
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable dispersivity and molecular-trapping performance of mesostructured CaCO3–thermosensitive polymer composite microspheres
Atsuharu
Inoue
a,
Hitoshi
Tamagawa
a,
Yuya
Oaki
a,
Sadahito
Aoshima
b and
Hiroaki
Imai
*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: hiroaki@applc.keio.ac.jp
bDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 26th March 2015
Abstract
Monodispersed thermosensitive microspheres were synthesized by the combination of poly(N-isopropylacrylamide) (PNIPAAm) and a mesostructured framework consisting of vaterite CaCO3 nanocrystals ∼20 nm in diameter. The switchable dispersivity in water and organic media was provided to the rigid porous framework by the soft shell of the thermosensitive organic component. The microspheres were shuttlecocked between water and hydrophobic organic media by a swing in temperature across the lower critical solution temperature (LCST). The composite microspheres performed as an active carrier of organic molecules due to their switchable property. Hydrophobic molecules were captured by the microspheres dispersed in an organic medium above LCST and were then delivered and released to another organic phase through a water phase by a swing in temperature.
Introduction
Biominerals are known as inorganic–organic composites that are elaborately organized from nanoscopic to macroscopic scales.1–9 In recent years, most CaCO3-based biominerals were revealed to have a hierarchical architecture composed of nanometric crystals.10–12 These specific nanostructures are categorized as a kind of mesocrystal, i.e., mesostructured crystals consisting of oriented nanoscale units that are covered with organic molecules. The architectures and formation processes of biominerals have provided considerable inspiration in the field of materials chemistry, leading to new fabrication techniques and the development of novel functionalities.13 Recently, well-defined microspheres of CaCO3 nanoparticles were produced in various solution systems.14–18 In our previous work,19 monodispersed microspheres having an oriented nanocrystal mosaic interior were obtained through the crystal growth of CaCO3 with polystyrene sulfonate (PSS) under specific conditions. The CaCO3-based mesocrystal is a potential host material because various guest molecules can be introduced in the interspatial organic domains between the oriented nanocrystals.20–23 From the standpoint of the exploration of their function, however, the CaCO3-based mesocrystals have hardly been studied for practical applications.Poly(N-isopropylacrylamide) (PNIPAAm) exhibits a lower critical solution temperature (LCST) transition from 32 to 34 °C.24 The polymer is hydrophilic and soluble in water below LCST but becomes hydrophobic and forms a macroscopic coacervate phase above that temperature due to the fluctuation of hydrophobic interactions and hydrogen bonding.25 The introduction of the polymer onto the nanoparticles of metals and other kinds of polymers provides their responsivity to temperature.26,27 For example, block copolymers including PNIPAAm formed a micelle and were applied as a drug delivery system (DDS).28 Gold29 and mesoporous silica30,31 nanoparticle composites with PNIPAAm are also applied as a DDS. Hydrogel composites containing PNIPAAm colloidal crystals were utilized as a photonic crystal.32
In the present study, we prepared the monodispersed thermosensitive microspheres of mesostructured CaCO3 that was covered with a shell of PNIPAAm. The original polymer (PSS) covering the specific crystalline structure of oriented vaterite CaCO3 nanoparticles was replaced by the functional molecule. The influence of temperature on the dispersivity in various media was investigated. Moreover, the release and retention abilities of the microspheres for a hydrophobic molecule were monitored according to the swing in temperature. The potential of the CaCO3-based thermosensitive microspheres as an active carrier is discussed on the basis of their switchable dispersivity and molecular-trapping performance by the tuning of temperature.
Experimental
We prepared mesostructured CaCO3 crystals by using a simple mixing method of two solutions at room temperature.19 Typically, 128 cm3 of 1.0 mol dm−3 CaCl2 solution was added to 4.0 dm3 of 16 mmol dm−3 sodium carbonate (Na2CO3, Junsei Chemical 99.8%) solution containing 1.0 g dm−3 PSS (Aldrich, Mw: 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). After mixing, the solution was stirred for 60 s, and then left for 24 h. The organic components included in the products were extracted by immersion in a 5 wt% NaClO aqueous solution for 48 h. The resultant samples were then washed extensively with purified water. N-Isopropylacrylamide (NIPAAm) as a monomer and methyrenebisacrylamide (MBA) as a crosslinker were introduced into the nanopores of the microspheres by impregnation of the mixture. The polymerization was performed in hexane containing 0.01 g of 2,2′-dimethoxy-2-phenylacetophenone (BDK) as an initiator under UV illumination for 3 h using Ushio OPM2-502XQ (3.8 W). Finally, we obtained the composite microspheres 2–3 μm in diameter through classification by dispersivity in water and a subsequent freeze–drying process.
000). After mixing, the solution was stirred for 60 s, and then left for 24 h. The organic components included in the products were extracted by immersion in a 5 wt% NaClO aqueous solution for 48 h. The resultant samples were then washed extensively with purified water. N-Isopropylacrylamide (NIPAAm) as a monomer and methyrenebisacrylamide (MBA) as a crosslinker were introduced into the nanopores of the microspheres by impregnation of the mixture. The polymerization was performed in hexane containing 0.01 g of 2,2′-dimethoxy-2-phenylacetophenone (BDK) as an initiator under UV illumination for 3 h using Ushio OPM2-502XQ (3.8 W). Finally, we obtained the composite microspheres 2–3 μm in diameter through classification by dispersivity in water and a subsequent freeze–drying process.
The dispersivity of 5 mg microspheres in 15 cm3 water was evaluated by changing the temperature between 25 and 60 or 90 °C. We observed the turbidity in a test tube and aggregation behavior on a glass slide under an optical microscope. A toluene–water–nitrobenzene system was used to examine the dispersivity of the composite microspheres. We put 15 dm3 nitrobenzene, 15 dm3 water containing 7 mg of the CaCO3–PNIPAAm composite powder, and 15 dm3 toluene into a glass vessel in order. The three-phase system was stirred at 20 or 90 °C for 72 h with ultrasonic agitation. The fixing ability of the microspheres was studied using a hydrophobic surface of a glass slide prepared by dipping of a toluene solution of 1 wt% dodecyltrichlorosilane. Alkyl chains were formed on the glass surface after subsequent drying.
Copper phthalocyanine (CuPh, a blue hydrophobic pigment) was introduced into the CaCO3–PNIPAAm composites by the following process. The composite powder (10 mg) was dispersed in 20 dm3 water at room temperature. Toluene (20 dm3) containing 0.2 mmol dm−3 of CuPh was poured on the aqueous dispersion. After the vessel containing the two-phase system was stirred at 90 °C for 72 h with ultrasonic agitation once in a while, the composite particles were transferred into the organic phase. By stirring at room temperature for 72 h with ultrasonic agitation once in a while, the pigment-containing composite particles were redispersed in water. The pigment was released from the composites in a fresh organic phase at 90 °C. The concentration of the pigment was monitored from the absorption spectra in the ultraviolet-visible region using a Jasco V-560. We also examined the transport of CuPh using a toluene–water–toluene system in a U-tube. The composite microspheres were shuttlecocked between the organic and water phases by a swing in temperature with ultrasonic agitation.
The morphology of the products was characterized using a field-emission scanning electron microscope (FESEM, Hitachi S-4700) and a field-emission transmission electron microscope (FETEM, FEI Tecnai F20). Optical images were taken using Keyence VHX 1000. The X-ray diffraction (XRD) patterns were recorded using Rigaku MiniFlex II with Cu Kα radiation. The pore-size distribution was evaluated from nitrogen adsorption–desorption isotherms obtained using Miromeritics Tristar 3000. The weight of the organic components was measured by thermogravimetry (TG) using Seiko Instruments TG/DTA 7200. The kind of organic components was measured by Fourier transform infrared spectroscopy (FT-IR) using Jasco FT-IR 4200.
Results and discussion
As shown in Fig. 1a, well-defined microspheres ca. 2.5 μm in diameter were produced in 24 h after the mixing of a calcium solution and a carbonate solution containing 1.0 g dm−3 PSS. In the SEM images of the cross section and surface of the CaCO3–PSS composite microspheres (Fig. 1b and c), the microspheres consisted of small particles with a diameter of ca. 20 nm, as illustrated in Fig. 1d. According to the XRD patterns (Fig. 2a), the microspheres were assigned to be pure vaterite. The PSS-mediated vaterite spheres were stable for a long time in water and an ambient atmosphere. The crystalline grains composed of the microspheres were clearly observed after the decomposition of PSS by using a NaClO solution (Fig. 1e–h). The pore volume as an interparticular space in the microspheres increased with the removal of the organic component after the treatment. | ||
| Fig. 1 SEM images and schematic illustrations of CaCO3–PSS (a–d), CaCO3–PSS after the NaClO treatment (e–h), CaCO3–PNIPAAm (i–l), and CaCO3–PNIPAAm after the HCl treatment (m–o). | ||
 | ||
| Fig. 2 XRD patterns (a), FT-IR spectra (b), and TG curves (c) of the CaCO3–polymer composite microspheres. | ||
After the polymerization of NIPAAm, the interparticular space was filled with the organic phase (Fig. 1i–l). After the dissolution of CaCO3 with an HCl solution, the ca. 300 nm thick shell of the polymer was clearly observed (Fig. 1m–o). The nanostructure was found on the surface of the polymer shell. Fig. 2b shows the FT-IR spectra of the CaCO3–polymer composite microspheres before and after treatment. We observed typical absorption bands of the vaterite phase at 744 (ν2 mode of CO32−) and 877 cm−1 (ν4 mode of CO32−) for all the samples. The CaCO3–PSS composites have strong peaks at 1130 and 1210 cm−1 due to SO3− and 1,4-substituted benzene of PSS, respectively. These absorption signals decreased with the NaClO treatment. The presence of amide II group absorption at 1655 cm−1 indicates the formation of PNIPAAm in the CaCO3 microspheres after the introduction of the monomer with a cross-linker and subsequent UV irradiation in hexane containing the initiator. The removal of PSS from the CaCO3 microspheres and the introduction of PNIPAAm into the interparticular space were supported by variation in the TG curves (Fig. 2c). The mass reduction in the range from 350 to 450 °C was ascribed to combustion of the polymers contained in the microspheres. The original microspheres had ca. 10 wt% of PSS, and half of them were lost by the NaClO treatment. The polymerization induced more than 20 wt% of PNIPAAm in the spheres. This means that the surface of the spheres was almost completely covered with the polymer, as shown in Fig. 1. The polymorph of CaCO3 was maintained after the removal of PSS and the introduction of PNIPAAm.
The microspheres were dispersed in water and slowly precipitated for several hours at 25 °C (Fig. 3a and c). On the other hand, they were immediately precipitated in a few minutes through aggregation at 60 °C (Fig. 3b and d). The monolayer assembly of the microspheres was achieved on the hydrophobic surface of an alkyl-modified glass substrate by dropping of the dispersion and subsequent drying (Fig. 4a). We observed a partial hexagonal arrangement of the microspheres connected with each other via the sticky polymer in the monolayer (Fig. 4c). The ordered structure of the microspheres was firmly fixed on the hydrophobic surface in water at 60 °C. On the other hand, the spheres were rapidly removed from the surface with ultrasonic agitation in water at 25 °C (Fig. 4b). The hydrophobic polymer chains of the shell are entangled with each other at the interface of the spheres and the alkyl chains of the hydrophobic surface.
 | ||
| Fig. 3 Macroscopic and microscopic images of the dispersion of CaCO3–PNIPAAm composites in water at 25 °C (a, c) and 60 °C (b, d). | ||
 | ||
| Fig. 4 Optical micrographs (a, b) and SEM image (c) of the CaCO3–PNIPAAm composites on an alkyl-modified glass substrate after washing in water by sonication at 60 °C (a, c) and 25 °C (b). | ||
We prepared a toluene–water–nitrobenzene system to clarify the property of the microspheres (Fig. 5). The water phase was turbid due to the dispersion of the microspheres at 20 °C. On the other hand, the aqueous part became clear and the toluene and nitrobenzene phases clouded at 90 °C. (The high temperature was used to induce the change of the system immediately.) This means that the microspheres that had moved into the organic phases were stably dispersed in the media above LCST. This behaviour was completely reversible by a swing in temperature. Because the PNIPAAm shell covering the porous carbonate body is hydrophilic and swells with water below LCST, the microspheres are dominantly dispersed in water. The hydrophobic shell above LCST induces aggregation in water and leads to a stable dispersion in organic media of the microspheres. Thus, the microspheres were shuttlecocked between water and hydrophobic media with switchable hydrophobicity of the shell layer by a swing in temperature. However it took several hours to achieve the transfer of the microspheres between two phases with ultrasonic agitation, although the microspheres were immediately precipitated in the original phase within a few minutes by increasing or decreasing the temperature. The gentle motion of the microspheres is ascribed to a low permeability of the dispersant.
 | ||
| Fig. 5 Photographs and schematic illustration of a toluene–water–nitrobenzene system containing the CaCO3–PNIPAAm composites with a swing in temperature. | ||
We prepared a toluene–water system containing a hydrophobic pigment, copper phthalocyanine (CuPh) (Fig. 6). The microspheres were dispersed in water below LCST and moved into the organic phases above LCST (90 °C). The microspheres moved back from the organic phase to water upon decreasing the temperature to 20 °C. We observed that the water phase became pale blue with the dispersion of the microspheres, indicating that the pigment was captured by the CaCO3–PNIPAAm microspheres in the toluene solution at 90 °C. The microspheres that captured the pigment were dispersive in water below LCST. The saturated CuPh content in the composite microspheres was estimated to be about 1.2 × 10−4 mol g−1. After the pigment-capturing microspheres were removed from the water, we redispersed them into a pure toluene–water system below LCST. When the temperature was increased, the microspheres were transported into the fresh toluene phase. After removal of the microspheres by decreasing the temperature, the toluene phase became blue due to the release of the pigment in the organic phase from the microsphere. The microspheres delivered and released the hydrophobic molecules to a pure organic medium above LCST. About 70% of the CuPh captured in the microspheres was released in the organic phase.
 | ||
| Fig. 6 Photographs of a toluene–water system containing the CaCO3–PNIPAAm composites and CuPh with a swing in temperature. CuPh was trapped by the CaCO3–PNIPAAm composites in a toluene phase transport at 90 °C and transported to water at 20 °C. The concentration of CuPh trapped in the dried composites was estimated to be 1.2 × 10−4 mol g−1. About 70% of CuPh in the CaCO3–PNIPAAm composites was released to another toluene phase with a swing in temperature. | ||
We prepared a toluene–water–toluene system containing CuPh in the left toluene phase using a U-tube (Fig. 7). The microspheres dispersed in water below LCST moved into the organic phases above LCST (90 °C). The microspheres moved back from the organic phase to water when the temperature was decreased to 20 °C. The CuPh concentration in the right toluene phase increased and became the same as that in the left toluene phase through 11 cycles of the shuttlecock behaviour. Therefore, the thermosensitive microspheres play a role as an active carrier delivering specific hydrophobic agents between two organic phases separated by water.
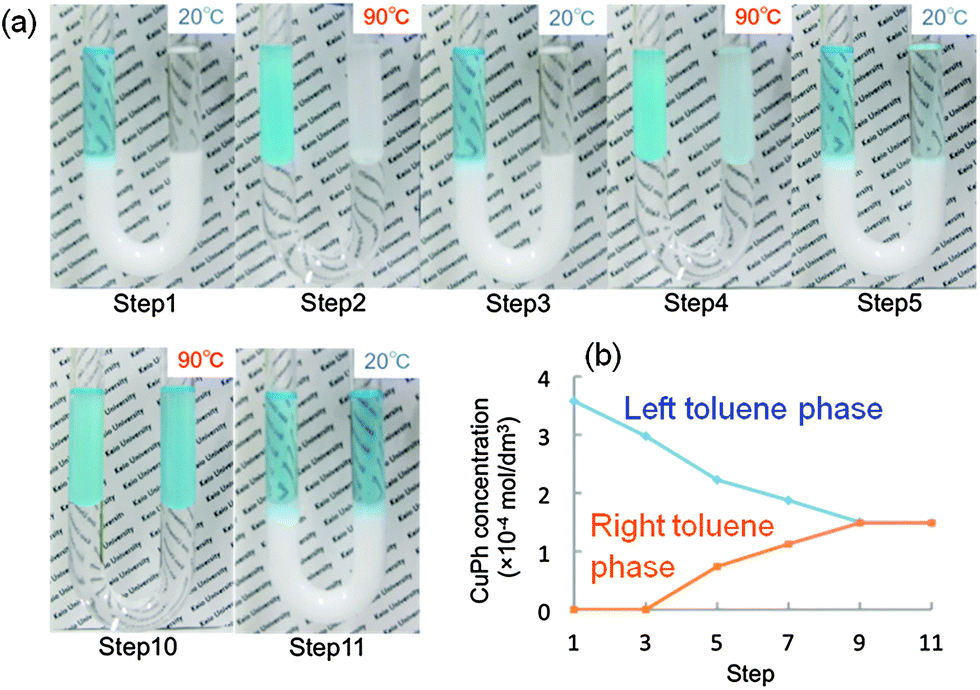 | ||
| Fig. 7 Photographs of a toluene–water–toluene system containing the CaCO3–PNIPAAm composites and CuPh in a U-tube (a). CuPh was transported with the CaCO3–PNIPAAm composites from the left toluene phase into the right toluene phase through the water phase by a swing in temperature. The CuPh concentration of the left and right toluene phases changed with the cycle of temperature (b). | ||
The functionality of the composite microspheres was confirmed using bare CaCO3 microspheres without PNIPAAm and pure PNIPAAm without CaCO3. We prepared the bare CaCO3 microspheres and pure PNIPAAm from the composite microspheres by the dissolution of PNIPAAm with NaClO and CaCO3 with diluted hydrochloric acid, respectively. The blue pigment stayed in the left toluene phase after the temperature swing in the toluene–water–toluene system. This means that the composite structure consisting of the mesostructured framework of CaCO3 and the polymer shell is essential for the thermosensitive trapping performance of hydrophobic molecules. The hydrophobic nature of the polymer shell attracts hydrophobic molecules in organic media above LCST. The hydrophobic molecules are trapped in the nanopores of the inorganic framework with the hydrophilic polymer shell below LCST. Thus, the composite microspheres containing hydrophobic molecules are dispersed in water. The hydrophobic molecules are released through the hydrophobic polymer shell to another organic medium above LCST. A series of trap-release mechanisms is illustrated in Fig. 8. Microspheres are stably dispersed in water because PNIPAAm swells below LCST. The hydrophobic pigment is captured by the polymer shell covering the CaCO3 framework above LCST. In water, the trapped hydrophobic pigment is protected in the nanopores of the framework below LCST. The pigment is released into the organic media with the swelling of the polymer above LCST. However, the pigments trapped in the deep region of the nanopores were not released in the organic media.
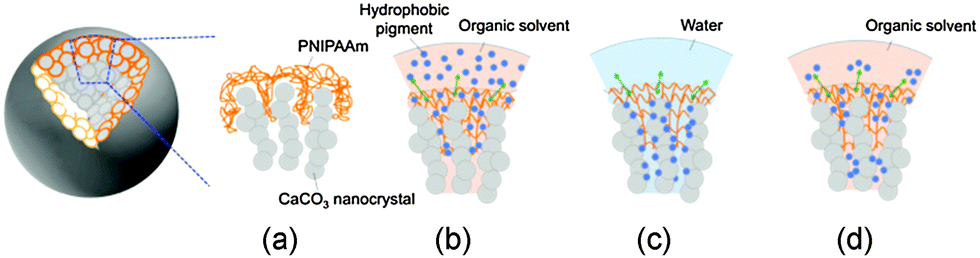 | ||
| Fig. 8 Schematic illustration of a trap–release mechanism of the hydrophobic pigment on the CaCO3–PNIPAAm composites (a). The hydrophobic pigment is trapped by PNIPAAm with swelling by the organic medium above LCST (b). The pigment is kept in the nanopores in water below LCST (c). The pigment is released into the organic phase with swelling by the organic medium above LCST (d). | ||
The dispersivity and the fixing ability of the microspheres are controllable with the swing in temperature. This suggests that specific organic agents can be delivered to hydrophobic media at a high temperature using the thermosensitive microspheres. Because the size of the CaCO3 spheres19 and the property of the thermosensitive polymer can be designed, the composite microspheres could be developed into a novel drug delivery system as an active carrier.
Conclusions
Monodispersed thermosensitive microcomposites consisting of a rigid inorganic porous core and a soft functional shell were synthesized by the combination of vaterite CaCO3 microspheres and poly(N-isopropylacrylamide). Their dispersivity in water, their fixing ability on a hydrophobic surface, and their molecular-trapping performance were switchable by temperature. The functional composites would be applicable to a novel drug delivery or transport system.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (no. 22107010) on Innovative Areas of “Fusion Materials: Creative Development of Materials and Exploration of their Function through Molecular Control” (area no. 2206) from the Ministry of Education, Culture, Sports, Science and Technology from Japan Society of the Promotion of Science.Notes and references
- S. Mann, Nature, 1988, 332, 119 CrossRef CAS.
- L. Addadi and S. Weiner, Angew. Chem., Int. Ed. Engl., 1992, 31, 153 CrossRef.
- S. Mann, Nature, 1993, 365, 499 CrossRef CAS.
- J. Aizenberg, J. C. Weaver, M. S. Thanawala, V. C. Sundar, D. E. Morse and P. Fratzl, Science, 2005, 309, 275 CrossRef CAS PubMed.
- S. I. Stupp and P. V. Braun, Science, 2007, 277, 1242 CrossRef.
- S. Weiner and L. Addadi, Annu. Rev. Mater. Sci., 2011, 41, 21 CrossRef CAS.
- A. W. Xu, M. Antonietti, S. H. Yu and H. Cölfen, Adv. Mater., 2008, 20, 1333 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161 CrossRef CAS PubMed.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461 CrossRef CAS.
- Y. Oaki, A. Kotachi, T. Miura and H. Imai, Adv. Funct. Mater., 2006, 16, 1633 CrossRef CAS.
- Y. Oaki and H. Imai, Angew. Chem., Int. Ed., 2005, 44, 6571 CrossRef CAS PubMed.
- Y. Oaki and H. Imai, Small, 2006, 2, 66 CrossRef CAS PubMed.
- T. Kokubu, Y. Oaki, E. Hosono, H. Zhou and H. Imai, Adv. Funct. Mater., 2011, 21, 3673 CrossRef CAS.
- K. Naka, S. C. Huang and Y. Chujo, Langmuir, 2006, 22, 7760 CrossRef CAS PubMed.
- X. H. Guo, S. H. Yu and G. B. Cai, Angew. Chem., Int. Ed., 2006, 45, 3977 CrossRef CAS PubMed.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576 CrossRef PubMed.
- M. G. Page and H. Cölfen, Cryst. Growth Des., 2006, 6, 1915 CAS.
- M. S. Mo, S. H. Lim, Y. W. Mai, R. K. Zheng and S. P. Ringer, Adv. Mater., 2008, 20, 339 CrossRef CAS.
- H. Imai, N. Tochimoto, Y. Nishino, Y. Takezawa and Y. Oaki, Cryst. Growth Des., 2012, 12, 876 CAS.
- Y. Oaki and H. Imai, Adv. Funct. Mater., 2005, 15, 1407 CrossRef CAS.
- Y. Oaki and H. Imai, Chem. Commun., 2005, 6011 RSC.
- L. Meijer, A. L. Skaltsounis, P. Magiatis, P. Polychronopoulos, M. Knockaert, M. Leost, X. P. Ryan, C. A. Vonica, A. Brivanlou, R. Dajani, C. Crovace, C. Tarricone, A. Musacchio, S. M. Roe, L. Pearl and P. Greengard, Chem. Biol., 2003, 10, 1255 CrossRef CAS PubMed.
- A. Varshney, B. Ahmad, G. Rabbani, V. Kumar, S. Yadav and R. H. Khan, Amino Acids, 2010, 39, 899 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627 CAS.
- S. Tsuji and H. Kawaguchi, Langmuir, 2004, 20, 2449 CrossRef CAS.
- S. Tsuji and H. Kawaguchi, Langmuir, 2005, 21, 8439 CrossRef CAS PubMed.
- M. Nakayama, T. Okano, T. Miyazaki, F. Kohori, K. Sakai and M. Yokoyama, J. Controlled Release, 2006, 115, 46 CrossRef CAS PubMed.
- J. Qin, Y. S. Jo, J. E. Ihm, D. K. Kim and M. Muhammed, Langmuir, 2005, 21, 9346 CrossRef CAS PubMed.
- Y. Z. You, K. K. Kalebaila, S. L. Brock and D. Oupicky, Chem. Mater., 2008, 20, 3354 CrossRef CAS.
- K. Ukigaya, Y. Oaki and H. Imai, Chem. Lett., 2012, 41, 507 CrossRef CAS.
- C. D. Jones and L. A. Lyon, J. Am. Chem. Soc., 2003, 125, 460 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |