
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface defect rich ZnO quantum dots as antioxidants inhibiting α-amylase and α-glucosidase: a potential anti-diabetic nanomedicine†
Adersh
Asok
a,
Sougata
Ghosh
b,
Piyush A.
More
b,
Balu A.
Chopade
c,
Mayuri N.
Gandhi
a and
Ajit R.
Kulkarni
*d
aCentre for Research in Nanotechnology and Science, Indian Institute of Technology Bombay, Mumbai-400076, Maharashtra, India
bInstitute of Bioinformatics and Biotechnology, University of Pune, Pune-411007, Maharashtra, India
cDepartment of Microbiology, University of Pune, Pune-411007, Maharashtra, India
dDepartment of Metallurgical Engineering and Materials Science, Indian Institute of Technology Bombay, Mumbai-400076, Maharashtra, India. E-mail: ajit.kulkarni@iitb.ac.in
First published on 6th May 2015
Abstract
Preventing chronic hyperglycaemia and associated oxidative stress is utmost important for the treatment and management of Type 2 Diabetes Mellitus (T2DM). Here we report the role of different size surface defect rich ZnO quantum dots (D-QDs) for inhibiting metabolic enzymes and scavenging free radicals, which plays a key role in reducing hyperglycaemia and oxidative stress. Quantitative analysis of radical scavenging and metabolic enzyme inhibition activity of D-QDs demonstrates a size dependent behaviour, where D-QDs with a smaller diameter shows superior activity compared to larger size D-QDs. Considering the size dependence in surface defect formation, the increased surface defect density in smaller size D-QDs can be considered as the reason behind this enhancement. Detailed studies establishing the underlying mechanism behind potent free radical scavenging and enzyme inhibition provides an intense scientific rationale for considering D-QDs to design safe and effective nanomedicine for T2DM.
1. Introduction
Type 2 Diabetes Mellitus (T2DM) is a persistent metabolic disorder characterised by chronic hyperglycaemia, which leads to an abnormally enhanced level of free radicals and concurrent decline of antioxidant defence mechanisms, finally resulting in oxidative stress (OS).1 Increasing evidence in both clinical and preclinical studies indicates that OS plays a major role in the disturbances of physiological homeostasis, manifested by an increase in lipid peroxidation, insulin resistance, damage of cellular organelles and enzymes.2–6 Hence, OS promotes the development of pathophysiological complications of T2DM like nephropathy, neuropathy, retinopathy and cardiomyopathy.7 These chronic diseases are responsible for majority of morbidity and mortality.Inhibition of α-glucosidase and α-amylase activity is reported as a suitable target for controlling postprandial hyperglycaemia in T2DM.8,9 Use of antioxidants is recommended for controlling OS and related complications.6,10,11 Therefore, designing a potential multifunctional therapeutic agent with enzyme inhibitory as well as antioxidant property is of utmost importance for the treatment of T2DM and prevention of associated problems.
“Jasada Bhasma” is a unique herbo-metallic formulation used in Indian traditional medicine (Ayurveda) for the treatment of diabetes and its progression.12 A comprehensive physico-chemical characterization of Jasada Bhasma by Bhowmick et al. reveals the presence of ZnO nanoparticles (NPs).13 These ZnO NPs present in Jasada Bhasma are predominantly of wurzite crystal structure and contains a considerable amount of oxygen deficiency.13 Hence, the presence of defect rich ZnO NPs in medicinal formulation like Jasada Bhasma strongly rationalises the possibility that surface oxygen vacancies (VO) can be the key attributing factor giving its medicinal value. Recently, in vivo evaluation of anti-diabetic activity and safety assessment of ZnO NPs were reported.14 However, to date, there have been no reports on the detailed mechanistic study of the therapeutic role for VO present in ZnO NPs in T2DM.
Recent experimental and theoretical work shows size dependence in the formation of defects in nanocrystals.15,16 These defects are found predominantly at the surface of nanocrystals and its density increases with decreasing nanocrystal size.15,17 In previous studies on defect rich ZnO quantum dots (D-QDs) synthesis, a similar trend for size dependence was observed for VO formation even when ZnO QDs are grown under defect promoting conditions.17,18 Thus, it is expected that the surface chemistry of D-QDs will vary with different sizes. Furthermore, the presence of this surface defects can contribute a net surface charge for D-QDs and the density of this surface charge will increase with decreasing particle size. Hence, this increase in surface charge density for smaller D-QDs can enhance charge based interaction of D-QDs with biomolecules. Earlier reports indicate that the NP surface charge is a contributing factor for protein adsorption onto NPs via electrostatic interaction.19–25 In ZnO NPs, VO is the most cited surface defects, and its presence results in a positively charged surface.26
A recent study on cerium oxide NPs by Lee et al. revealed that VO clustered at the surface of NPs have a size dependent antioxidant activity and smaller diameter NPs (containing more surface VO) were found to posses high antioxidant activity.27 This study suggests that size dependent VO formation in NPs may be the main reason for the observed increase in antioxidant activity for smaller NPs. However, the size dependent antioxidant behaviour of D-QDs towards reactive oxygen species (ROS) is least explored despite the presence of a high density of surface VO at smaller D-QDs.
In this article we report, for the first time, the rational designing of D-QDs of different size possessing well defined physico-chemical properties exhibiting dual potential as a metabolic enzyme inhibitor and as an antioxidant. For obtaining D-QDs of different sizes we have adopted defect rich seed mediated growth.17 We have grown three different sizes of monodispersed D-QDs from a defect rich seed QDs obtained by rapid microwave heating. Our experimental results suggest that both enzyme inhibition and antioxidant property have a good correlation with the size of D-QDs and hence the size dependent surface defect density.
2. Experimental details
Chemicals
All the precursors for synthesis of D-QDs and the assay chemicals are commercially available. The absolute ethanol (Analytical Reagent, China, assay: 99.99%) was the solvent used for D-QDs synthesis. The raw precursors Zn(OAc)2 (99.99%, metal basis) and LiOH (≥98%, reagent grade) from Sigma Aldrich, USA., were used for synthesis as received without further purification. The C-ZnO NPs were purchased from Sigma Aldrich, USA. Potassium dihydrogen phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4), sodium potassium tartarate, 2,2-diphenyl-1-picrylhydrazyl (DPPH), ascorbic acid, sodium hydroxide (NaOH), sodium chloride (NaCl) and porcine pancreatic α-amylase were bought from HiMedia Laboratories, Mumbai, India. α-Glucosidase, p-nitrophenyl α-D-glucopyranoside (pNPGP), 3,5-dinitrosalicylic acid (DNSA), riboflavin, ethylenediaminetetraacetic acid (EDTA), nitroblue tetrazolium (NBT) were bought from Sigma Aldrich, USA. The standard enzyme inhibitor, Acarbose, was procured from Bayer Pharmaceuticals, India.Synthesis of D-QDs
The D-QDs were synthesised as per the previous report.17 For a typical synthesis 0.05 M Zn(OAc)2 was mixed with 0.2 M LiOH (3 mL each) in a quartz reaction vial. A polymer cap was used to seal the reaction vial and a microwave reaction was initiated by using a microwave reactor (CEM Discover) with 2.45 GHz operating frequency. The reaction temperature (75 ± 5 °C) was maintained for one minute under high magnetic stirring followed by cooling to room temperature using compressed air flow. The solution obtained was used as a D-QDs seed and the different sizes of D-QDs were obtained by adding different concentrations of metal precursors Zn(OAc)2 to seed QDs followed by microwave heating up to 50 °C for one minute. All the samples for activity studies were prepared from the as prepared solution by appropriate dilution to achieve desired concentrations, which was expressed in μg mL−1.Characterization
Powder X-ray diffraction (XRD) data were collected using a Philips (PANalytical) powder diffractometer, with Cu Kα radiation as the X-ray source. The normal and high resolution transmission electron micrographs were recorded using a JEOL JEM-2100F high-resolution transmission electron microscope (HR-TEM) operating at 200 kV. PL and PLE spectra of D-QDs and enzymes were recorded at room temperature using a HORIBA Jobin Yvon Fluorolog 3 spectrofluorometer. Surface charges on the D-QDs and C-ZnO NPs were determined in terms of zeta potential using a zeta potential analyzer (ZetaPALS, Brookhaven Instruments Corporation, USA).DPPH radical scavenging assay
The scavenging of the DPPH radical was initiated by addition of 20 μL of different concentrations of each sample with 80 μL of ethanolic solution of 2,2-diphenyl-1-picrylhydrazyl (DPPH, 100 μM) in a 96 well plate which was incubated in a dark room at room temperature for 20 min.28 The change in absorbance value was measured at 517 nm. The radical scavenging activity of samples was determined using the following formula:| % Radical scavenging = (A517Control − A517Test)/A517Control × 100 | (1) |
EPR measurements
The DPPH˙ scavenging activity of D-QDs was measured using a previously described method.29 An ethanol solution of 200 μL of D-QDs (varying concentration) was added to 800 μL of DPPH (100 μM) in an ethanol solution (ethanol itself is used for collecting control spectra). The solutions were then transferred into a quartz capillary tube and fitted into the cavity of the electron paramagnetic resonance (EPR) spectrometer, after vigorous mixing for 10 s. The X-band (9.44 GHz) EPR trace of the DPPH˙ spin adduct was measured using a JES-FA200 EPR spectrometer (JEOL, Tokyo, Japan) exactly 1 min later at 25 °C. Measurement conditions: central field 3475 G, modulation frequency 100 kHz, modulation amplitude 2 G, microwave power 5 mW, gain 6.3 × 105 and sweep time of 2 min.Superoxide radical scavenging assay
Different concentrations of 100 μL of D-QDs sample were added to 100 μL riboflavin solution (20 μg), 200 μL EDTA solution (12 mM), 200 μL ethanol and 100 μL NBT solution (0.1 mg). To this, 3 mL of phosphate buffer (50 mM) was added to dilute the reaction mixture, which was further illuminated for 5 min. The absorbance of the solution was measured at 540 nm.30| % Radical scavenging = A540Control − A540Test/A540Control × 100 | (2) |
Porcine pancreatic α-amylase inhibition assay
The amylase activity was assayed using chromogenic DNSA as reported earlier.31 Different concentrations of each samples were incubated with porcine pancreatic α-amylase (50 μg mL−1) for 30 minutes at 37 °C. 1% starch solution was used as a substrate. Samples without α-amylase were taken as controls and test readings were subtracted from the absorbance of these controls. Reducing sugar was estimated by interpolation of the obtained absorbance value of DNSA for different concentrations of D-QDs at 540 nm (A540) in the standard graph. The percentage of enzyme inhibition was calculated using the formula:| % Enzyme inhibition = (A540Control − A540Test)/A540Control × 100 | (3) |
The Km and Vm for porcine pancreatic α-amylase inhibition by D-QDs and C-ZnO NPs were determined using Michaelis–Menten and Lineweaver–Burk equations. The different starch concentrations (1–5 mg mL−1) were incubated in the presence of samples and porcine pancreatic α-amylase for 10 min and DNSA was used for estimation of the residual enzyme activity.
Murine pancreatic and intestinal α-amylase inhibition assays
Swiss male mice were used as the source of murine enzymes with institutional animal ethical committee guidelines. In short, 10 week old mice with approximate weight of 20 g were starved for 12 h, followed by excision of small intestinal and pancreatic tissues which was homogenized in chilled phosphate buffer (10 mM containing 6 mM NaCl) containing protease inhibitors. The tissue homogenates were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 r.p.m for 10 min. The collected supernatant was diluted to get an absorbance of 0.4 (at 280 nm) which was used as a source of enzyme. Percentage inhibition of pancreatic α-amylase and small intestinal α-amylase was also calculated as described earlier.
000 r.p.m for 10 min. The collected supernatant was diluted to get an absorbance of 0.4 (at 280 nm) which was used as a source of enzyme. Percentage inhibition of pancreatic α-amylase and small intestinal α-amylase was also calculated as described earlier.
Porcine α-glucosidase inhibition assay
The glucosidase inhibitory activity was evaluated as per our earlier report.32 Typically, 100 μL of α-glucosidase (0.1 unit per mL) was mixed with 200 μL of different concentrations of D-QDs samples and incubated for 1 hour at 37 °C. Enzyme action was initiated by the addition of 10 mM p-nitrophenyl-α-D-glucopyranoside (p-NPGP) in 100 mM phosphate buffer of pH 6.8 and stopped after 10 minutes incubation at 37 °C by adding 2 mL of 0.1 M Na2CO3. Determination of the α-glucosidase activity was carried out by estimation of absorbance by p-nitrophenol released from pNPGP at 420 nm (A420) using a 96-well multiplate reader (SpectraMax M5, Molecular Devices Corporation, Sunnyvale, CA).| % Enzyme inhibition = (A420Control − A420Test)/A420Control × 100 | (4) |
Murine intestinal α-glucosidase inhibition assay
The intestinal extract of mice was prepared by the aforementioned process for evaluation of murine intestinal α-glucosidase inhibition. Percentage inhibition of murine intestinal α-glucosidase was determined using pNPGP as the substrate as per the above protocol.Statistical analysis
The statistical analysis was performed using one way analysis of variance (ANOVA) (P < 0.05). Results are expressed as means ± SEM (n = 3).Circular dichroism (CD) studies
The Jasco J-815 circular dichroism spectropolarimeter was used to record the CD spectra. The spectra were recorded at 25 °C using 1 nm slit width, scan speed = 40 nm min−1 and response time = 1 s. All the measurements were performed in a 2 mm path length quartz cell in the range of 190–300 nm. An IC50 of D-QDs(1) was incubated with a fixed enzyme concentration of 100 μg mL−1 in phosphate buffer. The CD spectrum of D-QDs in phosphate buffer acquired under the same conditions was used as baseline setting for acquiring the main CD spectra.Fluorescence study of enzymes
A HORIBA Jobin Yvon Fluorolog 3 spectrofluorometer was used to acquire the fluorescence spectra of tryptophan at 25 °C with a 1.0 cm path length quartz cuvette. A 3.0 nm slit width was set at the emission and excitation side. The emission spectra were collected in the range of 280 to 450 nm, with an excitation at 270 nm. The enzyme concentrations used are the same as that in the CD study, while D-QDs(1) concentrations were varied.3. Results and discussion
Physico-chemical properties of D-QDs
Near-spherical D-QDs with diameters ranging from 3 to 7 nm were prepared by seed mediated growth from defect rich seed QDs.17 A less defective ZnO NP procured from Sigma Aldrich, USA was used as a control. Fig. 1a–c, shows a transmission electron micrograph (TEM) of three different diameters of as grown D-QDs labeled as D-QDs(1): size ∼3.56 ± 0.25 nm, D-QDs(2): size ∼5.12 ± 0.25 nm, D-QDs(3): size ∼7.18 ± 0.25 nm and control ZnO NPs (C-ZnO NPs): size ∼50 ± 15 nm (Fig. 1d). The synthesized D-QDs have good monodispersity as confirmed by the particle size distribution profile (Fig. 1a–c, bottom right insets). All the samples and control were characterised for the purity and phase using X-ray diffraction (ESI,† Fig. S1) studies, which show peaks associated with the ZnO wurtzite crystal structure (JCPDS card no. 36-1451). A broadening in the X-ray diffraction peaks were observed for D-QDs, which can be attributed to a strained lattice due to the high density of surface defects and reduced crystal size. The high resolution transmission electron microscopy (HR-TEM) investigations of D-QDs (Fig. 1e) show a strained lattice with point defects, while the C-ZnO NPs (Fig. 1f) show a distinct lattice ordering associated with crystalline nanocrystals. This confirms that the D-QDs are rich in defects when compared with C-ZnO NPs. | ||
| Fig. 1 TEM images of different size D-QDs and C-ZnO NPs (a–d), the bottom right inset shows the size distribution histograms of the corresponding TEM images, (e) the HR-TEM image of D-QDs (dotted oval and circle show the exact location of some point defects, arrow serves as a guide to the eye) and (f) HR-TEM image of C-ZnO NPs. Particle size measurement of TEM images shown were analyzed using the Image J software from ∼100 NPs. | ||
The normalized photoluminescence (PL) spectra (I/Imax) of D-QDs of different size and C-ZnO NPs are shown in Fig. 2a. The observed PL emission spectra can be split into three regions: (1) an UVA emission from the ZnO band edge,33,34 (2) oxygen vacancy (VO) related visible emission35–38 and (3) zinc vacancy (VZn) related near infrared (NIR) emission.39 The emission spectra of D-QDs used in the current study possess a weak band edge emission and NIR emission, while a strong visible emission was observed. This confirms that the currently synthesized D-QDs are rich in VO. A bathochromic shift in D-QDs emission was observed with increasing D-QDs size, which can be attributed to the quantum confinement effect. The photoluminescence excitation (PLE) spectra (ESI,† Fig. S2) of D-QDs also follow a similar trend, which further confirm that the D-QDs were subjected to the quantum confinement effect. While in the case of C-ZnO NPs a strong band edge emission was observed, confirming its highly crystalline nature. The intensity ratio of λmax of defect emission (IDE) to λmax of UV emission (IBEE) could be utilized for semi-quantitative determination of defect density in ZnO NPs.33,34 The IDE/IBEE value shown in Fig. 2b shows a size dependent change in the case of D-QDs. The IDE/IBEE value is very low for C-ZnO NPs which signifies a very low concentration of radiative defect centres in C-ZnO NPs. Fig. 2c shows a photograph of C-ZnO NPs and D-QDs emitting a different colour under UV lamp excitation (365 nm, 4 watt). The zeta potential of D-QDs(1), D-QDs(2), and D-QDs(3) of same concentration (10 μg mL−1) in ethanol were −61.48 ± 9.1, −54.06 ± 5.06 and −41.39 ± 6.10 mV, which have a size dependent change. The increase in the value of negative zeta potential with decreasing size can be attributed to an increase in positive surface charge by increased surface VO density at smaller size (Fig. 2d). In contrast to this, C-ZnO NPs show a positive zeta potential (19.92 ± 5.02 mV), this can be due to the negative surface charge in the less defective oxygen terminated surface.
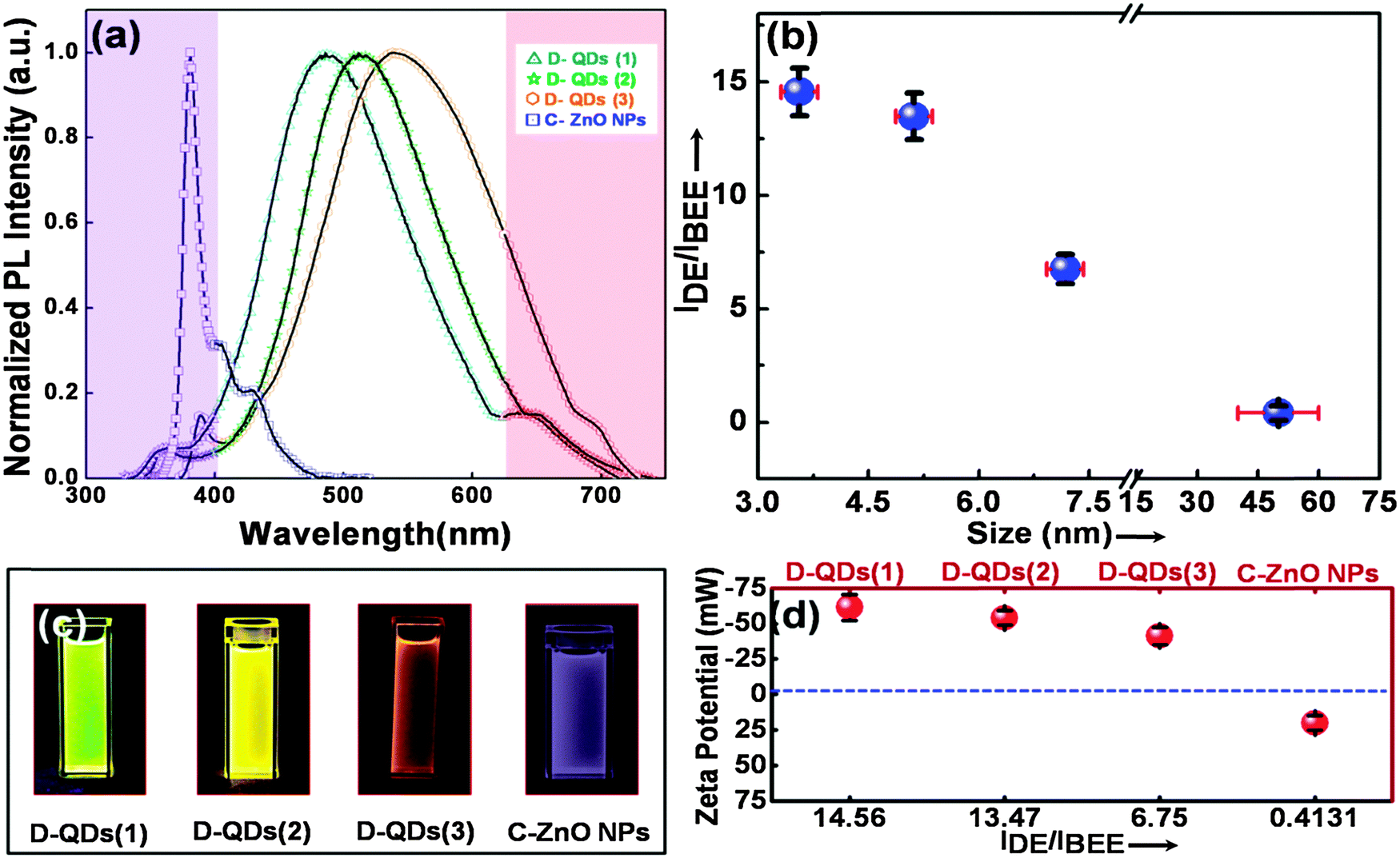 | ||
| Fig. 2 (a) Normalized photoluminescence emission spectra of D-QDs of different size and C-ZnO NPs, (b) the IDE/IBEE value for D-QDs of different size and C-ZnO NPs, (c) photographs of D-QDs of different size and C-ZnO NPs in ethanolic solution under irradiation by a 365 nm handheld UV lamp, (d) the zeta potential for D-QDs of different size and C-ZnO NPs and its corresponding IDE/IBEE value (x-axis is not scaled). | ||
Radical scavenging assays
The potential of D-QDs for reducing oxidative stress was determined by using 2,2-diphenyl-1-picrylhydrazyl radical (DPPH˙) scavenging assays. The half maximal inhibitory concentration (IC50) of different size D-QDs, C-ZnO NPs and a standard antioxidant ascorbic acid are shown in Fig. 3a. Scavenging of DPPH˙ by all tested ZnO NPs indicated their antioxidant potential which was significantly superior as compared to that of the standard antioxidant, ascorbic acid (IC50 ∼ 57.24 μg mL−1). D-QDs(1) (IC50 ∼ 1.59 μg mL−1) and D-QDs(2) (IC50 ∼ 1.77 μg mL−1) showed comparable activity while D-QDs(3) (IC50 ∼ 3.96 μg mL−1) showed slightly lower activity. C-ZnO NPs on the other hand showed a lower activity (IC50 ∼ 6.34 μg mL−1).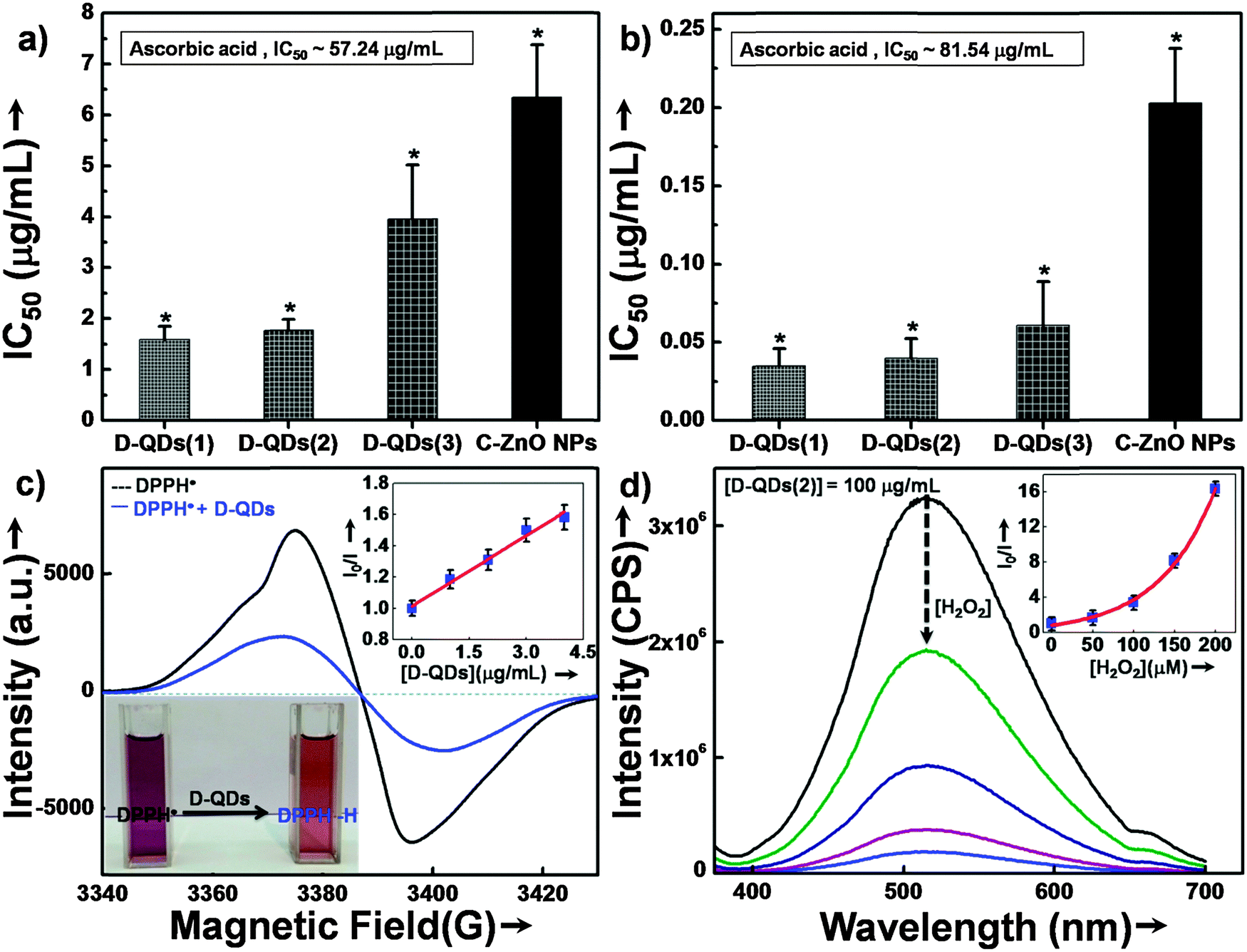 | ||
| Fig. 3 (a) DPPH˙ scavenging assay for different size D-QDs and C-ZnO NPs (b) superoxide radical scavenging assay for different size D-QDs and C-ZnO NPs. (c) A typical EPR spectrum for DPPH˙ with and without D-QDs (the bottom left inset shows the photograph for DPPH˙ in ethanol, with and without D-QDs and the upper right inset shows the SV plot for integrated EPR intensity with different concentration of D-QDs), (d) PL spectra showing defect luminescence quenching of D-QDs in the presence of different concentrations of H2O2 (the upper right inset, the SV plot for variation in integrated PL intensity with different H2O2 concentrations) (*p < 0.05, n = 3). | ||
The results of superoxide radical scavenging activity are shown in Fig. 3b. A similar trend was observed in the case of the superoxide radical scavenging activity as well where D-QDs(1) (IC50 ∼ 0.03 μg mL−1) and D-QDs(2) (IC50 ∼ 0.04 μg mL−1) showed comparable activity, which was superior to that of D-QDs(3) (IC50 ∼ 0.06 μg mL−1). However, all the tested D-QDs were found to be extremely potent compared to C-ZnO NPs (IC50 ∼ 0.20 μg mL−1) as well as ascorbic acid (IC50 ∼ 81.54 μg mL−1).
Mechanistic study of radical scavenging by D-QDs
The mechanism of the anti-radical property of D-QDs was studied using a stable DPPH˙.29 Here onwards for mechanistic study D-QDs(1) was used as a representative for D-QDs, unless otherwise mentioned. The addition of different concentrations of D-QDs ethanol solution to a 100 μM DPPH˙ in an ethanol solution resulted in the formation of 2,2-diphenyl-1-picrylhydrazine (DPPH-H). Electron paramagnetic resonance (EPR) spectroscopy was employed for monitoring the reduction of DPPH˙ to DPPH-H in the presence of D-QDs. The reduction in the integrated electron paramagnetic signal intensity of DPPH˙ in the presence of D-QDs was considered as direct evidence for the formation of DPPH-H. Fig. 3c shows the first derivative EPR spectrum without D-QDs and with D-QDs, its corresponding colour change from violet (DPPH˙) to pale orange (DPPH-H) is shown as the bottom left inset. A linear Stern–Volmer (SV) plot of integrated EPR absorption intensity (top right inset of Fig. 3c) with different concentrations of D-QDs shows a static nature for DPPH˙ quenching.This observation of anti-radical activity of D-QDs is similar to the recent report by Schrauben et al., where they observed that a solution containing reduced metal oxide NPs can transfer a proton (H+) and an electron (e−) to organic radicals, suggesting a proton coupled electron transfer (PCET) reaction.40 This observation of PCET in reduced metal oxide NP supports our observation on radical scavenging by D-QDs. In the case of D-QDs the oxygen vacancies may be present as singly ionized ( ) or doubly ionized (
) or doubly ionized ( ) states as shown below.41
) states as shown below.41
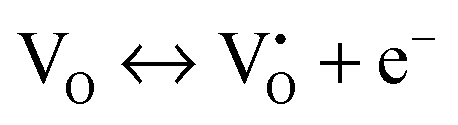 | (5) |
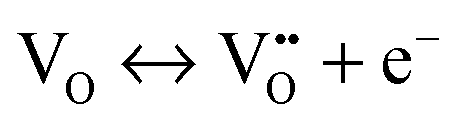 | (6) |
From eqn (5) and (6), it is clear that the existence of  or
or  will result in the generation of an electron. This e− along with the H+ from the solution matrix can be used for initiating PCET in the case of D-QDs. Thus the presence of oxygen vacancies in D-QDs will endow it with radical scavenging property via a PCET reaction.
will result in the generation of an electron. This e− along with the H+ from the solution matrix can be used for initiating PCET in the case of D-QDs. Thus the presence of oxygen vacancies in D-QDs will endow it with radical scavenging property via a PCET reaction.
Therefore, the above observation of anti-radical activity by D-QDs can follow the reaction scheme as shown in eqn (7), where R˙ = organic free radical.
| +H-ZnO1−x/e− + xR˙ → ZnO + xRH | (7) |
Furthermore we have checked the scavenging potential of D-QDs towards a non-radical reactive oxygen species, H2O2. The PL spectroscopy results show that in the presence of H2O2 a reduction in the defect emission was observed for D-QDs(2) (Fig. 3d).
At a higher concentration of H2O2 almost all the defect emission has vanished (ESI,† Movie 1). This implies the oxidation of VO by H2O2, which resulted in the healing of VO sites responsible for defect emission (eqn (8)).
| ZnO1−x + xH2O2 → ZnO + xH2O | (8) |
The SV plot for PL quenching by H2O2 shows an upward positive curve, which means the coexistence of both static and dynamic nature for luminescence quenching by H2O2.
Enzyme inhibition assays
The enzyme inhibition assays suggest a likely interaction of D-QDs with α-amylase and α-glucosidase. In porcine pancreatic α-amylase inhibition, all of the tested ZnO nanoparticle samples were found to be potent inhibitors of porcine pancreatic α-amylase (Fig. 4a). It is important to note that all of them exhibited superior activity as compared to acarbose, a standard inhibitor. Among this D-QDs show maximum inhibition activity, the activity of D-QD samples is in the order D-QDs(1) (IC50 ∼ 0.11 μg mL−1) > D-QDs(2) (IC50 ∼ 0.17 μg mL−1) > D-QDs(3) (IC50 ∼ 0.21 μg mL−1) which follows a decreasing trend with respect to reduction in particle size and increase in IDE/IBEE. However, C-ZnO NPs showed a lower enzyme activity (IC50 ∼ 0.34 μg mL−1). The effect of D-QDs on the inhibition kinetics of α-amylase was studied which was supported by a double reciprocal Lineweaver–Burk plot (Fig. 4b) revealing that the mode of D-QDs inhibition is uncompetitive (a type of reversible inhibition) with a decrease in both the apparent Km (Michaelis constant) and Vm (maximum enzyme reaction rate). Furthermore, the kinetic analysis when compared to the uninhibited enzyme activity (Vm of 2.82 and a Km of 79.84) was found to decrease upon incubation with D-QDs. The D-QDs(1) showed an enhanced decrease of Vm (0.11) and Km (2.54) while D-QDs(2) showed a relatively low decrease in the Vm (0.36) and Km (10.27). C-ZnO NPs exhibited a Vm and Km equivalent to 1.29 and 58.61 respectively. D-QDs(3) showed a relatively high Vm (1.10) and Km (57.36) as compared to D-QDs(1) and D-QDs(2). However, Vm and Km was found to be lower in all D-QD samples as compared to C-ZnO NPs. From the above results, it could be noted that incubation of α-amylase with D-QDs resulted in a size dependent inactivation (Fig. 4). Hence, the study suggests that D-QDs are promising candidates to control release of glucose by inhibiting the breakdown of starch by metabolic enzymes and could be developed as a lead anti-diabetic nanomaterial. | ||
| Fig. 4 (a) Enzyme inhibition assay of porcine pancreatic α-amylase, (b) kinetic analysis of porcine pancreatic α-amylase inhibition by different size D-QDs and C-ZnO NPs, (c) enzyme inhibition assay of murine pancreatic α-amylase and (d) murine intestinal α-amylase (*p < 0.05, n = 3). | ||
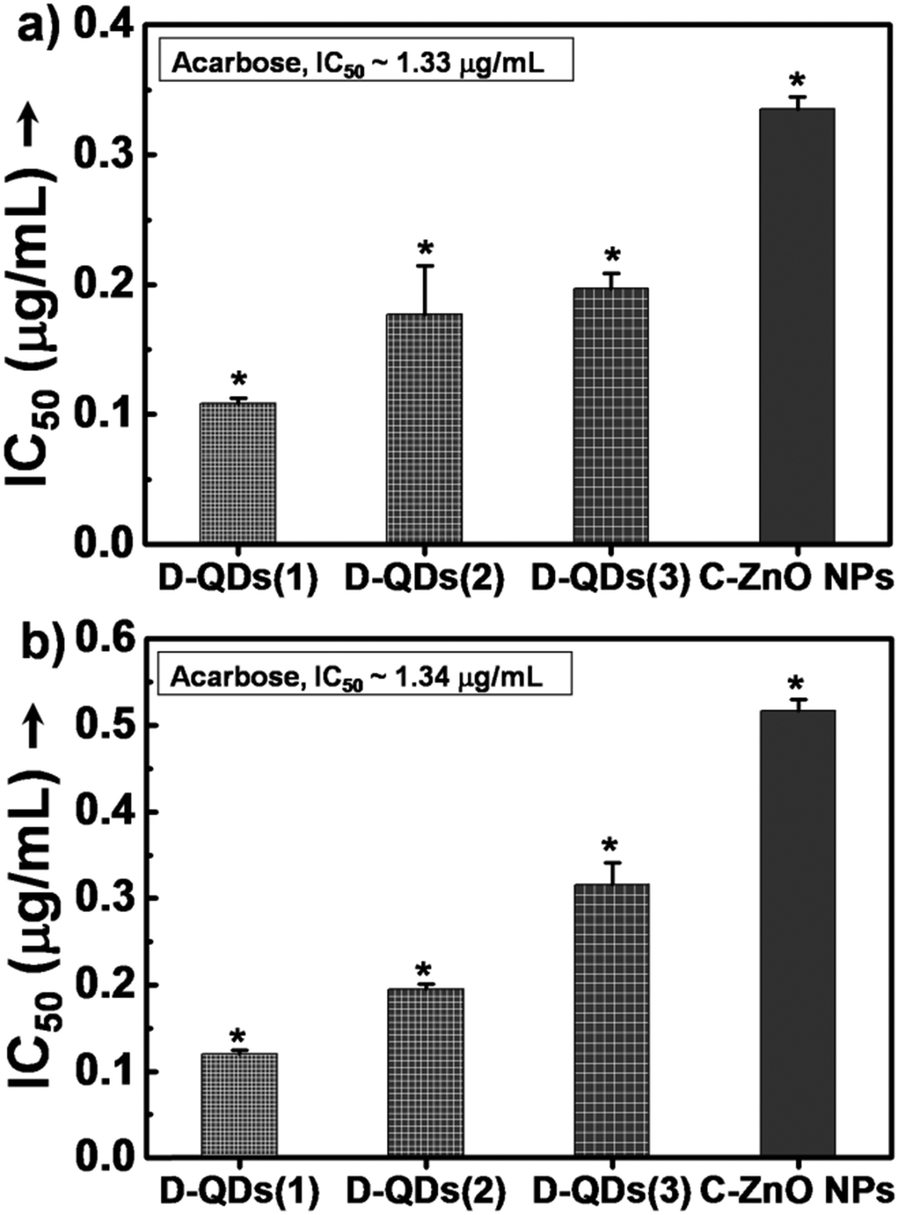 | ||
| Fig. 5 (a) Inhibition assay of porcine α-glucosidase and (b) murine intestinal α-glucosidase with different size D-QDs and C-ZnO NPs (*p < 0.05, n = 3). | ||
Mechanistic study of enzyme inhibition by D-ZnO QDs
Circular dichroism (CD) and fluorescence spectroscopy were used to investigate the nature of interaction of α-glucosidase and α-amylase with D-QDs. Initially, CD spectroscopy was used as it gave detailed secondary structure information of enzymes.42 Generally the α-helical content of an enzyme shows two characteristic minima at 208 and 222 nm.43 Any variation in the conformation of the α-helical content of enzyme can be found in CD spectra, either as a diminished minimum or a blue shift. Similarly, in the present study we observed a decrease in the negative humped peaks at 222 nm and 208 nm in the presence of D-QDs when compared with the control enzymes (Fig. 6a and b).These results clearly suggest a potential interaction of D-QDs with the α-helix of enzyme, which resulted in a conformational change in the enzyme secondary structure.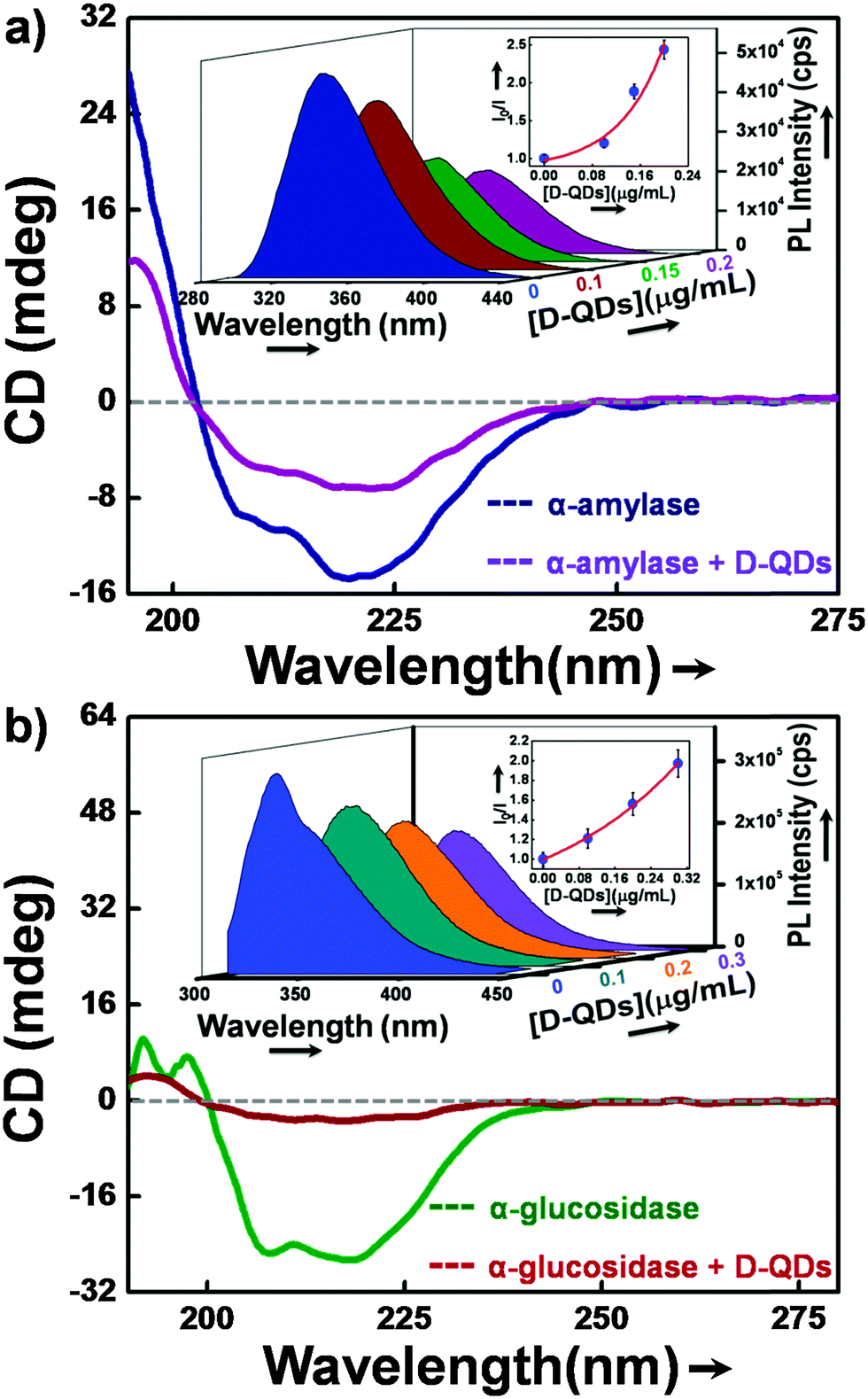 | ||
| Fig. 6 (a) CD spectra of α-amylase and (b) α-glucosidase with D-QDs and without D-QDs, the inset shows the quenching of tryptophan PL with different concentrations of D-QDs and the corresponding SV plot is given as the top right inset. | ||
In order to look into the nature of interaction of D-QDs with the enzymes, PL spectroscopy was used. The active site of both α-glucosidase and α-amylase contains residues of amino acid tryptophan (Trp).44,45 A strong intrinsic UVA fluorescence from these enzymes is an indication of the presence of a Trp residue at the active site of the enzyme. Thus the interaction of D-QDs with the enzyme can be determined from the reduction in the intrinsic UVA fluorescence.46 The SV plot is included in the inset of Fig. 6a and b, which shows decay in the Trp fluorescence intensity for α-glucosidase and α-amylase in the presence of different concentrations of D-QDs. The upward positive curve of the SV plot can be related to both dynamic and static interaction of D-QDs with α-glucosidase and α-amylase. The time-correlated single photon counting (TCSPC) for Trp emission at 345 nm is shown in Fig. S3 (ESI†). The biexponential fit shows a fast decay component of 1.45 ns for α-amylase in the presence of D-QDs(1), while for uninhibited α-amylase it was 1.67 ns. Similarly, in the case of the slow decay component, the presence of D-QDs with α-amylase shows an evident decrease in the lifetime, i.e. 3.78 ns when compared with the uninhibited α-amylase lifetime of 4.13 ns. These results were in correlation with the observed PL quenching, which could result in the reduction of the lifetime for Trp emission. Hence, it can be concluded that there was an apparent interaction of D-QDs with the enzyme. Therefore, CD, PL and TCSPC investigations together confirm the interaction of D-QDs with the enzymes which resulted in the observed enzyme inhibition.
The electrostatic interaction between positively charged VO and the negatively charged enzyme site can be the possible reason for the observed enzyme inhibition activity. The D-QDs used in the present study have size dependence in surface VO formation, where the surface defect density increases with particle size reduction. As a result of this an increased positive surface charge density was observed with a reduction in D-QDs particle size, as evidenced by the IDE/IBEEversus zeta potential value (Fig. 2d). Hence, an increase in the positive charge of the D-QD surface with decreasing size will enhance the electrostatic interaction of smaller D-QDs with negatively charged enzyme sites. This electrostatic interaction may result in the conformational change of the enzyme active site thereby inhibiting substrate binding. This could be the possible mechanism for enhanced enzyme inhibition activity with decreasing particle size of D-QDs.
A recent report on the inhibitory activity of surface-functionalized metal-chalcogenide QDs shows that nanoscale inhibitors are active even at very low concentrations, thereby eliminating their toxicity concerns.47 However, in order to consider D-QDs for biomedical applications, the biocompatibility study is inevitable. Hence, we have studied the biocompatibility of the current samples on the human embryonic kidney (HEK 293) cell line by using thiazolyl blue tetrazolium bromide (MTT) assay (see Fig. S4, ESI†). All the tested samples were found to show good cell viability at a very high concentration (∼100 μg mL−1), when compared with the very low IC50 value (<2 μg mL−1) for enzyme inhibition and radical scavenging activity reported in this paper.
The summary of physico-chemical properties of the sample studied in the current work (Table 1) shows the size dependence in the radical scavenging activity and enzyme inhibition activity. The reduction in IDE/IBEE with increasing size implies a size dependent VO formation. Similarly with the reduction in IDE/IBEE ratio an increase in the IC50 of D-QDs for radical scavenging and enzyme inhibition was observed. This suggests that the size dependence in surface VO formation can be the underlying reason for the observed increase in activity for smaller size D-QDs. Therefore, these D-QDs can be considered as an alternative strategy for efficient management and treatment of diabetes apart from the available anti-diabetic therapeutics.48
| Labels | Sizea (nm) | I DE/IBEE ratio | Zeta-potential* (mV) | IC50* for DPPH˙ (μg mL−1) | IC50* for α-amylaseb (μg mL−1) |
|---|---|---|---|---|---|
| a Particle size measured from TEM images shown in Fig. 1 (main text). b Enzyme source is porcine pancreas (*p < 0.05, n = 3). | |||||
| D-QDs(1) | 3.56 ± 0.25 | 14.56 | −61.48 ± 9.10 | 1.59 | 0.11 |
| D-QDs(2) | 5.12 ± 0.25 | 13.47 | −54.06 ± 5.06 | 1.77 | 0.17 |
| D-QDs(3) | 7.18 ± 0.25 | 10.75 | −41.39 ± 6.10 | 3.96 | 0.21 |
| C-ZnO NPs | ∼50 ± 15 | 0.41 | 19.92 ± 5.02 | 6.34 | 0.34 |
4. Conclusions
Therefore, the following conclusions were drawn from this study, (1) the antioxidant and enzyme inhibition activity of D-QDs have size dependence (i.e., it increases with decreasing D-QDs size), (2) the size dependent enzyme inhibition and antioxidant properties could be due to the size dependence in VO formation. In addition to this, our benchmark antioxidant and enzyme inhibition assay results revealed that smaller size D-QDs(1) are significantly superior, having four times lesser IC50 value (∼1.59 μg mL−1) for antioxidant activity and three times lesser IC50 value (∼0.11 μg mL−1) for enzyme inhibition activity when compared to C-ZnO NPs (Table 1). Thus the understanding gained from this study will pave the way towards rational design of safe and effective anti-diabetic nanomedicine through NP size and surface defect engineering.Contributions
A.A. and A.R.K. conceived the research and designed the experiments. A.R.K supervised the research. A.A. performed the synthesis and characterization of the materials. S.G. and P.A.M. performed the free radical scavenging, enzyme inhibition assays. A.A. and S.G. performed the mechanistic study. A.A., A.R.K., S.G., B.A.C., and M.N.G. analyzed the data. A.A., A.R.K., and S.G. wrote the paper.Competing financial interests
The authors declare no competing financial interests.Acknowledgements
We express our thanks to SAIF, IIT Bombay, for HR-TEM studies, Department of Chemistry, IIT Bombay, for CD studies and CDEEP, IIT Bombay, for supplementary video shooting. S.G. thanks Council of Scientific and Industrial Research (CSIR, Government of India) for Senior Research Fellowship (09/137(0516)/2012-EMR-I).Notes and references
- M. Brownlee, Diabetes, 2005, 54, 1615–1625 CrossRef CAS.
- J. W. Baynes, Diabetes, 1991, 40, 405–412 CrossRef CAS.
- P. A. Low, K. K. Nickander and H. J. Tritschler, Diabetes, 1997, 46, S38–S42 CrossRef CAS.
- D. Giugliano, A. Ceriello and G. Paolisso, Diabetes Care, 1996, 19, 257–267 CrossRef CAS.
- A. Ceriello and E. Motz, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 816–823 CrossRef CAS PubMed.
- A. Maritim, R. Sanders and R. J. Watkins, J. Biochem. Mol. Toxicol., 2003, 17, 24–38 CrossRef CAS PubMed.
- F. Giacco and M. Brownlee, Circ. Res., 2010, 107, 1058–1070 CrossRef CAS PubMed.
- S. E. Inzucchi, JAMA, J. Am. Med. Assoc., 2002, 287, 360–372 CrossRef CAS.
- W. Puls and U. Keup, Diabetologia, 1973, 9, 97–101 CrossRef CAS.
- P. Rosen, H. J. Tritschler, G. A. King and A. Azzi, Antioxidants in diabetes management, CRC Press, 2000 Search PubMed.
- J. S. Johansen, A. K. Harris, D. J. Rychly and A. Ergul, Cardiovasc. Diabetol., 2005, 4, 5 CrossRef PubMed.
- R. D. Umrani and K. M. Paknikar, J. Biomed. Nanotechnol., 2011, 7, 148–149 CrossRef CAS PubMed.
- T. K. Bhowmick, A. K. Suresh, S. G. Kane, A. C. Joshi and J. R. Bellare, J. Nanopart. Res., 2009, 11, 655–664 CrossRef CAS.
- R. D. Umrani and K. M. Paknikar, Nanomedicine, 2014, 9, 89–104 CrossRef CAS PubMed.
- M. Hegde, T. Wang, Z. L. Miskovic and P. V. Radovanovic, Appl. Phys. Lett., 2012, 100, 141903 CrossRef PubMed.
- G. Guisbiers, J. Phys. Chem. C, 2011, 115, 2616–2621 CAS.
- A. Asok, A. R. Kulkarni and M. N. Gandhi, J. Mater. Chem. C, 2014, 2, 1691–1697 RSC.
- A. Asok, M. N. Gandhi and A. R. Kulkarni, Nanoscale, 2012, 4, 4943–4946 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- A. Gessner, A. Lieske, B. R. Paulke and R. H. Müller, Eur. J. Pharm. Biopharm., 2002, 54, 165–170 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- J. Wang, J. D. Byrne, M. E. Napier and J. M. DeSimone, Small, 2011, 7, 1919–1931 CrossRef CAS PubMed.
- G. Su, H. Zhou, Q. Mu, Y. Zhang, L. Li, P. Jiao, G. Jiang and B. Yan, J. Phys. Chem. C, 2012, 116, 4993–4998 CAS.
- M. De, S. S. Chou and V. P. Dravid, J. Am. Chem. Soc., 2011, 133, 17524–17527 CrossRef CAS PubMed.
- C. A. Falaschetti, T. Paunesku, J. Kurepa, D. Nanavati, S. S. Chou, M. De, M. Song, J.-t. Jang, A. Wu and V. P. Dravid, ACS Nano, 2013, 7, 7759–7772 CrossRef CAS PubMed.
- H. Kaftelen, K. Ocakoglu, R. Thomann, S. Tu, S. Weber and E. Erdem, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014113 CrossRef.
- S. S. Lee, W. Song, M. Cho, H. L. Puppala, P. Nguyen, H. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS PubMed.
- W. Brand-Williams, M. Cuvelier and C. Berset, LWT–Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- F. Nanjo, K. Goto, R. Seto, M. Suzuki, M. Sakai and Y. Hara, Free Radical Biol. Med., 1996, 21, 895–902 CrossRef CAS.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- S. Ghosh, P. More, A. Derle, A. B. Patil, P. Markad, A. Asok, N. Kumbhar, M. L. Shaikh, B. Ramanamurthy, V. S. Shinde, D. D. Dhavale and B. A. Chopade, PLoS One, 2014, 9, e106039 Search PubMed.
- K. Tam, C. Cheung, Y. Leung, A. Djurišic, C. Ling, C. Beling, S. Fung, W. Kwok, W. Chan and D. Phillips, J. Phys. Chem. B, 2006, 110, 20865–20871 CrossRef CAS PubMed.
- S. Cho, J. Ma, Y. Kim, Y. Sun, G. K. Wong and J. B. Ketterson, Appl. Phys. Lett., 1999, 75, 2761–2763 CrossRef CAS PubMed.
- S. Monticone, R. Tufeu and A. Kanaev, J. Phys. Chem. B, 1998, 102, 2854–2862 CrossRef CAS.
- L. Zhang, L. Yin, C. Wang, N. Lun, Y. Qi and D. Xiang, J. Phys. Chem. C, 2010, 114, 9651–9658 CAS.
- K. Vanheusden, C. Seager, W. t. Warren, D. Tallant and J. Voigt, Appl. Phys. Lett., 1996, 68, 403–405 CrossRef CAS PubMed.
- A. Van Dijken, E. Meulenkamp, D. Vanmaekelbergh and A. Meijerink, J. Lumin., 2000, 90, 123–128 CrossRef CAS.
- A. J. Morfa, B. C. Gibson, M. Karg, T. J. Karle, A. D. Greentree, P. Mulvaney and S. Tomljenovic-Hanic, Nano Lett., 2012, 12, 949–954 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- G. Mahan, J. Appl. Phys., 1983, 54, 3825–3832 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2876–2890 CrossRef PubMed.
- K. M. Pan, M. Baldwin, J. Nguyen, M. Gasset, A. Serban, D. Groth, I. Mehlhorn, Z. Huang, R. J. Fletterick and F. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10962–10966 CrossRef CAS.
- M. Hermans, M. Kroos, J. Van Beeumen, B. Oostra and A. Reuser, J. Biol. Chem., 1991, 266, 13507–13512 CAS.
- N. Ramasubbu, C. Ragunath, P. J. Mishra, L. M. Thomas, G. Gyémánt and L. Kandra, Eur. J. Biochem., 2004, 271, 2517–2529 CrossRef CAS PubMed.
- H. M. Rawel, S. K. Frey, K. Meidtner, J. Kroll and F. J. Schweigert, Mol. Nutr. Food Res., 2006, 50, 705–713 CAS.
- S. Ghosh, M. Ray, M. R. Das, A. Chakrabarti, A. H. Khan, D. D. Sarma and S. Acharya, Phys. Chem. Chem. Phys., 2014, 16, 5276–5283 RSC.
- R. Mo, T. Jiang, J. Di, W. Tai and Z. Gu, Chem. Soc. Rev., 2014, 43, 3595–3629 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, PLE, supplementary video demonstrating defect luminescence quenching in the presence of H2O2. See DOI: 10.1039/c5tb00407a |
| This journal is © The Royal Society of Chemistry 2015 |