
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatially nanoscale-controlled functional surfaces toward efficient bioactive platforms
Dhruvajyoti
Roy
a and
Joon Won
Park
*ab
aNanogea Inc., 6162 Bristol Parkway, Culver City, CA 90230, USA
bDepartment of Chemistry, Division of Integrative Biosciences and Biotechnology, National Core Research Center for Systems Bio-Dynamics, Pohang University of Science and Technology, San 31 Hyoja-dong, Pohang, 790-784, Korea. E-mail: jwpark@postech.ac.kr; Fax: +82-054-279-0653
First published on 26th May 2015
Abstract
Interest in well-defined surface architectures has shown a steady increase, particularly among those involved in biological applications where the reactivity of functional groups on the surface is desired to be close to that of the solution phase. Recent research has demonstrated that utilizing the self-assembly process is an attractive and viable choice for the fabrication of two-dimensional nanoscale-controlled architectures. This review highlights representative examples for controlling the spatial placement of reactive functional groups in the optimization of bioactive surfaces. While the selection is not comprehensive, it becomes evident that surface architecture is one of the key components in allowing efficient biomolecular interactions with surfaces and that the optimized lateral spacing between the immobilized molecules is crucial and even critical in some cases.
 Dhruvajyoti Roy | Dhruvajyoti Roy obtained his BSc and MSc degrees in chemistry from the University of Calcutta and C.S.J.M University, respectively. He studied at Pohang University of Science and Technology (Korea) and completed his PhD under the mentorship of Professor Joon Won Park in 2011. His research is focused on the development of functionalized bioactive platforms, including functional SAMs, bioconjugation, single molecule detection, and bio-AFM. Since March 2011 he has been working as a senior scientist at Nanogea Inc., USA. |
 Joon Won Park | Joon Won Park obtained his PhD in 1988 at the California Institute of Technology under the supervision of Professor Robert H. Grubbs and carried out postdoctoral research at Professor Tobin J. Marks' lab of Northwestern University. He is currently a professor of Chemistry at Pohang University of Science and Technology (Korea) and a founder of NSB POSTECH Inc. and Nanogea Inc. Professor Park's research interests include molecular architecture on solid surfaces, microarrays, force-based atomic force microscopy, and single biomolecule analysis and manipulation. |
Introduction
In recent years, various structurally well-defined molecular surfaces have been actively studied as part of ongoing bio-interfacial exploration. For many applications, controlling the chemical and physical natures of the interfaces is of primary importance. In particular, there has been a great deal of interest in the development of molecularly ordered surface architectures that play a key role in a number of applications,1–4 including protein adsorption,5 clinical diagnostics,6 and cellular adhesion.7 A number of approaches have been investigated to replicate or mimic biochemical processes in life on solid supports. Recent advances in nanofabrication technologies have provided unique advantages associated with the “top down” approach, but their resolution is currently limited.8 On the other hand, the “bottom up” approach, including processes such as molecular self-assembly, has become a viable choice, and self-assembled monolayers (SAMs) have been widely employed because active functional groups may be intentionally placed precisely at the molecular scale. Various extended approaches have been examined to avoid congested functional surfaces because unwanted steric hindrance from the neighbouring molecules can be problematic. At the same time, the implementation of optimized spacing between the active groups is necessary as overly large spacing results in the reduction of signal intensity.Although rational approaches for the construction of surfaces spatially controlled at the nanoscale require further exploration, noticeable progress has been made in the field. In this review, recent progress in the development of nanoscale-controlled self-assembled molecular layers for application as biosensing interfaces are highlighted. While only a limited number of examples are presented here, cases representing a number of different approaches have been chosen. An overview of the techniques available for the control of spacing between functional groups on the molecular level and their potential applications are presented. We hope that this summary and discussion provide investigators in both the chemical and life sciences with a perspective on the field and insight into surface materials potentially suitable for their objectives.
Methods for the control of the spacing between functional groups on surfaces
Currently, there are several techniques available for the modification of biologically active platforms for biosensing and diagnostics. In particular, SAMs are of prime technological interest for the fabrication of interfacial architectures in molecular devices because the monolayer provides an opportunity to specifically place selected chemical functionalities on surfaces with molecular precision. Considerable attention has been given to the functionalization of a variety of substrates promoting the development of surface-bound assemblies and nanostructures. The most commonly employed substrates are gold and metal oxides. The self-assembly of alkylsilanes on oxide surfaces9 and sulphur-containing molecules, such as alkanethiolate and related molecules, on gold substrates has been extensively studied.10 The uses of SAMs have been thoroughly investigated for the development of sensors,11 organic thin film interfaces,12 and micro/nano device structures.13 For many applications, optimization of the density of functional groups, as well as the lateral spacing between them, is frequently vital.14 In the following sections, representative approaches for controlling these factors are introduced and some of their applications are included.Mixed self-assembled monolayers
To date, several methods have been applied to control the spacing of functional groups at regular intervals on material surfaces and tune the surface densities of surface-modifying probe molecules.15 Mixed SAMs have been the subject of considerable interest from the viewpoint of controlling surface properties such as wettability, friction, adhesion and the density of biomolecules on the surfaces to which they are applied.16 To adjust the lateral spacing of functional groups on a surface, multi-component self-assembled monolayers have been widely applied that involve the co-deposition of two (or more than two) adsorbates. Whitesides and co-workers utilized mixed SAMs consisting of alkanethiolates on gold surfaces to study lateral steric effects in the specific binding of carbonic anhydrase (CA) and arylsulfonamide. An approximately 10-fold decrease in the association constant was observed as the fraction of the surface covered by adsorbed CA increased from ∼0.15 to ∼0.35. In extended studies, they applied mixed SAMs to enable the hydrophobic adsorption of proteins such as β-galactosidase, carbonic anhydrase, lysozyme, and RNase A at solid–liquid interfaces. Mrksich and co-workers utilized a mixed SAM consisting of maleimide-terminated disulfide and penta(ethylene glycol) disulfide on gold surfaces for the immobilization of thiol-terminated ligands (L, Fig. 1).17 The maleimide moieties acted as active functional groups enabling ligand conjugation, whereas the penta(ethylene glycol) groups prevented the nonspecific adsorption of biomolecules to the substrate.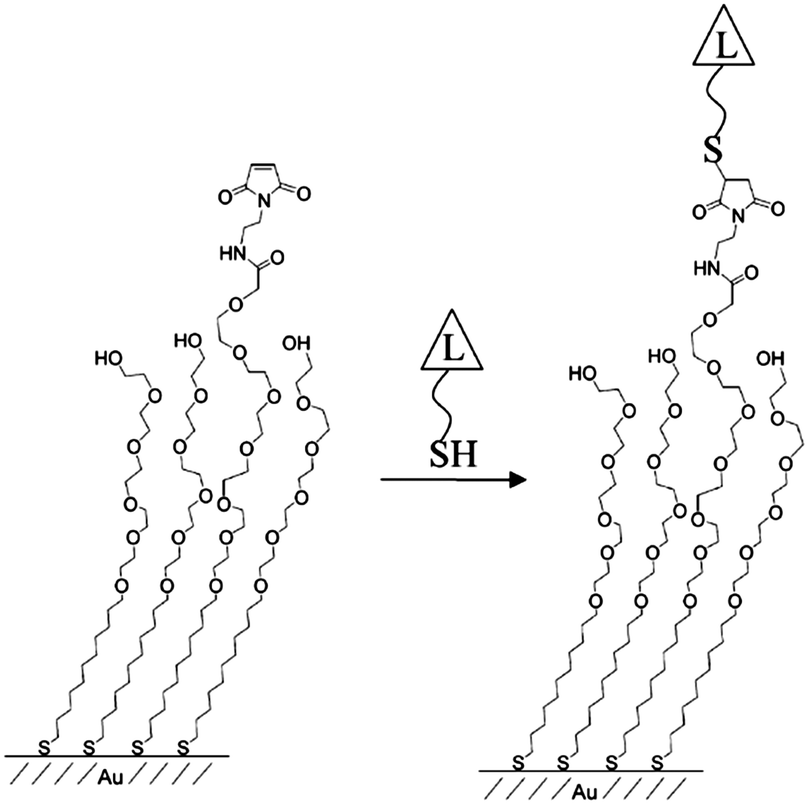 | ||
| Fig. 1 Schematic of the immobilized thiol-terminated ligand on a mixed-SAM (reprinted with permission from ref. 17. Copyright 2003, American Chemical Society). | ||
A mixed SAM consisting of alkanethiol derivatives of a single-stranded DNA (ssDNA) molecule and mercaptohexanol was utilized for the measurement of hybridization activity by Georgiadis and co-workers.18 In the course of their study, the mixed SAM was used to study the hybridization kinetics of ssDNA molecules complement to the surface confined tethered ssDNA19 and the effects of probe density on the hybridization efficiency of matched and mismatched targets.20 The mixed SAM consisting of a tri(ethylene glycol)OH (or (EG)3OH)-terminated thiol and a hexa(ethylene glycol)COOH (or (EG)6COOH)-terminated thiol was utilized for the covalent immobilization of antibodies.21 The treated surfaces were employed for the detection of pathogens by surface plasmon resonance (SPR). In another approach, Wang and co-workers used a similar mixed SAM consisting of N-hydroxysuccinimide thiol and oligoethylene-glycol-terminated thiol on a gold surface for the conjugation of antibodies via lysine residues.22 The prepared surface was utilized for single molecule spectroscopy.
To provide spacing between chemical functionalities, the use of the sequential adsorption approach has been investigated. In this scheme, the deposition of a large adsorbate is followed by backfilling with a second smaller adsorbate. This approach is exemplified by recent works where octadecyltrichorosilane (OTS) islands were generated on a silica surface, followed by the filling of the remainder of the bare substrate with bromine- or amino-terminated silane.23 In a further extension of this approach, Fadeev and McCarthy utilized the “holes” in an array of grafted bulky organosilanes to prepare a binary monolayer. After depositing a bulky silane, the holes were filled with organosilanes with smaller cross-sections to fit into the vacancies (Fig. 2).24 The size-exclusion contact angle hysteresis behaviour revealed the presence of nanopores at the surface with cross-sections of approximately 0.5 nm,2 confirming that the surface concentration of the functional silane was controlled primarily by the size of the bulky silane rather than the chemical nature of the functional silane. By selecting the appropriate functional silane, the binary monolayer may be further functionalized for biomolecule conjugation.
 | ||
| Fig. 2 Schematic of a sequential adsorption procedure (reprinted with permission from ref. 24. Copyright 1999, American Chemical Society). | ||
Several research groups have utilized protein molecules as scaffolds to provide lateral spacing on surfaces.25,26 In this context, streptavidin (or avidin), one of the most widely utilized protein scaffolds, along with mixed SAMs of biotinylated alkanethiol, has been employed to provide spacing between the probe DNA molecules on gold surfaces. Knoll and co-workers explored the use of mixed SAMs consisting of biotin-substituted and hydroxyl-substituted alkanethiols on gold surfaces, employing a protein to provide a multilayer sensor platform for molecular recognition.27 In another biosensor set-up, they utilized the mixed SAMs comprised of a biotinylated thiol and a hydroxyl-terminated thiol for the fabrication of a NeutrAvidin molecular layer, and the functionalized layer was further employed for the binding of biotin-β-lactamase on the chip surface (Fig. 3).28 The chip was then used for monitoring in situ enzymatic activity of the surface-bound enzyme by SPR.
 | ||
| Fig. 3 Schematic of the NeutrAvidin scaffold used for immobilizing biotinylated β-lactamase with controlled lateral spacing (redrawn from ref. 28). | ||
Zhang et al. fabricated an interface consisting of thiolated oligonucleotides and oligo(ethylene glycol)-terminated thiol and applied it as an electrochemical DNA sensor. The ample space provided by the oligo(ethylene glycol)-terminated thiol allowed facile detection of the reporter molecule (i.e., oxidized 3,3′,5,5′-tetramethylbenzidine (TMB)) with a gold electrode, exhibiting an impressive detection limit (10 copies of the target DNA strand).29 Analogously, Boozer et al. prepared a mixture of ssDNA and oligo(ethylene glycol) (OEG) adsorbate and utilized this mixed SAM platform for the site-directed immobilization of protein–DNA conjugates, which they utilized for the sensitive detection of human chorionic gonadotropin with SPR.30
Despite the popular use of mixed SAMs for the reduction of the steric constraints associated with probe–target binding, the approach does not guarantee regular and homogeneous spacing between adjacent active functional groups. In principle, the spacing provided by the approach is governed by statistics. In addition, aggregation, particularly in aged samples, was often observed, reflecting facile lateral mobility of alkanethiols on gold surfaces and a higher thermodynamic affinity among the thiols for molecules of their own type.31 Due to the spontaneous formation of molecular domains, optimal lateral spacing of functionalities cannot be guaranteed with the abovementioned approach; therefore, new methodologies have been examined.
Nanoparticles as templates
Recently, nanometric spacing on surfaces was achieved through the use of nanoparticles. Reinhoudt and co-workers reported the transfer of functional groups from gold nanoparticles to silicon oxide substrates, resulting in controlled spacing between the active functionalities on the substrate surfaces (Fig. 4).32 In their study, gold nanoparticles mono-functionalized with (mercaptopropyl)methyldimethoxysilane (MPMD), having diameters of ∼5 nm, were employed. A monolayer of the nanoparticles was formed on a clean silicon oxide substrate, and the gold nanoparticles were finally removed by iodine treatment to liberate the surface-bound silylpropanethiol functionalities on the oxide surface. The transferred functionality was subsequently used for the attachment of cytochrome C (cyt C) through the reaction of maleimide-derivatized-cyt C with the free thiol group, yielding a covalent thioether linkage. The approach was found to be capable of delivering tailored surface-spacing through the selection of nanoparticles of the appropriate size. The procedure developed by his group was not expected to form regular patterns, but it should allow a minimum spacing determined by nanoparticle sizes, which is important for single molecule studies. For the latter application, capability to transfer a single copy of the functional molecules or a defined copy number of the ink per pixel seems more desirable.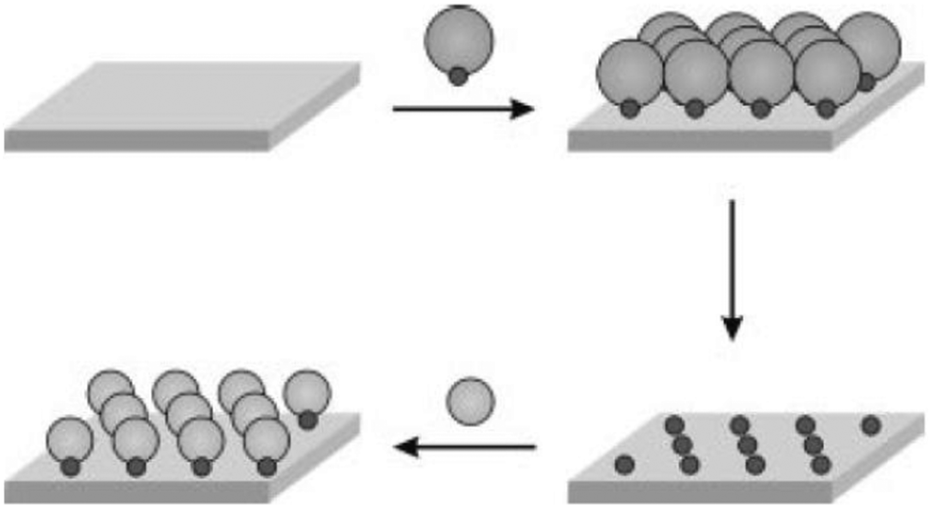 | ||
| Fig. 4 Scheme for functional group transfer from nanoparticles to substrate and subsequent attachment of Cyt C or gold nanoparticles through specific interactions (reprinted with permission from ref. 32. Copyright 2002, Wiley). | ||
In an integrated approach, Delcassian et al. utilized copolymeric micelles with cores loaded with gold(III) chloride for the fabrication of bioactive nanoscale arrays with controlled interparticle spacings in the range of 25–104 nm (Fig. 5).33 The dip-coating process provided two dimensional, close-packed layers of micelles on a substrate, with subsequent treatment by hydrogen plasma, generating patterned gold nanoparticle arrays. Two cell-stimulating ligands, one for the NK cell and the other for the T cell, were attached on the nanoarray and the response of the immune cells was investigated. Significant differences in signalling between two paradigmatic immune receptors (TCR and CD16) were observed as a result of proper spacing between the corresponding antibodies on the surface. The binding behaviour of the ligands on the patterned surface is informative for the design of therapeutics.
 | ||
| Fig. 5 Schematic of: (A) fabrication of a gold nanoparticle array utilizing block copolymer micellar nanolithography and (B) a biofunctionalized nanoarray with NK-cell-and T-cell-stimulating ligands (reprinted with permission from ref. 33. Copyright 2013, American Chemical Society). | ||
DNA nanostructures as templates
As the bottom-up approach has become increasingly popular and important, the use of the intrinsic functionalities of biomolecules to fabricate new nanostructures has gained considerable attention.34 Due to its particularly unique properties, DNA has become one such promising building block for self-assembled nanostructures.35 Since Seeman utilized DNA in the construction of well-designed DNA nanostructures,36 major advances have been made in the synthesis of more complicated DNA structures.37 More recently, Rothemund demonstrated that the approach can be expanded to materials of a larger scale.38 The “DNA origami” technique has become a promising method for the fabrication of two dimensional structures for a broad range of applications.39,40 DNA origami involves the folding of long single-stranded DNA scaffold strands into a variety of shapes using the self-assembly of a large number of short oligonucleotide staple strands. A variety of desirable functional groups can be conjugated to the selected staple strands, thereby incorporating the groups at predetermined positions with nanoscale precision. Several groups utilized the self-assembly of DNA nanostructures for the controlled placement of desired entities, in particular metallic nanoparticles, with a variety of patterns.41 Yan and co-workers demonstrated precise control of a pattern involving periodic spacing between individual proteins through the utilization of DNA tiles.42 Using two types of DNA nanoassemblies containing selectively modified biotinylated sites as templates, a set of programmed nanostructures were constructed, with subsequent binding of streptavidin at the biotin sites of the tiles verified by atomic force microscopy (AFM) (Fig. 6A). In another report, they utilized a DNA tile made of four four-arm DNA branched units.43 The nanogrid was constructed by self-assembly of the tiles through Watson–Crick base paring at the sticky ends. Because two biotins were placed at the centre of each 4 × 4 DNA tile by design, subsequent addition of streptavidin resulted in a programmed streptavidin nanoarray. Similar approaches have been developed for constructing highly ordered antibody arrays with controlled lateral spacing for the investigation of protein–protein interactions.44 Mao and co-workers fabricated antibody nanoarrays with a repeating distance of ∼19 nm using DNA nanostructures.44a They constructed periodic antigen 2D arrays by self-assembly of DNA tiles conjugated with antigens, followed by incubation with an antigen-specific antibody. AFM images confirmed the successful use of the antigen-modified DNA array in placing antibodies into their desired positions with a repeat distance of ∼19 nm (Fig. 6B). Rinker et al. demonstrated distance-dependent, multivalent ligand–protein binding through the placement of two ligands with different affinities into a DNA tile.45 The designed rectangular DNA nano-scaffolds, having two molecular recognition motifs composed of two DNA aptamers, were investigated for specific binding of the target protein. Thrombin was captured by only the bivalent aptamers, which were separated by 5–8 nm. Specificity was confirmed by AFM (Fig. 6C). These origami structures are perfectly suited for engineering multivalent 2D systems in which precise control of the spatial placement of key components is required.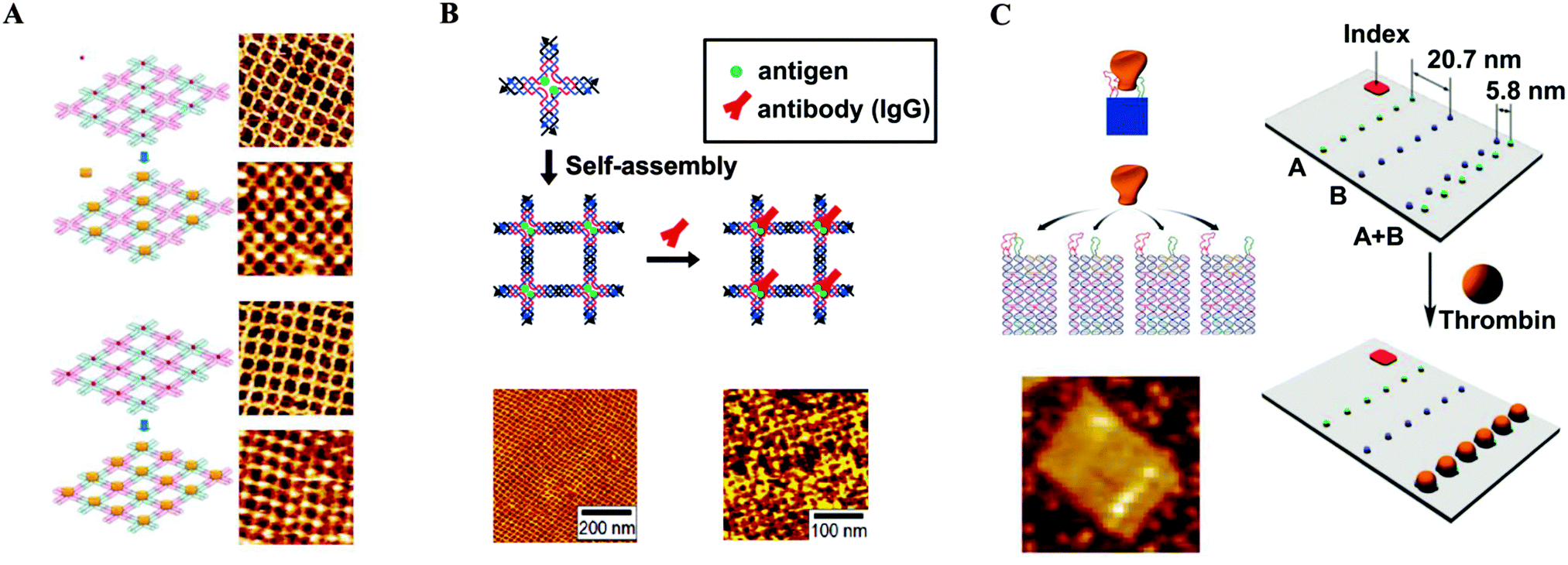 | ||
| Fig. 6 DNA nanostructures serving as templates for the controlled placement of desired entities. (A) Streptavidin arrays (reprinted with permission from ref. 42. Copyright 2005 American Chemical Society). (B) Antibody arrays (reprinted with permission from ref. 44a. Copyright 2006 American Chemical Society). (C) Distance-dependent, multivalent binding of thrombin on the origami surface (reprinted with permission from ref. 45. Copyright 2008 Macmillan Publishers Ltd). | ||
Recent advances in DNA origami techniques have enabled scientists to utilize these structures for various applications, including super-resolution microscopy. Tinnefeld and co-workers used the DNA origami structure labelled with fluorescent probes at defined positions as nanoscopic rulers for super-resolution microscopy and proposed that they are fluorescence and super-resolution standards for everyday use.46,47 Specific sites on the origami units were labelled with three different fluorescent dyes, and fluorescence images were consistent with separations of 78, 84, or 94 nm (Fig. 7A). In addition, to demonstrate the capability of DNA origami standards in covering the full range from the diffraction-limited regime to molecular dimensions, they constructed rectangular-shaped DNA origamis where two dye (ATTO647N) molecules were separated by distances of 6, 12, or 18 nm (Fig. 7B). Fluorescence transients were analysed after successive photobleaching, and the corresponding localization map showed clear separation of the two dye molecules. Distance histograms matched the programmed distance.
 | ||
| Fig. 7 DNA origami standards used for fluorescence microscopy. (A) Standards for stochastic switching-based microscopy. Sketch of the rectangular DNA origami with Alexa 488 (blue), Alexa 568 (yellow), and Alexa 647 (red) as nanorulers and 2D maps generated from successive localization of single blinking dyes. Scale bars, 100 nm. (B) Standards for molecular-scale resolution microscopy. Sketch of a DNA origami containing two ATTO647N molecules distances of d1 = 6 nm, d2 = 12 nm, or d3 = 18 nm and histograms corresponding to the distance measured from the three DNA origamis (reprinted with permission from ref. 46. Copyright 2012 Macmillan Publishers Ltd). | ||
One of the most thrilling advances in the positioning of different functional groups with nanoscale precision and the use of DNA origami scaffolds as highly versatile breadboards for chemical reactions was reported by Voigt et al.48 In their study, biotinylated linkers were positioned at predetermined positions of a rectangular DNA origami structure and conjugated to streptavidin at the prepositioned biotin molecules. The specific binding and two successive cleavage reactions were confirmed. Three different types of linkers were incorporated into the biotinylated staple stands: type A (non-cleavable), type B (having a disulfide moiety that can be cleaved by reduction), and type C (having an electron-rich 1,2-bis(alkylthio)ethene moiety that can be cleaved by singlet oxygen generated with UV light in the presence of a singlet oxygen photo-sensitizer). The selective disappearance of streptavidin from the origami scaffold, due to the selective cleavage of the B and C linkers, was confirmed by AFM (Fig. 8A). In addition, the DNA origami scaffold was utilized as a platform for single-molecule anchoring (Fig. 8B). The scaffold was functionalized with azide, amine, alkyne, and biotin groups at designated positions. The “click” reaction transformed the alkyne into a biotin tethering group and two streptavidin molecules were bound to two biotin sites (the new one and the index). An orthogonal reaction between the amine group and N-hydroxysuccinimide (NHS) allowed the placement of streptavidin at the amine position. Another reaction between the azide and the biotin-tethering alkyne group resulted in the successful placement of streptavidin at the azide position. AFM images confirmed the success of the chemical reactions by revealing the presence of streptavidin molecules in the predefined positions on the DNA origami scaffold with high selectivity. Direct monitoring of chemical reactions at the single-molecule level is a stunning example of the versatility of the DNA origami platform. Such an approach confirms the powerful potential of the DNA origami scaffold as a nanoscale template, a “nanobreadboard” for single molecule sciences.
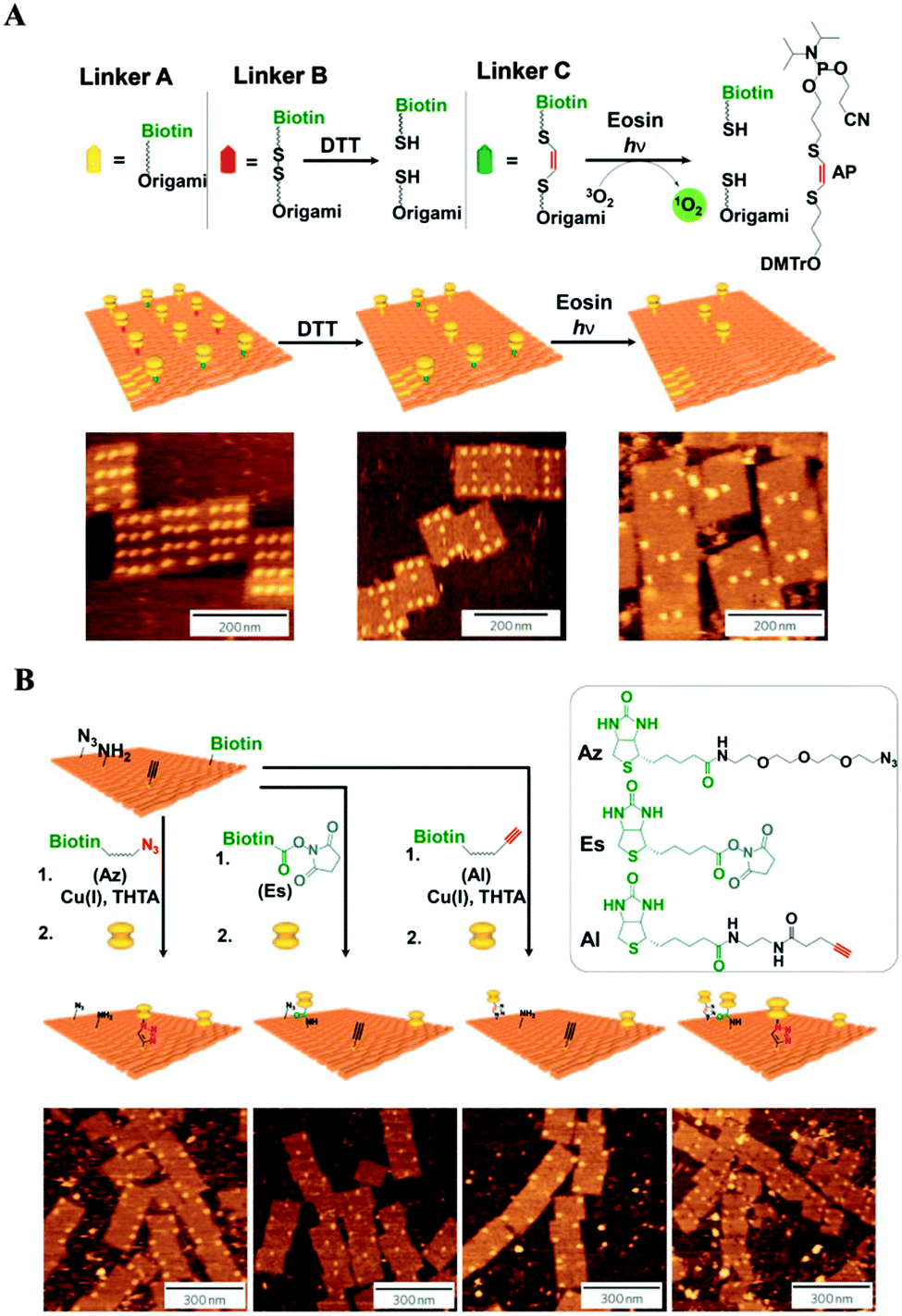 | ||
| Fig. 8 Single-molecule detection on DNA origami scaffolds. (A) Demonstration of single-molecule cleavage reactions. Bond cleavage was confirmed via AFM imaging after the selective disappearance of streptavidin from the origami surface after the corresponding cleavage conditions. (B) Demonstration of single-molecule bond formation reactions. Incoming functional groups were linked to biotin, and the bond formation was verified via AFM imaging after the addition of streptavidin (adapted with permission from ref. 48. Copyright 2010 Macmillan Publishers Ltd). | ||
In addition, 3D-structured DNA probes were utilized to minimize the lateral hindrance between the probes and to maximize the probe–target recognition performance. Fan and co-workers studied DNA nanostructure scaffold-based biosensors.49 They applied tetrahedron-shaped DNA to modify the working surface of a gold electrode for ultrasensitive detection of miRNA.50 In this process, the hybridization of the target miRNA acts as a linker between the rigid DNA tetrahedron spacer and an electrochemical probe, an avidin–horse radish peroxidase conjugate (avidin–HRP). This approach allowed positioning of the probe within the proximity of the electrode surface, concomitantly avoiding the crowding effect and improving mass transport at the water–electrode interface. The authors demonstrated enhanced binding recognition at the gold electrode surface and drastically improved detection sensitivity, up to attomole concentrations of miRNA with single base discrimination capability. In a related application, the DNA tetrahedron was utilized for the fabrication of microarrays on glass substrates.51 DNA tetrahedron nanostructures with three amine groups and one versatile single-stranded DNA extension were covalently immobilized on aldehyde-functionalized glass surfaces to fabricate the microarrays. A multiplex platform was developed for the recognition of a miRNA biomarker, a protein cancer biomarker, and a small molecule (Fig. 9). A parallel analysis of let-7a miRNA, prostate specific antigen, and cocaine was performed, showing detection limits of 10 fM, 40 pg mL−1 and 100 nM, respectively.
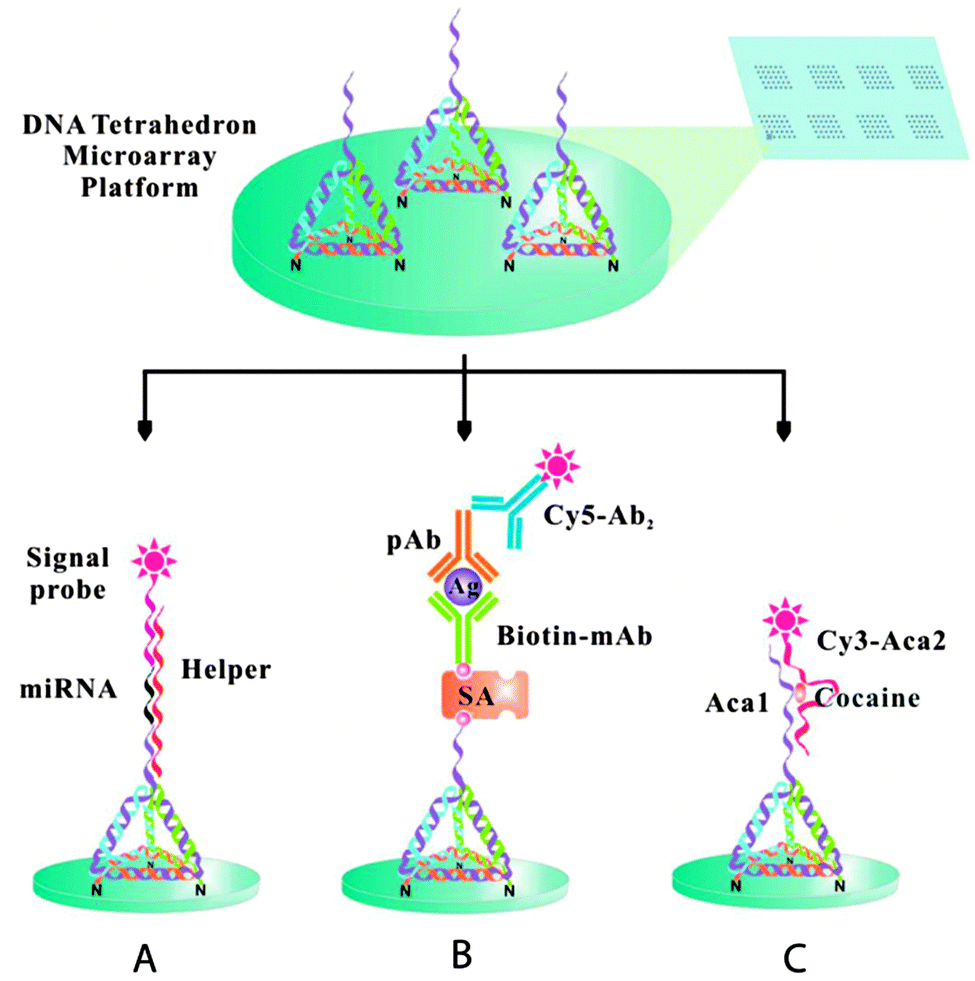 | ||
| Fig. 9 The tetrahedron 3D DNA nanostructured probe-based biosensing interface for fabricating multiplex microarrays useful in the detection of miRNA biomarkers (A), protein cancer biomarkers (B), and small molecules (C) (reprinted with permission from ref. 51. Copyright 2014 American Chemical Society). | ||
Recently, Leitner et al. used chemically tagged DNA tetrahedra containing three disulfide-modified vertices to achieve directed attachment on a gold surface. A heterobifunctional adapter was utilized to position individual protein molecules with defined lateral spacing on the gold surface.52 The directional surface attachment and the highly effective single-molecule binding of individual receptor molecules (streptavidin) to the ligand (biotin) were confirmed by simultaneous topographic and recognition imaging (TREC) using AFM (Fig. 10). The specificity of the recognition was also confirmed by blocking the biotin moieties by adding streptavidin to the tetrahedra surfaces. The DNA tetrahedra provides a higher surface ratio of bound streptavidin per biotin than available through conventional immobilization of molecular receptors at undefined spacings, where random orientation can result in poor recognition and specificity as a result of steric crowding. The power of the origami approach has been demonstrated in several studies where DNA nanostructures offer a unique opportunity to obtain unbiased intrinsic information about complex biomolecular networks on a variety of surfaces.
 | ||
| Fig. 10 Simultaneous topographic and recognition imaging of the specific receptor–ligand interaction on individual tetrahedra immobilized on a gold surface. (A) Schematic of the TREC imaging principle. (B) Simultaneously acquired topography (B-1) and recognition images (B-2), where the dotted circles correspond to tetrahedra with recognition and the solid circles represent tetrahedra without recognition, as schematically represented in B-3. An overlay of the topographic images (B-4, recognition signals in red) (reprinted with permission from ref. 52. Copyright 2011, American Chemical Society). | ||
The development in DNA-based nanomaterials has shown great potential in bioanalytical and biomedical applications. Nevertheless, certain progress must be made before its real-world applications can be realized. Factors such as robustness, durability, ease of design and ability to integrate with other materials must be addressed, and production in a cost-effective and scalable manner will be hailed by electronics, optics, and sensor fields.
Macromolecular architectures as templates
The assemblies of macromolecules on a variety of surfaces offer unique opportunities for providing functional molecular platforms with well-defined nanostructures. Various reports have exemplified the formation of macromolecular assemblies with controlled sizes and shapes on surfaces by utilizing non-covalent interactions between the surfaces and absorbed molecules as well as between the molecules themselves.53 For example, Yokoyama et al. reported the adsorption of substituted porphyrin molecules on a gold surface in a predictable fashion.54 Bonifazi et al. prepared patterned surfaces consisting of porphyrins and used them as templates for other molecules.55 In this study, both fused di-zincporphyrin (Fig. 11A) and nonfused porphyrin (Fig. 11B) were deposited onto an Ag(100) surface under ultrahigh vacuum, resulting in the formation of self-assembled monolayers of molecules, as confirmed by scanning tunneling microscopy (STM). The patterned surfaces were then used as templates for C60 molecules. The C60 molecules were placed atop the 3-cyanophenyl substituents of the porphyrins. This approach provided a laterally controlled platform for the assembly of fullerene at specific positions and demonstrates the potential usefulness of macromolecules in the fabrication of spatially addressable molecular architectures. In addition, immobilized macromolecules have frequently been utilized in bio-analytical applications. A good deal of research has focused on supramolecular functional polymeric materials tailored with desirable physical properties (e.g., nonlinear optical, photochromic) and with the capability to form well-organized superstructures at the interfaces.56,57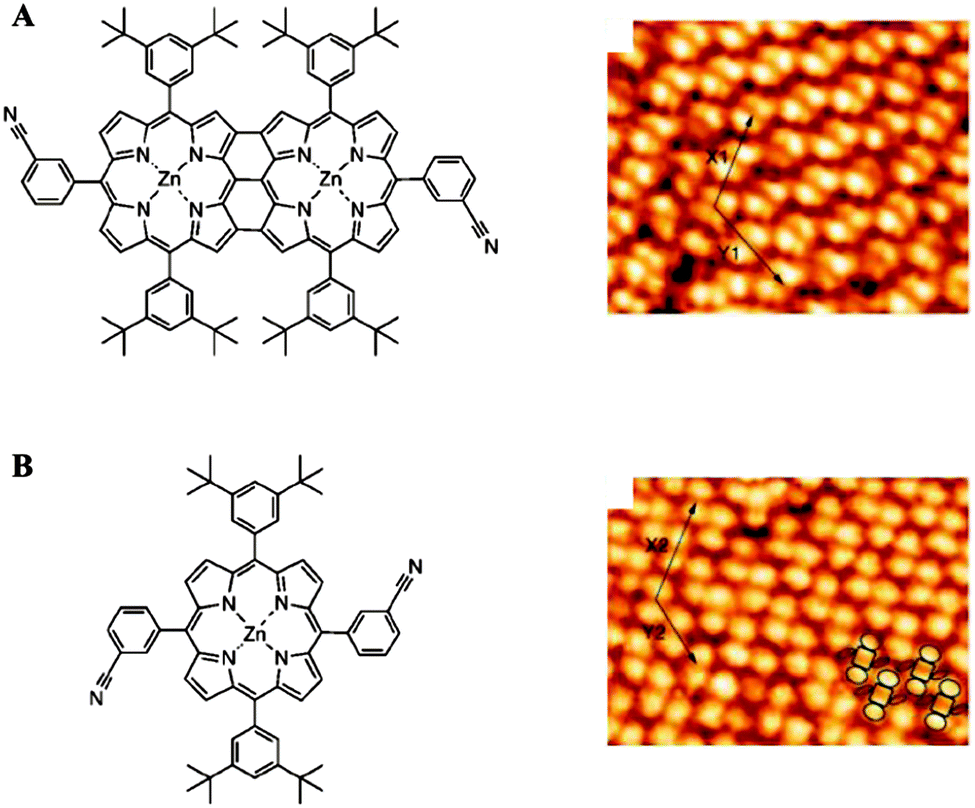 | ||
| Fig. 11 Chemical structures of the porphyrin derivatives and corresponding STM images of the full-coverage monolayers on Ag(100) (A and B) (reprinted with permission from ref. 55. Copyright 2004, Wiley). | ||
Baisch et al. utilized triazatriangulenium (TATA) ions as platforms. The various side chains on the ions serve to control the size of the platforms. These platforms act as pedestals, enforcing the controlled orientation of the functional groups relative to the surface. Their size dictates the spacing between the functional groups, resulting in arrays of anchored functional molecules on the surface (Fig. 12).58 Similar to the porphyrin or phthalocyanine monolayers, the TATA moiety forms hexagonally ordered adlayers via interactions between the π-system and the metal substrate. A surface-parallel assembly of TATA with the desired orientation of the functional group was confirmed by STM and photoelectrochemical measurements.
 | ||
| Fig. 12 Functional groups anchored on a surface with controlled lateral spacing. (A) Schematic of the approach. (B) STM images of TATA adlayers (reprinted with permission from ref. 58. Copyright 2009, American Chemical Society). | ||
The use of dendritic macromolecules (dendrimers) of well-defined geometrical shapes and of monodisperse molecular weights, bearing multiple functional groups at their peripheries, have been used for surface coating. Dendrimers, which are a class of materials with tree-like architectures and diameters normally spanning tens of nanometres, are capable of self-organizing into suprastructures. Crooks and co-workers reported the use of dendrimer monolayer films as chemically sensitive interfaces. A self-assembled mercaptoundecanoic acid (MUA) monolayer was modified by covalently conjugating hyperbranched dendrimers to the substrate surface.59 The dendrimers anchored on the surface provided multiple functional sites for further functionalization. It was demonstrated that a variety of structures could be built by varying the nature and concentration of the groups at the periphery, by modulating the attractive forces between molecule and surface, as well as between neighbouring molecules, and by varying the polymer backbone architecture.
 | ||
| Fig. 13 A molecular tripodal tip for ligand–receptor AFM force spectroscopy. (A) Structure of the biotinylated molecular tripod A. (B) Schematic of a tripodal tip interacting with a receptor on the substrate (reprinted with permission from ref. 62. Copyright 2010, American Chemical Society). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :<1). The authors reduced nonspecific binding through the judicious choice of the dendron skeleton. In addition, selectivity enhancement from 43–82 times greater was observed in comparison to high density surfaces (Fig. 14B).
:<1). The authors reduced nonspecific binding through the judicious choice of the dendron skeleton. In addition, selectivity enhancement from 43–82 times greater was observed in comparison to high density surfaces (Fig. 14B).
 | ||
| Fig. 14 Nanoscale spaced functional platforms using dendron spacers and their applications. (A) Schematic of dendron-based platforms for controlling the spacing between functional groups on surfaces. (B) DNA microarray platform (reprinted with permission from ref. 65. Copyright 2005, American Chemical Society). (C) DNA–DNA hybridization study with AFM (reprinted with permission from ref. 67. Copyright 2007, American Chemical Society). (D) Detection and counting individual immunocomplexes on microarray platform (reprinted with permission from ref. 70. Copyright 2010, American Chemical Society). (E) Nick-sealing of a single DNA duplex with AFM (reprinted with permission from ref. 71. Copyright 2012, American Chemical Society). (F) Picking up a single-molecule ferritin complex (reprinted with permission from ref. 72. Copyright 2012, American Chemical Society). | ||
In their studies of AFM, Park and group demonstrated that the dendron platform has great potential for the study of single-molecule interactions. Considering the ever-increasing interest in single-molecule analysis, it is noteworthy that the use of dendron molecules for AFM probes and substrates facilitates the detection of biomolecules at the single-molecule level. Jung et al. utilized dendron-functionalized substrates as well as AFM probes to study single molecular DNA–DNA interactions. A key to this study was the capacity of the dendron to provide ample spacing between the functional molecules to allow efficient DNA hybridization events (Fig. 14C).67 The nanoscale-controlled dendron surfaces enabled observation of force–distance curves for both attraction (or binding) and adhesion (or unbinding) and eliminated unwanted multiple interactions. The force histogram became sharp, and even the single base mismatched pair was able to be discriminated. In addition, the dendron-modified AFM probes were utilized to map the spatial distribution of mouse Pax6 mRNA on sectioned tissues of the mouse embryonic neocortex.69 A sample of 30-mer DNA was immobilized on the dendron-modified AFM probe to measure the specific adhesion force between it and the complementary part of the target mRNA on the tissue section. The distribution of mRNA was identified with nanometre resolution. Notably, the technique was sufficiently sensitive to allow the detection of mRNA in the ventricular zone, while the presence of Pax6 mRNAs was not identified by the fluorescence assay in the particular section.
In a more recent extension of their work, a dendron-modified surface has been employed for the detection of individual captured antigens on surfaces and mapping them, via force-based AFM, at 3 nm resolution.70 In this study, both the substrates and probes were modified with a 27-acid dendron molecule. The detection antibody (5A6) on the AFM probe visualized the PSA (prostate specific antigen) captured by the capture antibody (8A6) on the surface (Fig. 14D). Controlled spacing on both surfaces was believed to be the key for the ability to unambiguously “see” individual captured antigens within a selected area of a microarray spot. This approach was further utilized for the ligation of a single duplex with a nick (Fig. 14E).71 Bringing a template 71-mer DNA on the tip resulted in a DNA duplex on the surface with a nick between a captured 47-mer DNA and a 24-mer DNA immobilized on the surface. After nick-sealing with a DNA ligase, an increase in the unbinding force (from 24.0 ± 4.4 pN to 62.8 ± 14.6 pN) corresponding to the elongated DNA (71 bp) was observed. In separate work, controlled dendron surface functionality was employed for a single-molecule ferritin picking-up process through the use of AFM (Fig. 14F).72 In this approach, a biotinylated AFM probe was utilized to pick up a single avidin–ferritin–DNA conjugate from the surface. The strong binding interaction between the avidin and biotin, in comparison with the complementary DNA pair (20 mer), was exploited for transferring a single avidin–ferritin conjugate to the apex of the AFM tip. The single-molecule ferritin tip was used as a multifunctional magnetic force microscopy (MFM) probe for sensitive detection of the magnetic forces produced by magnetic materials, as well as for detecting the biomolecular interaction forces with DNA on surfaces. Apart from the abovementioned examples, Park and group utilized dendron-modified surfaces for various applications, including the study of single-molecule intramolecular interactions between single-transducing proteins,73 the mechanical unfolding of a foldamer,74 the nanoscale analysis of functionalized polymer surfaces,75 and sequencing single DNA using AFM.76 These examples are testament to the outstanding features of dendron-modified surfaces and their capacity for enhanced interactions and predominant single interaction in AFM studies.
Other groups have also utilized dendron-based templates to control the spacing between functionalities on surfaces. Cai and co-workers have employed dendron molecules to control lateral spacing on surfaces.77,78 They prepared organosiloxane thin films from SiCl3-terminated carbosilane dendrons of the second, third, and fourth generation, containing 9, 27, and 81 SiCl3 terminal groups, respectively. Otomo and co-workers used a dendron molecule with methylthiol groups at the peripheral sites and a rhodamine B (RhB) molecule at the focal point to prepare the fluorescent AFM probe (Fig. 15).79 The effective triplet–triplet energy transfer from a donor molecule attached to the dendron-modified probe to the acceptor in proximity on the substrate was demonstrated.
 | ||
| Fig. 15 Schematic of (A) an effective triplet–triplet (T–T) energy transfer from a donor on the dendron-functionalized AFM probe to an acceptor on the surface and (B) the nonproductive energy migration to neighbouring donor molecules on the probe when the donors are closely packed (redrawn from ref. 79). | ||
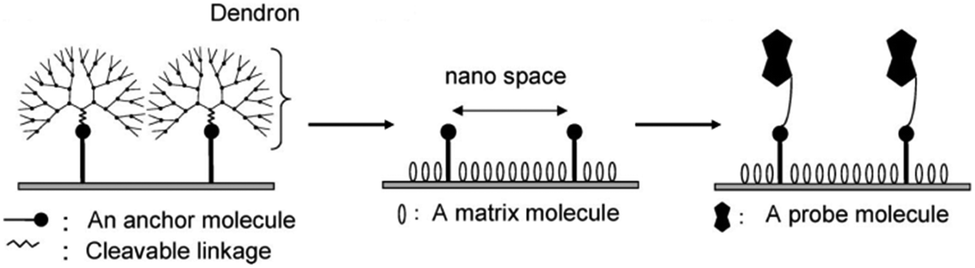 | ||
| Fig. 16 Fabrication of the nanoscale spaced functional platform using focal point anchoring of dendron spacers (reprinted with permission from ref. 81. Copyright 2009, American Chemical Society). | ||
In a hybrid approach, Iqbal et al. utilized supramolecular dendron-ligand assemblies for the controlled spatial distribution of chemical functionalities (Fig. 17).82 SAMs composed of pH-switchable, surface-appended pseudorotaxanes were constructed from a combination of a bulky macrocycle consisting of a combination of poly(aryl ether) dendron and dibenzo[24]crown-8 (DB24C8), and a ligand bearing a thiol group for the surface adsorption, a dibenzylammonium recognition site for the macrocycle, and a terminal fluorine for further modification. Following SAM formation, the dendron was released from the surface by switching off the noncovalent interaction by pH stimulation, generating surface materials with tailored physical and chemical properties.
 | ||
| Fig. 17 Schematic of the fabrication of a space-controlled surface by supramolecular dendron-ligand assemblies (reprinted with permission from ref. 82. Copyright 2014, American Chemical Society). | ||
Langer and co-workers used a bulky head group rather than a dendron, and their work is relevant in this context due to its originality and importance. They demonstrated the use of well-designed SAMs that were switchable under the influence of an electrochemical potential, with sufficient room to accommodate conformational changes of the immobilized molecules (Fig. 18).83 First, precursor molecules ((16-mercapto)hexadecanoic acid(2-chlorophenyl)diphenylmethyl ester, MHAE) having bulky head groups were self-assembled on the surface, resulting in a low-density SAM. Second, the bulky head groups were removed by hydrolysis, resulting in a low-density, but ‘standing-up’ layer of (16-mercapto)hexadecanoic acid (MHA). These low-density SAMs allowed chain mobility sufficient to accommodate reorientation from a hydrophilic (straight chains) to a hydrophobic (bent chains) state. The controlled surface density enabled the end groups of the MHA molecules to move freely. A surface with such appropriate spacing allows facile reversible conformational transitions of the single-layered molecules and thus effective switching of the surface from a hydrophilic to hydrophobic state (and vice versa) under electrical stimulus.
 | ||
| Fig. 18 Schematic of a spatially controlled SAM where spacing depends on the dimension of the precursor head group and the transition between straight (hydrophilic) and bent (hydrophobic) molecular conformations upon electrical stimulus (reprinted with permission from ref. 83. Copyright 2009, AAAS). | ||
In addition, surface materials with nanoscale-controlled shape, size, and morphology have been used for drug delivery and tissue engineering.84 Designed materials and formulations allow controlled release of drugs or small-molecules with minimal side effects. For example, composites containing supramolecular structures including nanoparticles, nanotubes, nanocapsules, nanogels, and dendrimers have been examined for drug delivery.85 In order to target cancer cells and control delivery of drugs, Langer and co-workers utilized a self-assembled polymeric nanoparticle platform.86 A10 aptamer immobilized on nanoparticles was used to target prostate cancer cells, and co-immobilized chemotherapeutic drugs (cisplatin and docetaxel) were delivered to the corresponding cells. Apart from the drug delivery, researchers engineered surface materials to mimic the morphological and chemical features of the natural extracellular matrix for the tissue regeneration.87 Natural macromolecules, such as collagen, fibrinogen, dextran, and chitosan, have been used to prepare nanofibrous matrices.88 Stupp and co-workers studied self-assembly of amphiphilic macromolecules that form cylindrical nanofibers with well-defined diameters and used them as tissue engineering scaffolds for bone.89 Furthermore, another group used a de novo designed peptide, a pH-responsive self-assembled β-hairpin in a nanofibrillar network, for in situ cell encapsulation.90
It is clear from these examples that the employment of macromolecular architectures, such as dendron molecules, for controlling spatial molecular spacing provides a useful route, not only to nanoscale-controlled placement of functionalities, but also to a wide range of applications requiring enhanced bio-recognition.
Suppression of nonspecific biomolecule adsorption
The reduction of nonspecific adsorption of biomolecules is also a key for high performance of surface-based bioanalytical and diagnostics devices.91 The most widely used materials for this goal are poly(ethylene glycol) [PEG] and oligo(ethylene glycol) [OEG].92 However, these materials are susceptible to oxidation in some biochemically relevant media,93 and in order to widen the choices, new materials including hydrophilic and zwitterionic materials have therefore been examined.94 In particular, the latter materials contain both positive and negative charged units can bind water molecules more tightly (in the thermodynamic sense) through electrostatically induced hydration than hydrophilic materials can. Therefore, suppression of nonspecific binding is more effective. Two types of zwitterionic materials, polybetaines and polyampholytes, were synthesized, and their capability for reducing nonspecific adsorption in various biological applications was investigated.95 Among polybetaines, poly(carboxybetaine) is most widely used for reducing the nonspecific protein adsorption in a single-protein medium,96 even in blood plasma97 and undiluted human blood serum.98 The observation is consistent with a previous report by Whitesides; polymers of alternating charges were more effective than polymers in which the charges were statistically distributed.99 Jiang and co-workers examined a related approach, and observed that peptides with alternating Glu/Lys or Asp/Lys showed noticeable resistance against the nonspecific adsorption of fibrinogen, lysozyme, and albumin.100 While use of PEG, OEG, zwitterionic materials, or alternatingly charged peptides is a good choice currently, it is desirable to find new materials of enhanced biochemical stability, even in hostile environments, shelf-life, and effectiveness.Conclusions and outlook
Herein, we reviewed the recent advances in a variety of approaches for controlling surface functionalities, with a particular focus on lateral spacing with nanoscale precision. The approaches include the use of mixed self-assembled monolayers, protein motifs, nanoparticles, DNA nanostructures, macromolecular architectures, including porphyrins and dendrons as templates. The use of mixed monolayers or protein motifs is common and straightforward, and the exploitation of DNA nanostructures, in particular DNA origami, is noteworthy. The capability for programmed placement of functionalities on a surface is obviously a unique advantage. Interesting progress has also been witnessed with the use of dendrons, which are especially useful if coating of a large area is required. While we have not covered analytical tools for examining surface materials, importance of thorough characterization is beyond a doubt. Commonly employed methods include ellipsometry, infrared spectrometry, Raman spectrometry, X-ray diffractometry, contact angle goniometry, mass spectrometry, X-ray photoelectron spectrometry, atomic force microscopy, scanning tunnelling microscopy, scanning electron microscopy, and transmission electron microscopy.101Most of the applications involving spatially controlled platforms provided enhanced biomolecular recognition. Their efficacies have been demonstrated with biosensors such as SPR, immunoassay, electrochemical sensors, microarrays, and single molecular spectroscopy including atomic force microscopy, scanning tunneling microscopy, and super-resolution spectroscopy. Due to its highly parallel nature and convenience, DNA microarray techniques have been extended to other microarrays such as aptamer, RNA, protein, and carbohydrate. Fine spatial (or lateral) control of surfaces at the nanoscale resulted in high performance and reliability in their use as microarrays. As many investigators have demonstrated, precise control of the surface is of prime importance. In addition, the degree to which surface control helped single molecular spectroscopy is particularly notable. Such control allowed scanning probe microscopes to reproducibly observe true single-molecule interactions at interfaces and provided super-resolution spectroscopy with the valuable standards.
Moreover, there are features yet to be achieved to fully satisfy the requirements of each specific application. Effective suppression of non-specific binding is one example. Although many approaches are already reasonably good in this regard, to utilize state-of-the-art instruments and spectrometers maximally, enhanced suppression of non-specific binding is no doubt required due to the persistent existence of noise (or background) signals resulting from non-specific binding. In addition, increasing cost-effectiveness of material production and easy accessibility of target surface materials will lead to wide spread uses. Advancements in controlling the physical and chemical nature of surfaces for use as efficient bioactive platforms are expected. We envision that through proper collective efforts, spatially controlled bio-interfaces will be further refined and we will find a myriad of sensing and bio-medical applications including stem cell research. Furthermore, they have the capacity to perfect single molecular spectroscopy, thereby elucidating the fundamental properties of single molecules and the behaviour of single biomolecules both in vitro and in vivo.
Acknowledgements
The authors acknowledge financial support of POSCO/POSTECH.Notes and references
- G. B. Sigal, C. Bamdad, A. Barberis, J. Strominger and G. M. Whitesides, Anal. Chem., 1996, 68, 490–497 CrossRef CAS.
- G. P. López, M. W. Albers, S. L. Schreiber, R. Carroll, E. Peralta and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 5877–5878 CrossRef.
- M. C. Pirrung, Angew. Chem., 2002, 114, 1326–1341 ( Angew. Chem., Int. Ed. , 2002 , 41 , 1276–1289 ) CrossRef.
- D. S. Wilson and S. Nock, Angew. Chem., 2003, 115, 510–517 ( Angew. Chem., Int. Ed. , 2003 , 42 , 494–500 ) CrossRef PubMed.
- (a) A. J. Pertsin, M. Grunze, H. J. Kreuzer and R. L. C. Wang, Phys. Chem. Chem. Phys., 2000, 2, 1729–1733 RSC; (b) R. L. C. Wang, H. J. Kreuzer and M. Grunze, J. Phys. Chem. B, 1997, 101, 9767–9773 CrossRef CAS; (c) R. L. C. Wang, H. J. Kreuzer, M. Grunze and A. J. Pertsin, Phys. Chem. Chem. Phys., 2000, 2, 1721–1727 RSC.
- (a) K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164–1167 CrossRef CAS; (b) J. Frommer, R. Luthi, E. Meyer, D. Anselmetti, M. Dreier, R. Overney, H.-J. Guntherodt and M. Fujihira, Nature, 1993, 364, 198 CrossRef PubMed.
- (a) C. Roberts, C. S. Chen, M. Mrksich, V. Martichonok, D. E. Ingber and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 6548–6555 CrossRef CAS; (b) D. A. Stenger, J. H. Georger, C. S. Dulcey, J. J. Hickman, A. S. Rudolph, T. B. Nielson, S. M. McCort and J. M. Calvert, J. Am. Chem. Soc., 1992, 114, 8435–8442 CrossRef CAS.
- (a) Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS; (b) X. M. Zhao, Y. Xia and G. M. Whitesides, J. Mater. Chem., 1997, 7, 1069–1074 RSC.
- (a) J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92–98 CrossRef CAS; (b) J. D. Legrange, J. L. Markham and C. R. Kurkjian, Langmuir, 1993, 9, 1749–1753 CrossRef CAS.
- (a) R. G. Nuzzo and D. Allara, J. Am. Chem. Soc., 1983, 105, 4481–4483 CrossRef CAS; (b) P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS.
- (a) B.-H. Huisman, R. P. H. Kooyman, F. C. J. M. van Veggel and D. N. Reinhoudt, Adv. Mater., 1996, 8, 561–564 CrossRef CAS PubMed; (b) K. D. Schierbaum, T. Weiss, E. U. Thoden van Velzen, J. F. J. Engbersen, D. N. Reinhoudt and W. Gopel, Science, 1994, 265, 1413–1415 CAS.
- (a) Y. S. Shon, S. Lee, R. Colorado Jr., S. S. Perry and T. R. Lee, J. Am. Chem. Soc., 2000, 122, 7556–7563 CrossRef CAS; (b) R. M. Crooks and A. J. Ricco, Acc. Chem. Res., 1998, 31, 219–227 CrossRef CAS.
- (a) D. J. Díaz, J. E. Hudson, G. D. Storrier, H. D. Abruña, N. Saudararajan and C. K. Ober, Langmuir, 2001, 17, 5932–5938 CrossRef; (b) C. Alain, M. Geissler, H. Schmid, B. Michel and E. Delamarche, Langmuir, 2002, 18, 2406–2412 CrossRef; (c) G. P. Lopez, H. A. Biebuyck, C. D. Frisbie and G. M. Whitesides, Science, 1993, 260, 647–649 CAS; (d) S. Xu, S. Miller, P. E. Laibinis and G. Y. Liu, Langmuir, 1999, 15, 7244–7251 CrossRef CAS; (e) G.-Y. Liu, S. Xu and Y. Qian, Acc. Chem. Res., 2000, 33, 457–466 CrossRef CAS PubMed.
- S. K. Shin, H. J. Yoon, Y. J. Jung and J. W. Park, Curr. Opin. Chem. Biol., 2006, 10, 423–429 CrossRef CAS PubMed.
- (a) P. Harder, M. Grunze, R. Dahint, G. M. Whitesides and P. E. Laibinis, J. Phys. Chem., 1998, 102, 426–436 CrossRef CAS; (b) E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3–30 CrossRef CAS.
- (a) J. Lahiri, L. Isaacs, J. Tien and G. M. Whitesides, Anal. Chem., 1999, 71, 777–790 CrossRef CAS; (b) P. M. Mendes, C. L. Yeung and J. A. Preece, Nanoscale Res. Lett., 2007, 2, 373–384 CrossRef CAS PubMed; (c) M. Mrksich, MRS Bull., 2005, 30, 180–184 CrossRef CAS; (d) M. Mrksich, Curr. Opin. Chem. Biol., 2002, 6, 794–797 CrossRef CAS; (e) A. Pranzetti, S. Mieszkin, P. Iqbal, F. J. Rawson, M. E. Callow, J. A. Callow, P. Koelsch, J. A. Preece and P. M. Mendes, Adv. Mater., 2013, 25, 2181–2185 CrossRef CAS PubMed; (f) K. C.-F. Leung, S. Xuan and C.-M. Lo, ACS Appl. Mater. Interfaces, 2009, 1, 2005–2012 CrossRef CAS PubMed; (g) A. Stephenson-Brown, H. C. Wang, P. Iqbal, J. A. Preece, Y. T. Long, J. S. Fossey, T. D. James and P. M. Mendes, Analyst, 2013, 138, 7140–7145 RSC; (h) J. Lahiri, L. Isaacs, B. Grzybowski, J. D. Carbeck and G. M. Whitesides, Langmuir, 1999, 15, 7186–7198 CrossRef CAS; (i) W. Yeo, M. N. Yousaf and M. Mrksich, J. Am. Chem. Soc., 2004, 125, 14994–14995 CrossRef PubMed; (j) S. Husale, H. Persson and O. Sahin, Nature, 2009, 462, 1075–1078 CrossRef CAS PubMed.
- B. T. Houseman, E. S. Gawalt and M. Mrksich, Langmuir, 2003, 19, 1522–1531 CrossRef CAS.
- K. A. Peterlinz, R. Georgiadis, T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119, 3401–3402 CrossRef CAS.
- R. Georgiadis, K. P. Peterlinz and A. W. Peterson, J. Am. Chem. Soc., 2000, 122, 3166–3173 CrossRef CAS.
- A. W. Peterson, L. K. Wolf and R. M. Georgiadis, J. Am. Chem. Soc., 2002, 124, 14601–14607 CrossRef CAS PubMed.
- A. Subramanian, J. Irudayaraj and T. Ryan, Sens. Actuators, B, 2006, 114, 192–198 CrossRef CAS PubMed.
- V. K. Yadavalli, J. G. Forbes and K. Wang, Langmuir, 2006, 22, 6969–6976 CrossRef CAS PubMed.
- J. Buseman-Williams and J. C. Berg, Langmuir, 2004, 20, 2026–2029 CrossRef CAS.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 1999, 15, 7238–7243 CrossRef CAS.
- L. Häussling, H. Ringsdorf, F. J. Schmitt and W. Knoll, Langmuir, 1991, 7, 1837–1840 CrossRef.
- W. Müller, H. Ringsdorf, E. Rump, G. Wildburg, X. Zhang, L. Angermaier, W. Knoll, M. Liley and J. Spinke, Science, 1993, 262, 1706–1708 Search PubMed.
- J. Spinke, M. Liley, H.-J. Guder, L. Angermaier and W. Knoll, Langmuir, 1993, 9, 1821–1825 CrossRef CAS.
- F. Xu, G. Zhen, F. Yu, E. Kuennemann, M. Textor and W. Knoll, J. Am. Chem. Soc., 2005, 127, 13084–13085 CrossRef CAS PubMed.
- J. Zhang, R. Lao, S. Song, Z. Yan and C. Fan, Anal. Chem., 2008, 80, 9029–9033 CrossRef CAS PubMed.
- (a) C. L Boozer, J. Ladd, S. Chen, Q. Yu, J. Homola and S. Jiang, Anal. Chem., 2004, 76, 6967–6972 CrossRef PubMed; (b) C. Boozer, J. Ladd, S. Chen and S. Jiang, Anal. Chem., 2006, 78, 1515–1519 CrossRef CAS PubMed.
- (a) S. J. Stranick, A. N. Parikh, Y.-T. Tao and D. L. Allara, J. Phys. Chem., 1994, 98, 7636–7645 CrossRef CAS; (b) N. Camillone, P. Eisenberger, T. Y. B. Leung, P. Schwartz, G. Scoles, G. E. Poirier and M. J. Tarlov, J. Chem. Phys., 1994, 101, 11031–11036 CrossRef PubMed; (c) R. K. Smith, S. M. Reed, J. D. Monnell, P. A. Lewis, R. S. Clegg, K. F. Kelly, L. A. Bumm, J. E. Hutchison and P. S. Weiss, J. Phys. Chem. B, 2001, 105, 1119–1122 CrossRef CAS; (d) S. J. Stranick, A. N. Parikh, Y. T. Tao, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 7636–7646 CrossRef CAS; (e) F. Fan, C. Maldarelli and A. Couzis, Langmuir, 2003, 19, 3254–3265 CrossRef CAS.
- V. Paraschiv, S. Zapotoczny, M. R. Jong de, G. J. Vancso, J. Huskens and D. N. Reinhoudt, Adv. Mater., 2002, 14, 722–726 CrossRef CAS.
- D. Delcassian, D. Depoil, D. Rudnicka, M. Liu, D. M. Davis, M. L. Dustin and I. E. Dunlop, Nano Lett., 2013, 13, 5608–5614 CrossRef CAS PubMed.
- (a) D. Wilson, R. Martin, S. Hong, M. Golomb, C. Mirkin and D. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13660–13664 CrossRef CAS PubMed; (b) L. M. Demers, D. S. Ginger, S. J. Park, Z. Li, S. W. Chung and C. A. Mirkin, Science, 2002, 296, 1836–1838 CrossRef CAS PubMed; (c) J. Hyun, S. J. Ahn, W. K. Lee, A. Chilkoti and S. Zauscher, Nano Lett., 2002, 2, 1203–1207 CrossRef CAS.
- (a) N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed; (b) R. P. Goodman, I. A. T. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- (a) N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS; (b) N. R. Kallenbach, R.-I. Ma and N. C. Seeman, Nature, 1983, 305, 829–831 CrossRef CAS PubMed.
- (a) N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed; (b) Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. Bidault, F. J. Garcia de Abajo and A. Polman, J. Am. Chem. Soc., 2008, 130, 2750–2751 CrossRef CAS PubMed.
- K. Keren, R. S. Berman, E. Buchstab, U. Sivan and E. Braun, Science, 2003, 302, 1380–1382 CrossRef CAS PubMed.
- (a) S. Xiao, F. Liu, A. E. Rosen, J. E. Hainfeld, N. C. Seeman, K. Musier-Forsyth and R. A. Kiehl, J. Nanopart. Res., 2002, 4, 313–317 CrossRef CAS; (b) J. D. Le, Y. Pinto, N. C. Seeman, K. Musier-Forsyth, T. A. Taton and R. A. Kiehl, Nano Lett., 2004, 4, 2343–2347 CrossRef CAS; (c) J. Zhang, Y. Liu, Y. Ke and H. Yan, Nano Lett., 2006, 6, 248–251 CrossRef CAS PubMed; (d) B. Ding, Z. Deng, H. Yan, S. Cabrini, R. N. Zuckermann and J. Bokor, J. Am. Chem. Soc., 2010, 132, 3248–3249 CrossRef CAS PubMed.
- S. H. Park, P. Yin, Y. Liu, J. H. Reif, T. H. LaBean and H. Yan, Nano Lett., 2005, 5, 729–733 CrossRef CAS PubMed.
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, Science, 2003, 301, 1882–1884 CrossRef CAS PubMed.
- (a) Y. He, Y. Tian, A. E. Ribbe and C. Mao, J. Am. Chem. Soc., 2006, 128, 12664–12665 CrossRef CAS PubMed; (b) B. A. R. Williams, K. Lund, Y. Liu, H. Yan and J. C. Chaput, Angew. Chem., Int. Ed., 2007, 46, 3051–3054 CrossRef CAS PubMed.
- S. Rinker, Y. G. Ke, Y. Liu, R. Chhabra and H. Yan, Nat. Nanotechnol., 2008, 3, 418–422 CrossRef CAS PubMed.
- J. J. Schmied, A. Gietl, P. Holzmeister, C. Forthmann, C. Steinhauer, T. Dammeyer and P. Tinnefeld, Nat. Methods, 2012, 9, 1133–1134 CrossRef CAS PubMed.
- C. Steinhauer, R. Jungmann, T. L. Sobey, F. C. Simmel and P. Tinnefeld, Angew. Chem., Int. Ed., 2009, 48, 8870–8873 CrossRef CAS PubMed.
- N. V. Voigt, T. Tørring, A. Rotaru, M. F. Jacobsen, J. B. Ravnsbæk, R. Subramani, W. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, Nat. Nanotechnol., 2010, 5, 200–203 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Pan, J. Shi, Q. Huang and C. Fan, NPG Asia Mater., 2013, 5, 1–9 Search PubMed.
- Y. Wen, H. Pei, Y. Shen, J. Xi, M. Lin, N. Lu, X. Shen, J. Li and C. Fan, Sci. Rep., 2012, 2, 867 Search PubMed.
- Z. Li, B. Zhao, D. Wang, Y. Wen, G. Liu, H. Dong, S. Song and C. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 17944–17953 CAS.
- M. Leitner, N. Mitchell, M. Kastner, R. Schlapak, H. J. Gruber, P. Hinterdorfer, S. Howorka and A. Ebner, ACS Nano, 2011, 5, 7048–7054 CrossRef CAS PubMed.
- J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS PubMed.
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CrossRef CAS PubMed.
- D. Bonifazi, H. Spillmann, A. Kiebele, M. de Wild, P. Seiler, F. Cheng, H.-J. Güntherodt, T. Jung and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 4759–4763 CrossRef CAS PubMed.
- A. Ulman, Introduction to Ultrathin Organic Films, Academic Press, San Diego, CA, 1991 Search PubMed.
- (a) J. H. Fendler and F. C. Meldrum, Adv. Mater., 1995, 7, 607–632 CrossRef CAS PubMed; (b) D. A. Tomalia, Adv. Mater., 1994, 6, 529–539 CrossRef CAS PubMed; (c) J. F. Jansen, E. M. de Brabander-van den Berg and E. W. Meijer, Science, 1994, 266, 1226–1229 CAS; (d) V. Percec and M. Kawasumi, Macromolecules, 1994, 27, 4441–4453 CrossRef CAS.
- B. Baisch, D. Raffa, U. Jung, O. M. Magnussen, C. Nicolas, J. Lacour, J. Kubitschke and R. Herges, J. Am. Chem. Soc., 2009, 131, 442–443 CrossRef CAS PubMed.
- M. Wells and R. M. Crooks, J. Am. Chem. Soc., 1996, 118, 3988–3989 CrossRef CAS.
- J. K. Whitesell and H. K. Chang, Science, 1993, 261, 73–76 CAS.
- J.-S. Park, A. N. Vo, D. Barriet, Y.-S. Shon and T. R. Lee, Langmuir, 2005, 21, 2902–2911 CrossRef CAS PubMed.
- M. E. Drew, A. Chworos, E. Oroudjev, H. Hansma and Y. Yamakoshi, Langmuir, 2010, 26, 7117–7125 CrossRef CAS PubMed.
- D. Takamatsu, K.-I. Fukui, S. Aroua and Y. Yamakoshi, Org. Biomol. Chem., 2010, 8, 3655–3664 CAS.
- B. J. Hong, J. Y. Shim, S. J. Oh and J. W. Park, Langmuir, 2003, 19, 2357–2365 CrossRef CAS.
- B. J. Hong, S. J. Oh, T. O. Youn, S. H. Kwon and J. W. Park, Langmuir, 2005, 21, 4257–4261 CrossRef CAS.
- B. J. Hong, V. Sunkara and J. W. Park, Nucleic Acids Res., 2005, 33, e106 CrossRef PubMed.
- Y. J. Jung, B. J. Hong, W. Zhang, S. J. B. Tendler, P. M. Williams, S. Allen and J. W. Park, J. Am. Chem. Soc., 2007, 129, 9349–9355 CrossRef CAS PubMed.
- D. Roy, J.-W. Kwak, W. J. Maeng, H. Kim and J. W. Park, Langmuir, 2008, 24, 14296–14305 CrossRef CAS PubMed.
- Y. J. Jung, Y. S. Park, K.-J. Yoon, Y.-Y. Kong, J. W. Park and H. G. Nam, Nucleic Acids Res., 2009, 37, e10 CrossRef PubMed.
- D. Roy, S. H. Kwon, J.-W. Kwak and J. W. Park, Anal. Chem., 2010, 82, 5189–5194 CrossRef CAS PubMed.
- E. S. Kim, J. S. Kim, Y. Lee, K. Y. Choi and J. W. Park, ACS Nano, 2012, 6, 6108–6114 CrossRef CAS PubMed.
- D. Kim, N. K. Chung, S. Allen, S. J. B. Tendler and J. W. Park, ACS Nano, 2012, 6, 241–248 CrossRef CAS PubMed.
- I. H. Kim, M. N. Lee, S. H. Ryu and J. W. Park, Anal. Chem., 2011, 83, 1500–1503 CrossRef CAS PubMed.
- J. S. Kim, Y. J. Jung, J. W. Park, A. D. Shaller, W. Wan and A. D. Q. Li, Adv. Mater., 2008, 20, 1–4 CrossRef PubMed.
- J.-E. Lee, J.-W. Kwak, J. W. Park, S.-C. Luo, B. Zhu and H. Yu, Anal. Chem., 2014, 86, 6865–6871 CrossRef CAS PubMed.
- Y. Kim, E. S. Kim, Y. Lee, J. H. Kim, B. C. Shim, S. M. Cho, J. S. Lee and J. W. Park, J. Am. Chem. Soc., 2014, 136, 13754–13760 CrossRef CAS PubMed.
- Z. Xiao, C. Cai, A. Mayeux and A. Milenkovic, Langmuir, 2002, 18, 7728–7739 CrossRef CAS.
- Z. Xiao and C. Cai, Langmuir, 2005, 21, 5019–5025 CrossRef CAS PubMed.
- A. Otomo, H. Miki, I. Aoki and S. Mashiko, Thin Solid Films, 2006, 499, 35–39 CrossRef CAS PubMed.
- H. Tokuhisa, T. Kubo, E. Koyama, K. Hiratani and M. Kanesato, Adv. Mater., 2003, 15, 1534–1538 CrossRef CAS PubMed.
- H. Tokuhisa, J. Liu, K. Omori, M. Kanesato, K. Hiratani and L. A. Baker, Langmuir, 2009, 25, 1633–1637 CrossRef CAS PubMed.
- P. Iqbal, F. J. Rawson, W. K.-W. Ho, S.-F. Lee, K. C.-F. Leung, X. Wang, A. Beri, J. A. Preece, J. Ma and P. M. Mendes, ACS Appl. Mater. Interfaces, 2014, 6, 6264–6274 CAS.
- J. Lahann, S. Mitragotri, T.-N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G. A. Somorjai and R. Langer, Science, 2003, 299, 371–374 CrossRef CAS PubMed.
- (a) M. Goldberg, R. Langer and X. Jia, J. Biomater. Sci., Polym. Ed., 2007, 18, 241–268 CrossRef CAS PubMed; (b) A. Tiwari and A. Tiwari, Nanomaterials in Drug Delivery, Imaging, and Tissue Engineering, Wiley-Scrivener Publishing LLC, USA, 2013 Search PubMed; (c) A. Tiwari, Y. Sharma, S. Hattori, D. Terada, A. K. Sharma, A. P. F. Turner and H. Kobayashi, Biopolymers, 2012, 99, 334–341 CrossRef PubMed.
- (a) K. Y. Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS PubMed; (b) V. A. Kickhoefer, Y. Garcia, Y. Mikyas, E. Johansson, J. C. Zhou, S. Raval-Fernandes, P. Minoofar, J. I. Zink, B. Dunn, P. L. Stewart and L. H. Rome, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4348–4352 CrossRef CAS PubMed; (c) C. R. Martin and P. Kohli, Nat. Rev. Drug Discovery, 2003, 2, 29–37 CrossRef CAS PubMed; (d) S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS PubMed.
- N. Kolishetti, S. Dhar, P. M. Valencia, L. Q. Lin, R. Karnik, S. J. Lippard, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17939–17944 CrossRef CAS PubMed.
- (a) R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS; (b) R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- (a) J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS PubMed; (b) G. E. Wnek, M. E. Carr, D. G. Simpson and G. L. Bowlin, Nano Lett., 2003, 3, 213–216 CrossRef CAS; (c) K. Ohkawa, D. I. Cha, H. Kim, A. Nishida and H. Yamamoto, Macromol. Rapid Commun., 2004, 25, 1600–1605 CrossRef CAS PubMed; (d) H. L. Jiang, D. F. Fang, B. S. Hsiao, B. Chu and W. L. Chen, Biomacromolecules, 2004, 5, 326–333 CrossRef CAS PubMed.
- (a) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed; (b) J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS PubMed.
- (a) J. M. Harris, Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Press, New York, 1992 Search PubMed; (b) E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- M. C. Shen, L. Martinson, M. S. Wagner, D. G. Castner, B. D. Ratner and T. A. Horbett, J. Biomater. Sci., Polym. Ed., 2002, 13, 367–390 CrossRef CAS PubMed.
- (a) M. Wyszogrodzka and R. Haag, Biomacromolecules, 2009, 10, 1043–1054 CrossRef CAS PubMed; (b) S. L. McArthur, K. M. McLean, P. Kingshott, H. A. W. St John, R. C. Chatelier and H. J. Griesser, Colloids Surf., B, 2000, 17, 37–48 CrossRef CAS; (c) S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS PubMed.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- Z. Zhang, H. Vaisocherová, G. Cheng, W. Yang, H. Xue and S. Jiang, Biomacromolecules, 2008, 9, 2686–2692 CrossRef CAS PubMed.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS PubMed.
- W. Yang, H. Xue, W. Li, J. Zhang and S. Jiang, Langmuir, 2009, 25, 11911–11916 CrossRef CAS PubMed.
- R. E. Holmlin, X. Chen, R. G. Chapman, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 2841–2850 CrossRef CAS.
- S. F. Chen, Z. Q. Cao and S. Y. Jiang, Biomaterials, 2009, 30, 5892–5896 CrossRef CAS PubMed.
- A. Ulman, Characterization of Organic Thin Films, Butterworth-Heinemann, Stoneham, MA, 1995 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |