI†). HAP microtwins are favored in the presence of anion substituents in the lattice. Particularly, the substitution of phosphate ions by CO32− leads to a twinned structure.49 With proceeding transformation the symmetry change of the crystals also causes a deformation of the compliant TC and microfibril structures. The angle between neighboring α chains shifts to 120°, which in the case of mineralized individual TC molecules leads to prismatic HAP-TC nanorods with a triangular cross section. The TC molecule is completely enclosed by the three single crystalline HAP platelets. In the case of a mineralized microfibril, the hexagonal symmetry of the HAP lattice leads to an overall shape change (Fig. 5). Stress-driven microtwinning is probably the dominating stress relaxation mechanism in HAP crystals. The shape instability of axially compressed microfibrils has been already identified in thicker non-mineralized collagen fibrils by Hulmes et al.50 They have studied the radial packing and axial order of non-mineralized collagen macrofibrils using X-ray diffraction. A satisfying simulation of the experimental diffraction patterns has demanded the implementation of compressed microfibrils in order to reproduce the observed combination of sharp Bragg peaks and underlying diffuse scatter.
It is not obvious that the OCP structure always transforms completely into a HAP structure as assumed in the schematics given in Fig. 4b and 5. There are at least two reasons which can hinder the complete transition. First, HPO42− ions are deprotonated during the OCP–HAP transition.
| HPO42− + OH− = PO43− + H2O | (2) |
5. Discussion
The analysis of the crystallographic structure of OCP and HAP as well as of collagen shows that the molecular structures of the TC molecule and the five-stranded microfibril of collagen type 1 perfectly fulfill the conditions needed for biologically controlled precipitation of collagen reinforced HAP. That is the result of the successful evolutionary selection of the biomolecular template directed by the metastable OCP as well as the stable HAP phase. The selection process is governed by triangular disc-like post-nucleation complexes Ca2(HPO4)32− and triangular Ca3(OH)5+ motifs. Their interacting assemblies had been the ‘blueprints’ for the optimized selection of the heterotrimeric structure of tropocollagen. There is a direct crystallographic correspondence of the structure of OCP as well as HAP to the molecular composition of the chains α1, α2 and α3 consisting of the repeat of amino acid triplets Gly–X–Y. The (Gly–X–Y) motifs enable a controlled arrangement of (001) oriented crystalline OCP units in three-fold symmetry. It leads to an alignment of the major helix of TC along the c-axis of OCP caused by a zippered binding of the TC molecule at triangular ‘apatitic’ phosphate stacks in the ribbons or at ‘non-apatitic’ phosphate ions in the ‘water layer’ of the OCP crystal.The repeat of (Gly–X–Y) motifs causes the fivefold symmetry of the cross section of the microfibril constituting the basic structural unit of thicker collagen type 1 fibers. As noted already by Smith,43 the minimum number of five TC molecules arranged as a hollow cylinder yields a structure with the highest ratio of molecules to lumen, which provides the high stability under bending stress. The formation of long strong fibers composed of TC molecules of finite lengths demands that the five TC molecules are arranged in a staggered structure with a repeat length D. Due to symmetry reasons each TC molecule consists of four segments equal to D and a shorter fifth segment (in the case of TC only 0.4 D long). Insofar not only the local, but also the long-range structure of the TC molecule is determined to a great extent by OCP crystallography. As shown by Buehler the segment length D can be derived from additional selection criteria optimizing the mechanical response of the collagen type 1 fibrils.51 Optimization of strength and of energy dissipation results in a length on the order of a few hundred nanometers. This model is supported by the observed experimental values of the TC length of about ≈300 nm, which corresponds to a D value of about D ≈ 67 nm.
Obviously, the basic structural motif for the selection of the collagen structure is related to the ribbon-like assemblies of Ca2(HPO4)32− NCs governing the precipitation kinetics of calcium phosphate phases in an aqueous solution. The comparison of Fig. 2b, c and 3b shows that the TC molecule offers appropriate adsorption sites for the tαtβ ribbon arranged along the (010) face of OCP. Adsorbed tαtβ ribbons can act as progenitors for the nucleation of OCP crystals at individual α chains. In the OCP mineralization model studied in Sections 3.2 and 3.3 it is assumed that the OCP unit cells are radially oriented with their [100] axis along both the TC helix and the microfibril, whereas the [001] axis is directed parallel to the fiber axis. The main growth directions of the OCP platelets are the [010] and [001] directions.
The models of a mineralized tropcollagen molecule and microfibril shown in Fig. 4b and 5 could find their experimental correspondence in structures produced by biomimetic mineralization of gelatin or low-concentrated suspensions of non-cross-linked microfibrils of collagen type 1.52,53 For instance, the fast Fourier Transform of HRTEM images of fluorapatite–gelatin composite show superstructure streaks with a periodicity of about 5 nm caused by a patterned array of individual FAP-coated tropollagen molecules. This a highly mosaic-controlled nanocomposite superstructure, which can be modeled by prismatic rod-like units with a diameter of about 10 nm.52,53 The individual unit agrees fairly well with the relaxed HAP-TC structure shown in Fig. 4b. Recently, Li et al.54 have reported the subfibrillar structure of mineralized collagen fibrils. Mineralized microfibrils as well as densely-packed macrofibrils have been observed by HRTEM. Transversely sectioned mineralized individual compressed microfibrils had been described as a core–shell structure with a collagen core (a ≈ 3.20 nm, b ≈ 2.16 nm) and a HAP shell (≈3 nm thick). The collagen spacing changed from about 1.55 nm in non-mineralized fibrils to 1.24 nm in mineralized microfibrils. Such a compression of the TC molecules in mineralized microfibrils results also in the above derived model of the OCP–HAP transition as shown in Fig. 5. The hypothesized optimized fit of the collagen structure as a precondition for regular biomineralization of HAP is supported by the observation of a direct connection between anomalous growth of calcium phosphate phases and a disturbed molecular structure of collagen in genetic studies of bone diseases as Osteogenesis imperfecta (OI), a heritable disease in children characterized by high fragility of bone. Mutations in the two structural genes for type 1 procollagen (Coll1A1 and Coll1A2) have been identified in probands suffering under OI. The devastating effect of such mutations seems to be linked with disturbance of the folding of the triple-helical TC molecules or of their self-assembly in fibrils. As described by Kuivaniemi et al.,55 more than 70 mutations in type 1 genes are involved in OI. Particularly, mutations are observed in which glycine is substituted with bulkier amino acids as arginine, valine, glutamic acid, aspartic acid for triple helix N-terminal positions >178 (i.e. 178th residue starting from N-terminal), and with cysteine for triple-helix N-terminal positions >688.56 This glycine substitution prevents the correct folding of triple-helical TC molecules. Unfolded molecules accumulate in osteoblasts and are degraded during secretion. There are other glycine substitutions which do not disturb the TC folding, however they can hinder the nucleated growth of the collagen fibrils. For instance, substitution cysteine for glycine at position α1-748 causes cysteine kinks in the TC molecule. The morphology of the grown fibrils is strongly changed. The fibrils are highly branched.55 The loss of well-organized TC molecules and the formation of disturbed collagen fibrils lead to serious disturbances of bone mineralization. For instance, Fratzl et al.57 have shown in an OI mouse model that already a homotrimer produced with only α1 chains provides remarkable disturbances of the shape and size of the mineral crystals. Large mineral blocks (with all dimensions typically exceeding 50 nm) and a family of thin, possibly needle-like crystals were observed in cortical bone. Furthermore, differences in the chemical composition of the mineral phase in comparison to the normal bone were detected by Fourier transform infrared microscopy.58 X-ray diffraction studies on non-mineralized OI mouse tendon have indicated that the α1 chain substitution for the α2 chain results in the loss of crystalline lateral packing of the TC molecules in the macrofibers.59 The lower hydrophobic content of the α1 chain results in an increase in the molecular spacing in the collagen fibrils. By atomistic simulation Chang et al. have shown that the persistence length of the homotrimer is reduced in comparison to the heterotrimer, and an enhanced kinking of the collagen molecules in the microfibril is observed.60 Obviously, the incorporation of the α2 chain is critical for the crystalline lateral packing of fibrillar collagen which results in the optimized control of the growth of an ordered mesocrystalline HAP reinforcement.
The close structural relationship between collagen type 1 and OCP as well as HAP can be used also for the design of optimized biomimetic precursors for the in vitro formation of defined HAP nanostructures. A first example has been given by Zhan et al.,32 who produced single-crystalline HAP nanorods. Bundles of single-crystalline OCP nanorods were produced by precipitation of OCP in gelatin under acidic conditions in the presence of urea. Thus a porous biocomposite was formed. The nanorods, about 100 nm wide, had a length of hundreds of micrometers. By subsequent decomposition of urea, the pH rose above 7.4, which led to the topotaxial transformation into c-axis oriented single-crystalline HAP nanorods without change in the morphology of the initial OCP structure. Owing to the small diameter of the nanorods, mismatch strains formed by the transformation can relax as explained already in Section 4. The authors have in vision a possible use of such bundles of HAP nanorods for bone replacement.
The formation of optimized protein structures by evolutionary selection driven through a structural motif of inorganic components seems to be a general principle. Therefore, it is not surprising to find further examples in connection with the creation of structural components in living organisms. One remarkable example is given with the structure of osteocalcin (OC). OC is a multifunctional protein which plays a major role in the regulation of osteoblast and osteoclast activity, in insulin secretion, glucose metabolism, and fat mass production.61 It is the most abundant non-collagenous protein in bone. As proven by immunochemistry, OC is present at the surface of mineralized collagen macrofibrils as well as within them.62 Recently it has been shown that OC together with osteopontin forms dilatational bands connecting extrafibrillar HAP platelets.63 This mechanism gives an essential contribution to the high fracture toughness of bone. The mechanism is based on a high binding efficiency of OC to HAP. Inside the collagen fibrils it has been detected in part of the gap region and the neighboring overlap zone. Thus it is assumed that OC mediates nucleation, growth, and development of the platelet-shaped HAP crystals.62 OC contains 46–50 amino acids, depending on the species. It folds into two antiparallel α helices framed by β sheet structures. Three γ-carboxyglutamic acids (Gla) placed in the α1 helix and Asp–Glu residues of the α2 helix possess a strong binding efficiency to Ca2+ ions. As shown by molecular modeling, the three Gla-residues together with the Asp-residue can coordinate Ca2+ ions on the (100) face of HAP, with minor differences in the displacements needed for a coordination binding, and also on the secondary prism face (110) of HAP.64 In view of the high structural similarity of the ‘apatitic layers’ of OCP with the HAP structure, a similar optimized fit of OC with the OCP structure can be assumed. Indeed, the distance of neighboring Ca ions of the ribbon pattern on the (100) face of OCP of about 0.56 nm (as shown in Fig. 2c) fits fairly well with the distance of neighboring Gla-residues in the α1 helix of OC of about 0.54 nm.
The selection of optimized macromolecular templates by the symmetry of the targeted inorganic component seems to be a promising approach for the design of biomimetic processes in biomineralization. Recently, an indirect example of such strategy has been provided for template controlled nucleation of calcite on self-assembled monolayers (SAMs) by Hamm et al.65 By an appropriate choice of the functional group chemistry (COOH, PO4, SH or OH) of the head groups of the SAMs and the conformation of their molecules (length of alkanethiol chains) the stereochemical matching has been changed. As shown by atomistic simulation,66 the structural motifs of the growing calcite nuclei cause a distinctive change in the arrangement of the head groups of the SAM. The self-organized structure selection of the macromolecular template results in different shapes of calcite crystals grown on the SAMs.
6. Conclusions
The molecular structure of collagen type 1 can be understood as the result of evolutionary selection in the process of formation of calcium phosphate based biocomposites acting as load bearing components in living organisms. Disk-like post-nucleation complexes of Ca2(HPO4)32− organized in ribbon-like assemblies in the metastable OCP phase and Ca3 triangles in the stable HAP phase had formed the crystallographic motifs in this selection process. The rotational as well as the translational symmetry of the major TC helix agree nearly perfectly with the corresponding symmetries of the OCP structure. The sequence of (Gly–X–Y) motifs of the three α chains constituting the TC molecule enables an optimized structural fit for the nucleation of Ca3 triangles, the directed growth of nanostructured OCP and the subsequent formation of HAP in collagen macrofibrils by a topotaxial transition. The known connection between genetic defects of collagen type 1 and Osteogenesis imperfecta should motivate the search for similar dependences of other bone diseases on a disturbed molecular structure of collagen on the genetic scale. It can be assumed that the evolutionary selection of biomolecular templates governed by metastable inorganic crystalline phases is a general feature of biomineralization processes. For the biomimetic formation of biohybrid crystalline inorganic materials, it could be a useful approach to derive the structure of an appropriate biomolecular template from characteristic symmetry motifs of the crystalline structure of the intended inorganic target material.Acknowledgements
We thank Günter Reich and Michael Meyer for helpful discussions of details of the molecular structure of collagen, as well as Michael Ruhnow and Karsten Böhme for their support in the deeper understanding of crystallographic aspects of the interaction of OCP with tropocollagen. We also thank Peter Fratzl and Hans-Jürgen Weiss for critical reading of the final manuscript and valuable discussions of experimental details of the mineralized nanostructures.References
- A. H. Heuer, D. J. Fink, V. J. Laraia, J. L. Arias, P. D. Calvert, K. Kendall, G. L. Messing, J. Blackwell, P. C. Rieke, D. H. Thompson, A. P. Wheeler, A. Veis and A. I. Caplan, Science, 1992, 255, 1098–1105 CAS.
- S. Mann, ed. Biomimetic Materials Chemistry, VCH, New York, 1996 Search PubMed.
- S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 41, 21–40 CrossRef CAS.
- M. J. Olszta and X. Cheng, et al. , Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef PubMed.
- E. Baeuerlein, ed. Biomineralization - From Biology to Biotechnology and Medical Application, Wiley-VCH, Weinheim, 2000 Search PubMed.
- S. Mann, Biomineralization: principles and concepts in bioinorganic materials chemistry, Oxford University Press, Oxford, 1st edn, 2001 Search PubMed.
- L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4110–4114 CrossRef CAS.
- S. Mann, D. D. Archibald, J. M. Didymus, T. Douglas, B. R. Heywood, F. C. Meldrum and N. J. Reeves, Science, 1993, 261, 1286–1292 CAS.
- H. Cölfen and S. Mann, Angew. Chem., 2003, 42, 2350–2365 CrossRef PubMed.
- F. Nudelman, K. Pieterse, A. George, P. H. Bomans, H. Friedrich, L. J. Brylka, P. A. Hilbers, G. de With and N. A. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- R. Kniep and P. Simon, Angew. Chem., 2008, 47, 1405–1409 CrossRef CAS PubMed.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS PubMed.
- S. Mann, Nature, 1988, 332, 119–124 CrossRef CAS PubMed.
- S. Mann, Angew. Chem., 2008, 47, 5306–5320 CrossRef CAS PubMed.
- E. N. Trifonov and I. N. Berezovsky, Curr. Opin. Struct. Biol., 2003, 13, 110–114 CrossRef CAS.
- H. Ehrlich, R. Deutzmann, E. Brunner, E. Cappellini, H. Koon, C. Solazzo, Y. Yang, D. Ashford, J. Thomas-Oates, M. Lubeck, C. Baessmann, T. Langrock, R. Hoffmann, G. Worheide, J. Reitner, P. Simon, M. Tsurkan, A. V. Ereskovsky, D. Kurek, V. V. Bazhenov, S. Hunoldt, M. Mertig, D. V. Vyalikh, S. L. Molodtsov, K. Kummer, H. Worch, V. Smetacek and M. J. Collins, Nat. Chem., 2010, 2, 1084–1088 CrossRef CAS PubMed.
- A. Kawska, O. Hochrein, J. Brickmann, R. Kniep and D. Zahn, Angew. Chem., 2008, 47, 4982–4985 CrossRef CAS PubMed.
- W. Z. Ostwald, Z. Phys. Chem., 1897, 22, 289–330 CAS.
- P. Raiteri and J. D. Gale, J. Am. Chem. Soc., 2010, 132, 17623–17634 CrossRef CAS PubMed.
- A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12096–12101 CrossRef CAS PubMed.
- A. E. Nielsen and O. Söhnel, J. Cryst. Growth, 1971, 11, 233–242 CrossRef CAS.
- M. I. Kay, R. A. Young and A. S. Posner, Nature, 1964, 204, 1050–1052 CrossRef CAS PubMed.
- M. S. Johnsson and G. H. Nancollas, Crit. Rev. Oral Biol. Med., 1992, 3, 61–82 CAS.
- S. Recillas, V. Rodriguez-Lugo, M. L. Montero, S. Viquez-Cano, L. Hernandez and V. M. Castano, Ceram. Process., 2012, 13, 5–10 Search PubMed.
- W. J. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. Sommerdijk, Nat. Commun., 2013, 4, 1507–1519 CrossRef PubMed.
- A. Chughtai, R. Marshall and G. H. Nancollas, J. Phys. Chem., 1968, 72, 208–211 CrossRef CAS.
- X. Lu and Y. Leng, Biomaterials, 2005, 26, 1097–1108 CrossRef CAS PubMed.
- C. Ohtsuki, T. Kokubo and T. Yamamuro, J. Non-Cryst. Solids, 1992, 143, 84–92 CrossRef CAS.
- M. E. Fernandez, C. Zorrilla-Cangas, R. Garcia-Garcia, J. A. Ascencio and J. Reyes-Gasga, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 175–181 CrossRef CAS PubMed.
- W. E. Brown, Nature, 1962, 196, 1050–1055 CrossRef CAS PubMed.
- Y. Wang, S. Von Euw, F. M. Fernandes, S. Cassaignon, M. Selmane, G. Laurent, G. Pehau-Arnaudet, C. Coelho, L. Bonhomme-Coury, M. M. Giraud-Guille, F. Babonneau, T. Azais and N. Nassif, Nat. Mater., 2013, 12, 1144–1153 CrossRef CAS PubMed.
- J. Zhan, Y.-H. Tseng, J. C. Chan and C.-Y. Mou, Adv. Funct. Mater., 2005, 15, 2005–2010 CrossRef CAS PubMed.
- D. G. Nelson and J. D. McLean, Calcif. Tissue Int., 1984, 36, 219–232 CrossRef CAS.
- F. Nudelman, A. J. Lausch, N. A. Sommerdijk and E. D. Sone, J. Struct. Biol., 2013, 183, 258–269 CrossRef CAS PubMed.
- W. J. Landis and R. Jacquet, Calcif. Tissue Int., 2013, 93, 329–337 CrossRef CAS PubMed.
- Y. Wang, T. Azais, M. Robin, A. Vallee, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M. M. Giraud-Guille and N. Nassif, Nat. Mater., 2012, 11, 724–733 CrossRef CAS PubMed.
- J. Brinckmann, H. Notbohm and P. K. Müller, Eds. Collagen: Primer in Structure, Processing and Assembly, Springer, Berlin, Heidelberg, 2005 Search PubMed.
- M. Celerin, J. M. Ray, N. J. Schisler, A. W. Day, W. G. Stetler-Stevenson and D. E. Laudenbach, EMBO J., 1996, 15, 4445–4453 CAS.
- H. Ehrlich, Int. Geol. Rev., 2010, 52, 661–699 CrossRef PubMed.
- V. Ottani, D. Martini, M. Franchi, A. Ruggeri and M. Raspanti, Micron, 2002, 33, 587–596 CrossRef CAS.
- J. K. H. Bamford and D. H. Bamford, Virology, 1990, 177, 445–451 CrossRef CAS.
- A. Bhattacharjee and M. Bansal, IUBMB Life, 2005, 57, 161–172 CrossRef CAS PubMed.
- J. W. Smith, Nature, 1968, 219, 157–158 CrossRef CAS PubMed.
- U. Freudenberg, S. H. Behrens, P. B. Welzel, M. Muller, M. Grimmer, K. Salchert, T. Taeger, K. Schmidt, W. Pompe and C. Werner, Biophys. J., 2007, 92, 2108–2119 CrossRef CAS PubMed.
- H. Cölfen and M. Antonietti, Angew. Chem., 2005, 44, 5576–5591 CrossRef PubMed.
- Y. H. Tseng, C. Y. Mou and J. C. Chan, J. Am. Chem. Soc., 2006, 128, 6909–6918 CrossRef CAS PubMed.
- N. J. Crane, V. Popescu, M. D. Morris, P. Steenhuis and M. A. Ignelzi Jr., Bone, 2006, 39, 434–442 CrossRef CAS PubMed.
- M. Bennet, A. Akiva, D. Faivre, G. Malkinson, K. Yaniv, S. Abdelilah-Seyfried, P. Fratzl and A. Masic, Biophys. J., 2014, 106, L17–L19 CrossRef CAS PubMed.
- M. E. Fleet, X. Liu and P. L. King, Am. Mineral., 2004, 89, 1422–1432 CAS.
- D. J. Hulmes, T. J. Wess, D. J. Prockop and P. Fratzl, Biophys. J., 1995, 68, 1661–1670 CrossRef CAS.
- M. J. Buehler, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12285–12290 CrossRef CAS PubMed.
- P. Simon, W. Carrillo-Cabrera, P. Formanek, C. Göbel, D. Geiger, R. Ramlau, H. Tlatlik, J. Budera and R. Kniep, J. Mater. Chem., 2004, 14, 2218–2224 RSC.
- P. Simon, U. Schwarz and R. Kniep, J. Mater. Chem., 2005, 15, 4992–4996 RSC.
- Y. Li and C. Aparicio, PLoS One, 2013, 8, e76782 CAS.
- H. Kuivaniemi, G. Tromp and D. J. Prockop, FASEB J., 1991, 5, 2052–2060 CAS.
- D. K. Dubey and V. Tomar, Appl. Phys. Lett., 2010, 96, 023703 CrossRef PubMed.
- P. Fratzl, O. Paris, K. Klaushofer and W. J. Landis, J. Clin. Invest., 1996, 97, 396–402 CrossRef CAS PubMed.
- N. P. Camacho, W. J. Landis and A. L. Boskey, Connect. Tissue Res., 1996, 35, 259–265 CrossRef CAS.
- D. J. McBride Jr., V. Choe, J. R. Shapiro and B. Brodsky, J. Mol. Biol., 1997, 270, 275–284 CrossRef CAS PubMed.
- S. W. Chang, S. J. Shefelbine and M. J. Buehler, Biophys. J., 2012, 102, 640–648 CrossRef CAS PubMed.
- A. Neve, A. Corrado and F. P. Cantatore, J. Cell. Physiol., 2013, 228, 1149–1153 CrossRef CAS PubMed.
- L. Chen, R. Jacquet, E. Lowder and W. J. Landis, Bone, 2015, 71, 7–16 CrossRef CAS PubMed.
- O. Nikel, D. Laurencin, S. A. McCallum, C. M. Gundberg and D. Vashishth, Langmuir, 2013, 29, 13873–13882 CrossRef CAS PubMed.
- Q. Q. Hoang, F. Sicheri, A. J. Howard and D. S. Yang, Nature, 2003, 425, 977–980 CrossRef CAS PubMed.
- L. M. Hamm, A. J. Giuffre, N. Han, J. Tao, D. Wang, J. J. De Yoreo and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1304–1309 CrossRef CAS PubMed.
- C. L. Freeman, Q. Hu, M. H. Nielsen, J. Tao, J. J. De Yoreo and J. H. Harding, J. Phys. Chem. C, 2013, 117, 5154–5163 CAS.
- W. Kraus and G. Nolze, PowderCell for Windows Version 2.4, BAM, Berlin, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00673b |
| This journal is © The Royal Society of Chemistry 2015 |