
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide-directed crystal growth modification in the formation of ZnO†
Anna
Sola-Rabada
a,
Mei-Keat
Liang
a,
Martin J.
Roe
b and
Carole C.
Perry
*a
aBiomolecular and Materials Interface Research Group, Interdisciplinary Biomedical Research Centre, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK. E-mail: Carole.Perry@ntu.ac.uk
bAdvanced Materials Research Group, X-ray Photoelectron Spectroscopy, Faculty of Engineering, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 2nd April 2015
Abstract
Biomolecule-mediated synthesis is fascinating in terms of the level of control and the intricate hierarchical structures of the materials that can be produced. In this study we compare the behavior of a phage display identified peptide, EAHVMHKVAPRP (EM-12) with that of a mutant peptide EAHVCHKVAPRP (EC-12), having additional complexation capability, on the formation of ZnO from solution. The synthesis conditions (Zn(CH3COO)2–NH3 hydrothermal method at 50 °C) were chosen to generate rod-shaped ZnO via layered basic zinc salts (LBZs) as intermediates. Both peptides affected the crystal formation process by moderating the amount of Zn2+ ions in solution (EC12 having a greater effect than EM12) but only EC12 was shown to interact with the solid phase(s) formed during the reaction. Depending on the peptide concentration used, EM-12 was shown to delay and/or suppress ZnO formation. In contrast, additions of EC-12, although leading to the retention of higher levels of Zn2+ ions in solution did not similarly delay the transformation of the intermediate phases to ZnO but were found to dramatically modify the morphology of ZnO crystallites with mushroom shaped crystals being formed. From the results of detailed materials characterization and changes in the morphology observed, the interactions between the peptide(s) and solution and solid state species present during the process of ZnO crystal formation in the presence of EM-12 and EC-12 are proposed.
Introduction
The identification of biomineral-associated biomolecules has shed light on how biominerals are formed in living organisms. This has also led to the use of the bioinspired/biomimetic approach for materials synthesis,1–4 even for non-biologically produced minerals such as ZnO.5 Despite ZnO not being found in living organisms, to date, more than twenty ZnO-binding peptides (ZnO-BPs) have been identified from either cell surface display6,7 or phage display libraries.8–11 However, only a few peptides such as GLHVMHKVAPPR (G-12),12,13 EAHVMHKVAPRP (EM-12)9,14 and derivatives GLHVMHKVAPPR-GGGC (GT-16)12,15 and EAHVMHKVAPRP-GGGSC (ET-17)9,15 have been studied further. The addition of GT-16 during ZnO synthesis has been shown to reduce the aspect ratio of the ZnO crystals formed.8 In order to unravel the cause(s) of aspect ratio reduction by this ZnO-BP, our group conducted some studies using G-1212,13 and/or GT-16.12 The direct evidence obtained suggested that (i) ZnO aspect ratio reduction occurs via an adsorption-growth inhibition mechanism, (ii) G-12 adsorbing on both (0001) and (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes of ZnO is sufficient to reduce ZnO aspect ratio (GGGC-tag is not essential); and, (iii) that the GGGC-tag is responsible for the selective adsorption of GT-16 on the (0001) plane of ZnO crystals. Also, the addition of G-12 in different ZnO experimental systems led to a modification of the growth mechanisms.13 Studies by others have shown that the addition of ET-17 into stable Zn(OH)2 sols catalyzes ZnO formation, directing a flower-type anisotropic crystal growth.9 The catalytic effect of ET-17 is generated either by the presence of the EM-12 segment or GGGSC-tag.9 The catalytic effect of ET-17 was further confirmed as ET-17 attached to a genetically modified-collagen template was able to generate ZnO nanowires.16 A study proposed to understand the interactions taking place between the ZnO surface and EM-12 showed that conformational changes as well as charged amino acids and residues adjacent to the charged aminoacids are critical to the binding to ZnO.17 It was concluded that the electrostatic interaction between ZnO particles and ZnO-BPs was enthalpy-driven, and involved hydrogen bonding, as also suggested by Patwardhan et al.18 The specific binding sites of the EM-12 peptide have been explored,19 with the shorter sequences; HVMHKV and HKVAPR having either high specificity and/or affinity for ZnO nanoparticles. Additionally, it was proposed that ZnO binding did not depend on hydrophobicity or other physical parameters but ZnO recognition of the specific amino acid alignment of these peptides.19 Morphology changes observed for ZnO growth in the presence of ZnO-BPs did not lead to random morphologies. For example, the addition of EM-12 or dipeptides from its sequence (i.e. Met–His or His–Lys) during solution synthesis using Zn(OH)2 as a precursor, suppressed crystal growth of ZnO in the (0001) direction.14
0) planes of ZnO is sufficient to reduce ZnO aspect ratio (GGGC-tag is not essential); and, (iii) that the GGGC-tag is responsible for the selective adsorption of GT-16 on the (0001) plane of ZnO crystals. Also, the addition of G-12 in different ZnO experimental systems led to a modification of the growth mechanisms.13 Studies by others have shown that the addition of ET-17 into stable Zn(OH)2 sols catalyzes ZnO formation, directing a flower-type anisotropic crystal growth.9 The catalytic effect of ET-17 is generated either by the presence of the EM-12 segment or GGGSC-tag.9 The catalytic effect of ET-17 was further confirmed as ET-17 attached to a genetically modified-collagen template was able to generate ZnO nanowires.16 A study proposed to understand the interactions taking place between the ZnO surface and EM-12 showed that conformational changes as well as charged amino acids and residues adjacent to the charged aminoacids are critical to the binding to ZnO.17 It was concluded that the electrostatic interaction between ZnO particles and ZnO-BPs was enthalpy-driven, and involved hydrogen bonding, as also suggested by Patwardhan et al.18 The specific binding sites of the EM-12 peptide have been explored,19 with the shorter sequences; HVMHKV and HKVAPR having either high specificity and/or affinity for ZnO nanoparticles. Additionally, it was proposed that ZnO binding did not depend on hydrophobicity or other physical parameters but ZnO recognition of the specific amino acid alignment of these peptides.19 Morphology changes observed for ZnO growth in the presence of ZnO-BPs did not lead to random morphologies. For example, the addition of EM-12 or dipeptides from its sequence (i.e. Met–His or His–Lys) during solution synthesis using Zn(OH)2 as a precursor, suppressed crystal growth of ZnO in the (0001) direction.14
The coordination of Zn2+ with proteins plays an important role for the structural stability and functionality of proteins in living organisms as evidenced in metalloproteins such as metallothionine and zinc fingers of DNA-binding proteins.20 Zn2+–protein complexation occurs at specific domain(s) via the side chains of amino acid residues namely; sulphur atoms of cysteine and methionine, nitrogen atoms of histidine, and oxygen atoms of aspartic acid and glutamic acid.20 In experimental studies using a Zn(NO3)2–HMTA hydrothermal reaction, Gerstel et al. demonstrated that the presence of aspartic acid, glutamic acid, and their corresponding dipeptides produced layered basic zinc salts (LBZs) as opposed to the expected ZnO.21 They also observed the formation of soluble complexes when higher concentrations of these additives were used and proposed that Zn2+-additive complexation via electrostatic interaction was responsible for LBZs or soluble complex formation.21 More recently, it has been shown that the presence of peptides with different isoelectric points (in the acid, basic and neutral range) using a Zn(NO3)2–NaOH hydrothermal reaction also formed LBZs as a metastable phase during the precipitation process. It was suggested that negatively charged peptides intercalated easily in metastable LBZs.22
Previous studies have shown that the formation of ZnO (without additives) occurs via intermediate(s), LBZs, that only later transform to the oxide phase (ZnO).23–29 This crystallization process usually occurs by a phenomenon known as Ostwald's Law of phases30 in which a metastable phase is formed prior to the formation of the stable phase. In a recent study conducted by our group, the ZnO growth mechanism without the presence of additives has been described and compared for two different hydrothermal methods: (a) zinc acetate (Zn(CH3COO)2) and ammonia (NH3) and (b) zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and 1,3-hexamethylenetetramine (HMTA).31 Both synthesis routes supported the growth of twinned hexagonal rods. However, the progression of solid phase transformation from LBZs to ZnO showed a dissolution/reprecipitation process in the Zn(CH3COO)2–NH3 system, which did not occur for Zn(NO3)2·6H2O–HMTA system.31
In the current study, the effect of the peptides EM-12 (identified by phage display) and EC-12‡ during ZnO formation was investigated using a Zn(CH3COO)2–NH3 hydrothermal method.32 A specific aim of the investigation was to explore the effect of increasing number of potential complexing groups on ZnO formation, for instance, the replacement of methionine (M5) with cysteine (C5) in the peptide sequence.
Results and discussion
Delay/suppression of ZnO Formation by EM-12
The role of EM-12 in affecting ZnO formation was explored by characterization of the precipitates and analysis of components present in solution at a series of reaction times. Previous studies have shown that the formation of ZnO occurs via intermediate(s), layered structures that only later transform to the oxide phase (ZnO).23–29 In this system (ZnAc2–NH3), the intermediate compounds have been identified as Layered Basic Zinc Acetate (LBZA)23–29 and Layered Basic Zinc Acetate Carbonate (LBZAC)27,28,31 with formulae, Zn5(OH)8(CH3COO)2·2H2O and Zn5(OH)x(CH3COO)y(CO3)z·nH2O (x + y + z = 10), respectively. Based on the characterization of the 24 hour precipitates using XRD and SEM (Fig. 1), the addition of 0.1 mM of EM-12 (Zn-to-peptide ratio = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) did not affect ZnO formation significantly. This was inferred from the observation of hexagonal rods (Fig. 1a(i)) and the detection of characteristic diffraction peaks of ZnO (Fig. 1a(i)) at 31.98°, 34.63°, and 36.48° assigned to the (100), (0002), and (101) planes, respectively.9,25,33 However, in the presence of EM-12 at higher concentrations, either a delay or suppression of ZnO formation was observed. In the presence of 0.2 mM EM-12 and above (Fig. 1a(iii)–(vi)), a layered structure with irregular shapes was observed, with the main diffraction peak at 6.86 ± 0.03°, attributed to the (001) plane of LBZA32–34 with an interlayer spacing of 12.94 ± 0.05 Å. This is in agreement with previously reported interlayer spacing values of ∼12–15 Å for LBZA.25,29,35,36 The presence of LBZAC was discounted as 00l reflections would have had a larger interlayer spacing (∼19 Å) produced by the co-intercalation of both acetates and carbonate ions.27,28 For samples with 0.2 mM and 0.3 mM of peptide, the detection of ZnO as the major component was delayed to 48 hours (Fig. 1b(iii)–(iv)). This effect will be referred to as “delayed-formation” hereafter. In the presence of 0.4 mM EM-12, both LBZA and ZnO were detected in the sample at 48 hours (Fig. 1b(v)) and for the highest EM-12 concentration studied; 0.5 mM, LBZA remained the major phase at 48 hours (Fig. 1b(vi)) suggesting the suppression of ZnO formation. The change in the twinning behavior of ZnO crystals (SEM images, Fig. 1b) with increasing EM-12 concentration was also observed, and will be discussed later in this article.
:1) did not affect ZnO formation significantly. This was inferred from the observation of hexagonal rods (Fig. 1a(i)) and the detection of characteristic diffraction peaks of ZnO (Fig. 1a(i)) at 31.98°, 34.63°, and 36.48° assigned to the (100), (0002), and (101) planes, respectively.9,25,33 However, in the presence of EM-12 at higher concentrations, either a delay or suppression of ZnO formation was observed. In the presence of 0.2 mM EM-12 and above (Fig. 1a(iii)–(vi)), a layered structure with irregular shapes was observed, with the main diffraction peak at 6.86 ± 0.03°, attributed to the (001) plane of LBZA32–34 with an interlayer spacing of 12.94 ± 0.05 Å. This is in agreement with previously reported interlayer spacing values of ∼12–15 Å for LBZA.25,29,35,36 The presence of LBZAC was discounted as 00l reflections would have had a larger interlayer spacing (∼19 Å) produced by the co-intercalation of both acetates and carbonate ions.27,28 For samples with 0.2 mM and 0.3 mM of peptide, the detection of ZnO as the major component was delayed to 48 hours (Fig. 1b(iii)–(iv)). This effect will be referred to as “delayed-formation” hereafter. In the presence of 0.4 mM EM-12, both LBZA and ZnO were detected in the sample at 48 hours (Fig. 1b(v)) and for the highest EM-12 concentration studied; 0.5 mM, LBZA remained the major phase at 48 hours (Fig. 1b(vi)) suggesting the suppression of ZnO formation. The change in the twinning behavior of ZnO crystals (SEM images, Fig. 1b) with increasing EM-12 concentration was also observed, and will be discussed later in this article.
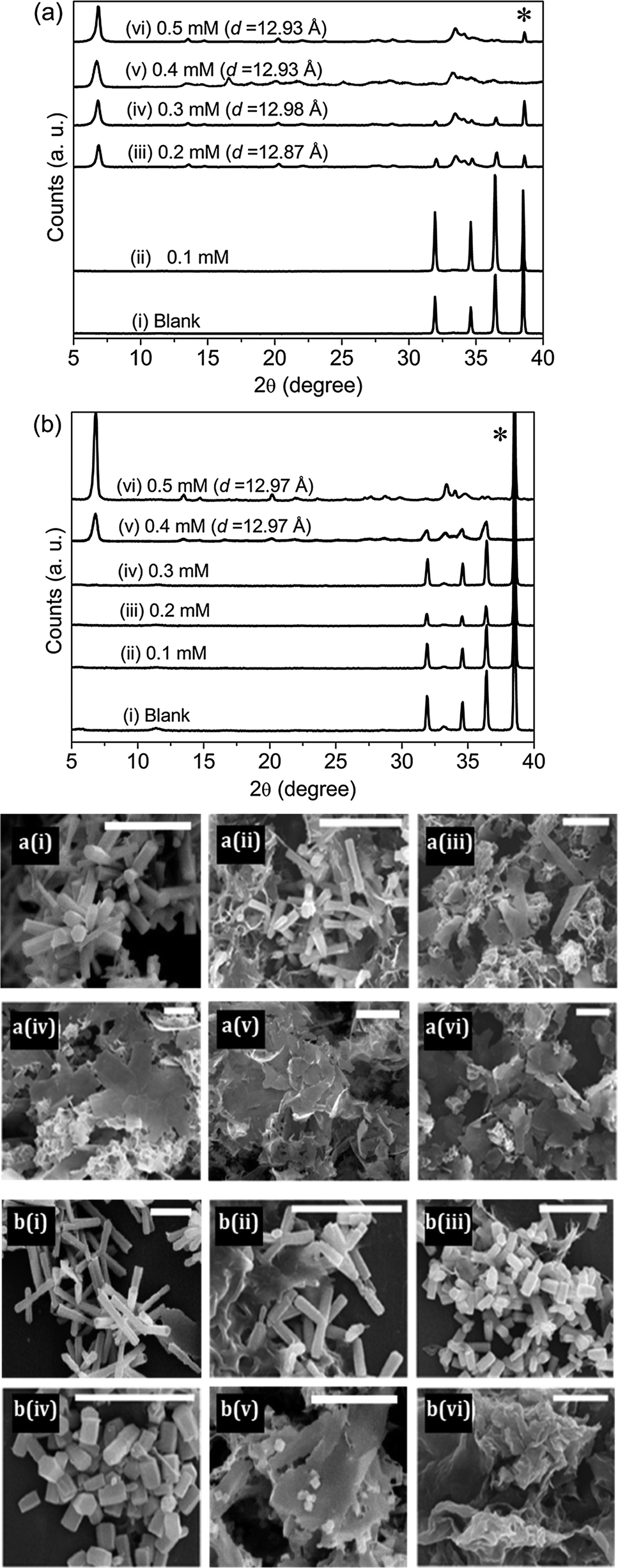 | ||
| Fig. 1 XRD diffractograms of precipitates collected in the presence of different concentrations of EM-12 at (a) 24 hours and (b) 48 hours. The diffraction peak arising from the aluminum sample holder is marked with an asterisk (*). d denotes the interlayer spacing of LBZA formed. The corresponding SEM images of the precipitates are shown. All scale bars shown are 5 μm and (i) to (vi) correspond to Blank, 0.1 mM, 0.2 mM, 0.3 mM, 0.4 mM and 0.5 mM of EM-12, respectively. | ||
The delayed-formation of ZnO was also supported by subjecting the precipitates to thermal decomposition (TGA) which resulted in three stages of weight loss (Fig. 2a); first, the release of physisorbed water at temperatures below 100 °C.25,29 Second, the dehydration of intercalated water (100 °C–168 °C) and/or the dehydroxylation of zinc hydroxide layers in LBZA (168 °C–250 °C). Finally, the decomposition of acetate groups at temperatures from 250 to 400 °C.25,29 Total weight loss values of 31–38% have been reported for LBZA in the literature.26,29,31,37 In this study, the weight losses observed in the presence of 0.2 mM EM-12 and above, supported the presence of LBZA in the samples (Fig. 2b). The reduction in weight loss from 24 to 48 hours was due to the phase transformation from LBZA to ZnO. For the highest concentration of peptide studied; 0.5 mM, LBZA remained the major phase up to 168 hours (∼34%) (Fig. 2b). Hence, the addition of 0.5 mM suppressed ZnO formation to the extent that LBZA remained stable up to 168 hours while lower EM-12 concentrations only slowed down the rate of conversion of LBZA to ZnO (Fig. 2b).
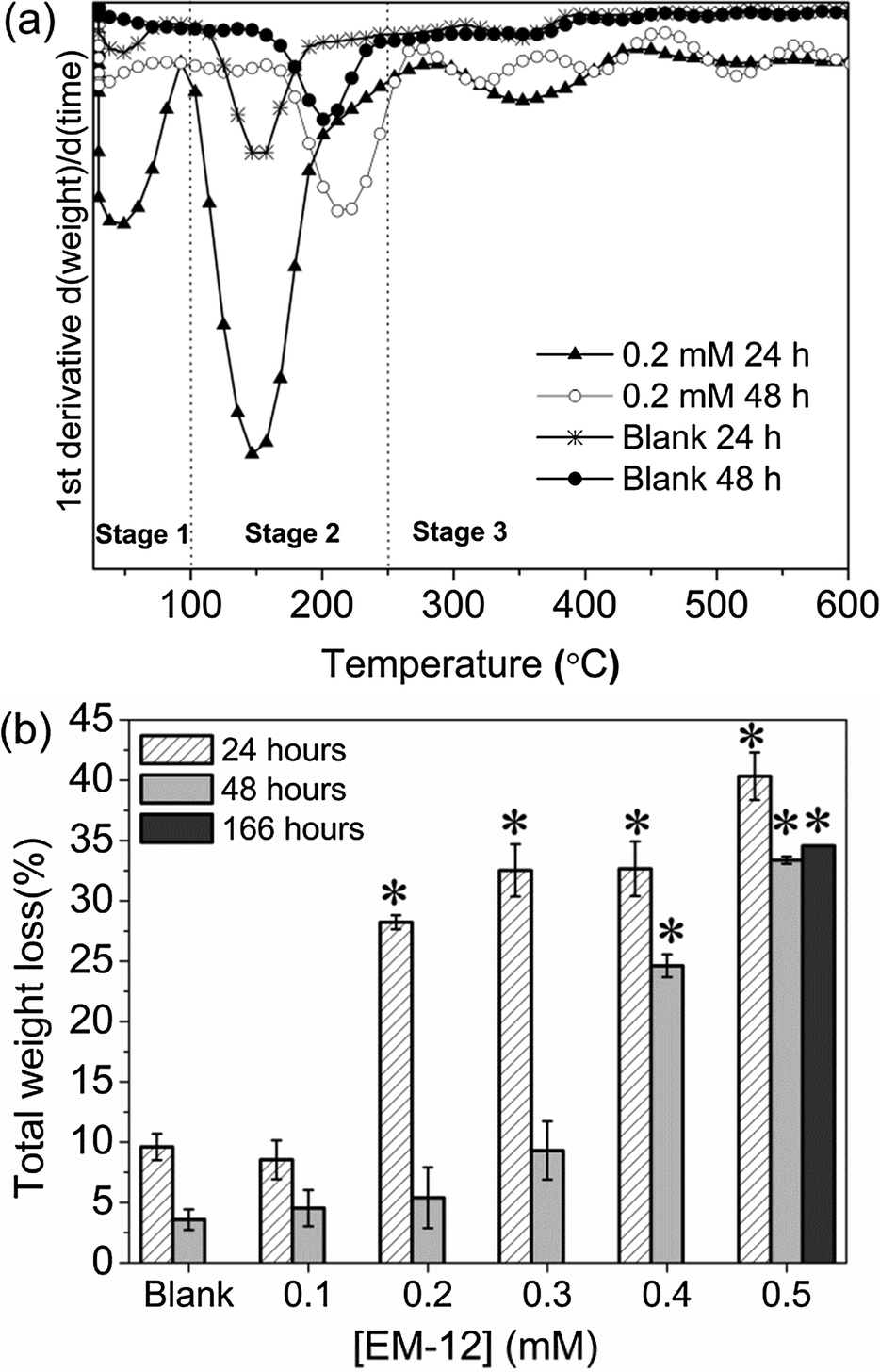 | ||
| Fig. 2 (a) Representative TGA 1st derivative weight loss curves of precipitates collected at 24 and 48 hours with [EM-12] of 0 mM (Blank) and 0.2 mM; and (b) total weight loss (%) of precipitates collected in the presence of different concentrations of EM-12 (0.1–0.5 mM) at 24 hours, 48 hours and 168 hours (0.5 mM EM-12 only). The starred bars correspond to samples that are mainly LBZA as observed by XRD. | ||
Further evidence for the various phases came from XPS analysis of precipitated material. The survey spectra showed the presence of Zn, O and C in all the precipitates studied (Blanks, 0.2 mM and 0.4 mM of EM-12), with peaks in the Zn 2p3/2, O 1s and C 1s regions; having BE values of 1021.7 ± 0.2 eV, 531.6 ± 0.1 eV, 285.0 ± 0.0 eV, respectively; (Fig. 3). From the plot of % area of the C 1s and Zn 2p3/2 peaks at different concentrations of peptide, an increase of the C 1s region with peptide addition can be observed (Fig. 3) supporting the supposition that the presence of EM-12 slowed down the rate of conversion of LBZA to ZnO, as more C is expected with the presence of the intermediate. Even though the data obtained include traces of “adventitious carbon”, usually present in the XPS experiment (ESI 1†), the remaining carbon was thought to be from LBZA, as identified by XRD and TGA and further evidenced by the analysis of core-level spectra for Zn 2p3/2, C 1s and O 1s regions, Fig. 4. The presence of Zn metal was discarded in our system as the Zn LMM peak was shown to be at 988.4 eV attributable to ZnO,38 whereas the kinetic energy peak for metallic Zn would be expected at 992 eV.38 The different local chemical states of each element in the precipitates indicated the presence of such element in a mixed environment, as is the case for LBZA with formula Zn5(OH)8(CH3COO)2·2H2O. The peak position values, the full width half maximum (FWHM) and its identification are given in ESI 2.†
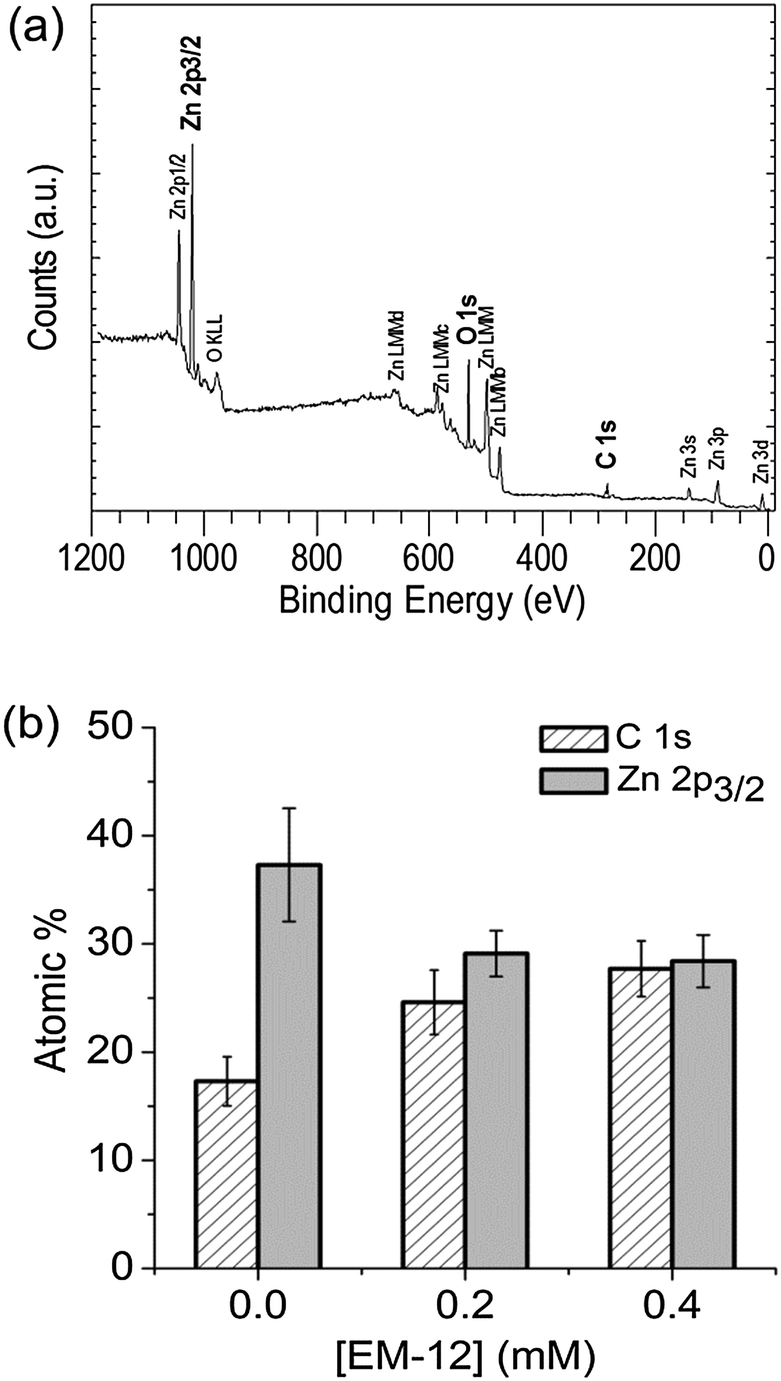 | ||
| Fig. 3 (a) XPS survey spectrum recorded from an as-deposited precipitate collected at 24 hours with [EM-12] of 0.4 mM and (b) atomic % of carbon and zinc for the 48 hour precipitate with 0.0 mM (Blank), 0.2 mM and 0.4 mM of EM-12. | ||
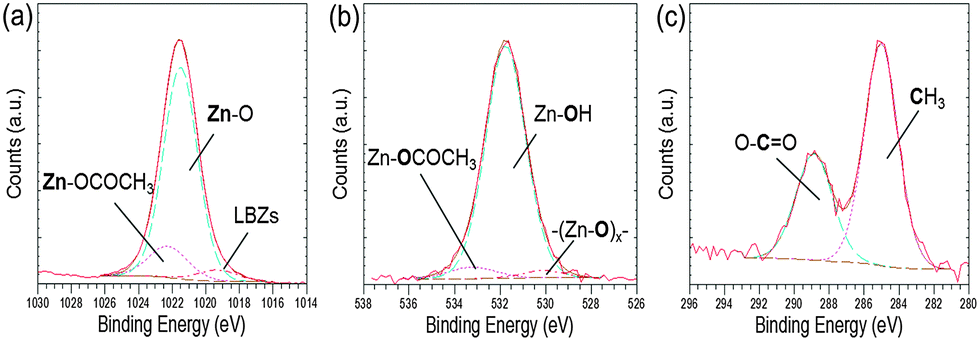 | ||
| Fig. 4 High resolution core level spectra of an as-deposited precipitate collected at 24 hours with [EM-12] of 0.2 mM for (a) Zn 2p3/2, (b) O 1s and (c) C 1s. | ||
ZnO formation in the ZnAc2–NH3 system goes through several intermediates: LBZA and LBZAC.23–29,31 It is believed that the acetate groups in LBZA are gradually replaced by carbonate groups originating from dissolved CO2 present in solution, hence a new intermediate can be observed, LBZAC. At the later stages of ZnO formation (i.e. 48 hours) carbonate species can be dominant over acetate species, albeit in traces amounts relative to the amount of ZnO present.31 Although XRD data did not show the presence of these intermediates, traces of carbonates and acetates were detected by FTIR, Fig. 5. For the sample prepared in the presence of 0.2 mM EM-12, the peaks identified were similar to those for the Blank, and arose from traces of carbonate species present in the sample even though ZnO was the dominant compound detected by XRD. For the highest concentration of peptide studied, 0.5 mM, characteristic acetate peaks were detected (Table 1), confirming the delayed-formation of ZnO. This effect (delay-formation) can be due to either EM-12 stabilized LBZA or retained Zn2+ ions in solution; in both cases LBZA is favored and is discussed in more detail later in this paper.
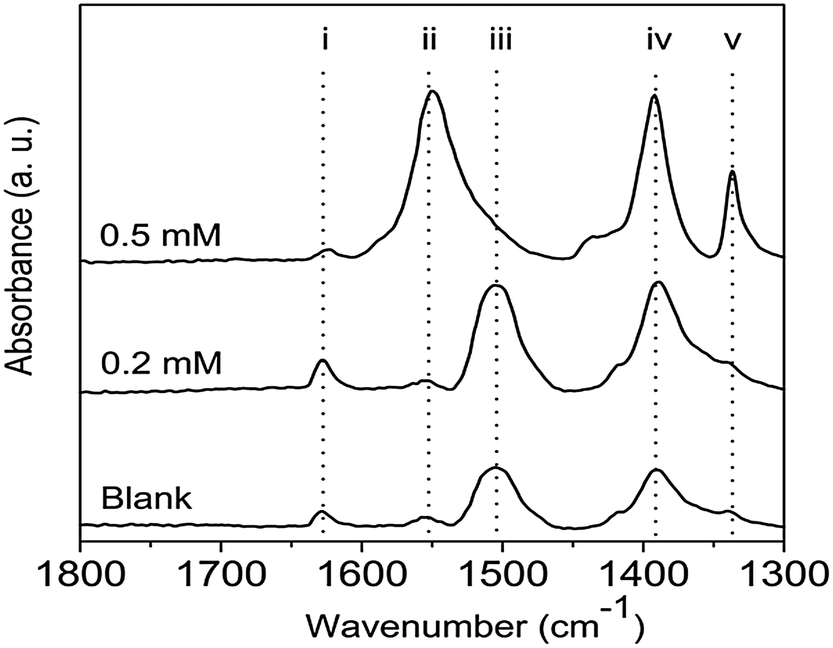 | ||
| Fig. 5 FTIR of 48 hour precipitates collected at different concentrations of EM-12. The identification of the peaks (i) through to (v) is given in Table 1. | ||
| Peak | Wavenumber (cm−1) | Assignment |
|---|---|---|
| a The carbonate group has two ν3 modes assigned to the νas of CO32− band.27 | ||
| i | ∼1620 | O–H vibrational mode of the hydroxide slab in LBZA24,25 and/or O–H bending mode of water.39 |
| ii | ∼1550 | Antisymmetric stretchinga (νas) of CO2−25,27,40 |
| iii | ∼1505 | Antisymmetric stretching (νas) of CO32−27,39 |
| iv | ∼1390 | Symmetric stretching (νs) of CO2−25,27,40 and/or ν3 of CO32−27,39 |
| v | ∼1340 | In-plane bending (δs) or deformation of CH327 |
Zn2+-EM-12 complexation and delayed/ suppression of ZnO formation
The presence of EM-12 extended the period where LBZA was stable as the intermediate phase, delaying or suppressing the formation of ZnO. According to Gerstel et al.,21 amino acids having side chain functionality that interact electrostatically with Zn2+ in solution or on a crystal seed interface, would impose a strong influence on the morphology or the type of precipitate formed.21 Since amino acids capable of complexing with Zn2+, such as glutamic acid, histidine and methionine, are present in EM-12, they may reduce the amount of free Zn2+ in solution available for ZnO formation. Thus, the delay/suppression of ZnO formation was hypothesised to be linked with changes in the amount of Zn2+ in solution when EM-12 was present. This was confirmed from measurement of the Zn2+ concentration in the supernatants using ICP-OES. As previously mentioned, in this particular system (ZnAc2–NH3) the progression of solid phase transformation from LBZs to ZnO goes through a dissolution/reprecipitation process in which precipitation of LBZA occurs immediately after mixing of reagents.31 The dissolution process takes place within 16 hours of reaction when Zn2+ are released back into solution, hence showing an increase of Zn2+ over time, followed by stabilization of Zn2+ in solution after 24 hours of reaction.31 As shown in Fig. 6a, this phenomenon is also observed in the presence of EM-12.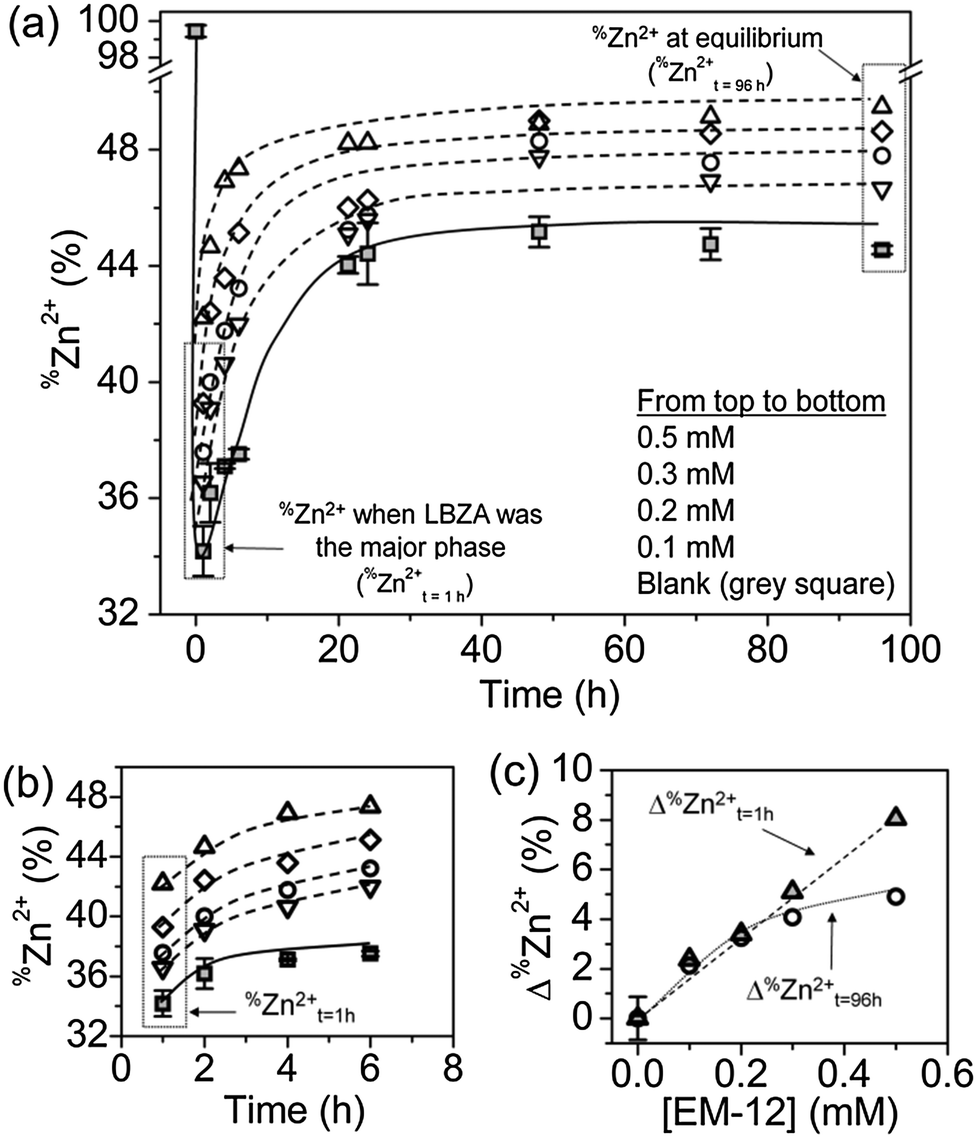 | ||
| Fig. 6 %Zn2+, the percentage of Zn2+ in solution with respect to the initial Zn2+ concentration, for different EM-12 concentrations: %Zn2+ as a function of reaction time (a) up to 96 hours and (b) up to 7 hours, and (c) Δ%Zn2+, the difference between %Zn2+ for the EM-12 added reaction and the blank, at t = 1 hour (%Zn2+t=1h where LBZA was the major phase, grey triangle symbols) and t = 96 hours (%Zn2+t=96h at equilibrium, circle symbols). | ||
The addition of EM-12 increased the % of Zn2+ (%Zn2+) in solution throughout the reaction (Fig. 6a and b). For data analysis, t = 1 hour was used to represent the stage where LBZA was the major phase and t = 96 hours to represent equilibrium. The increase of %Zn2+ with respect to the Blank for these two selected time points (Δ%Zn2+t=1h and Δ%Zn2+t=96h) was calculated (Fig. 6c). Δ%Zn2+t=1h and Δ%Zn2+t=96h increased as a function of EM-12 (initial) concentration. The increase of %Zn2+ in the presence of EM-12 started from the early stages of reaction while a positive correlation was observed between Δ%Zn2+ and EM-12 concentration. These two observations suggest that EM-12 stabilized high concentrations of Zn2+ in solution with the stabilization starting from the beginning of the reaction.
If the higher concentration of Zn2+ present in solution was due to Zn2+ and EM-12 complexation, retention of EM-12 in solution would also be expected. The concentration of EM-12 in solution was measured using UV spectrometry. Absorbance calibration curves of the ZnAc2–NH3 system and for the peptides; EM-12 and EC-12, are available in ESI 3.† Almost complete retention of EM-12 in solution throughout the reaction was observed, (Fig. 7a), implying that either EM-12 only interacts with Zn2+ species in solution or interacts weakly (i.e. van der Waals forces) with zinc species in the solid phase (LBZs/ZnO), being released back into solution when a small force is applied to the system (such as centrifugation). The retention of EM-12 in the solution was further supported using FTIR analysis, since the spectra of 24 hour lyophilized precipitates did not show the characteristic peak of the amide-I band of EM-12 (Fig. 7b), in contrast to the behavior of EC-12 which will be discussed later.
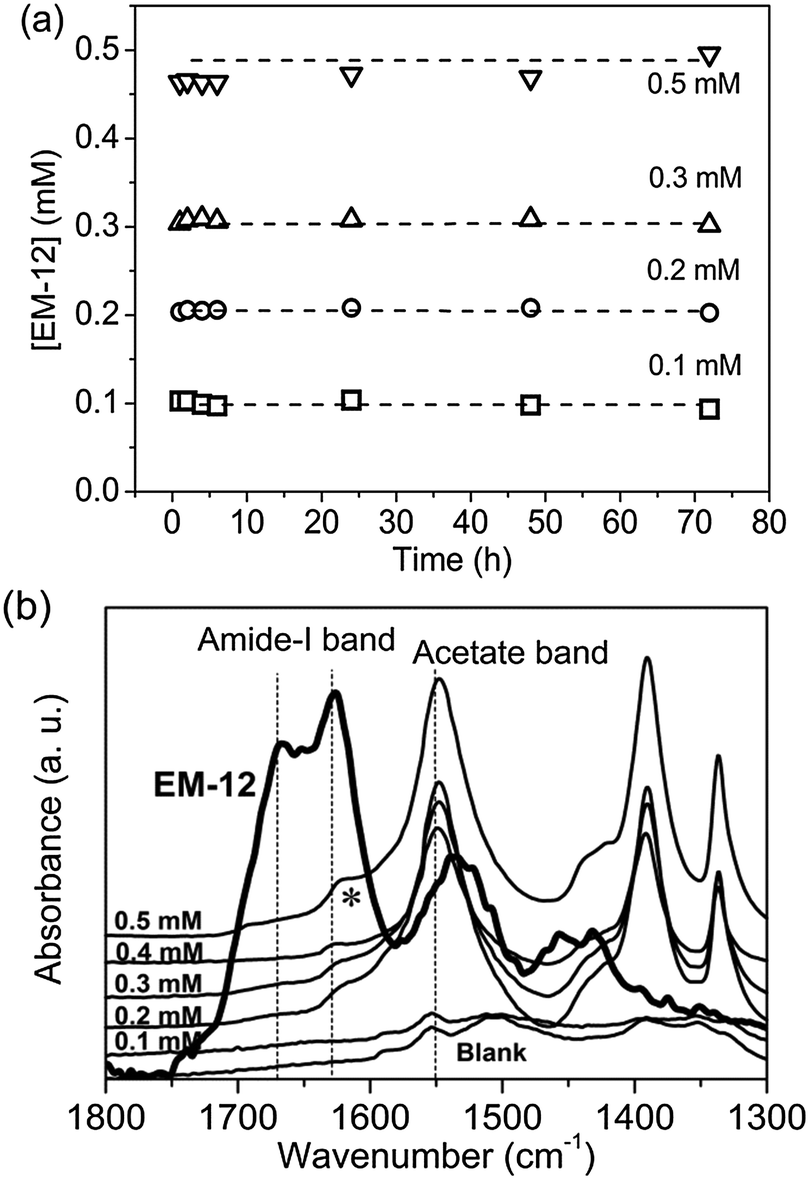 | ||
| Fig. 7 (a) From UV-Vis, EM-12 concentration, [EM-12], in supernatants collected at different reaction time from reactions with initial [EM-12] of 0.1 mM, 0.2 mM, 0.3 mM, 0.4 mM and 0.5 mM and (b) FTIR spectra of lyophilized precipitates collected at 24 hours. The peak arising with an asterisk (*) is attributed to the O–H vibrational mode (corresponding to the hydroxide slab in LBZA).24 | ||
Comparison with EC-12; a peptide with higher complexation capability
Up to this point, the delay formation effect shown by the addition of EM-12 to the ZnO-forming reaction was very different from the ZnO catalytic effect shown by GGGSC-tagged EM-12.9,16 This difference is believed to be due to the additional GGGSC tag present. Based on the side chain pKas calculated using Marvin,41 the charges of EM-12 and EC-12 side chains at pH 7.0 ± 0.1 (ESI 4†) are summarized in Table 2. For simplicity, only charges involving at least 50% ionization of the side chain of a peptide residue are shown. Over this narrow range of pH values, the side chain and termini charges of both peptides are nearly identical and their net charges are the same. Although only E1 and P12 are anionic, other residues such as histidine (H), cysteine (C), and methionine (M) are able to coordinate with Zn2+.20,42,43 Histidine can bind to metal ions through its imidazole Nε2 or Nδ1 and become a bridging ligand when present in its deprotonated state. Although cysteine binds to metal ions most frequently in its deprotonated cysteinate form (i.e. deprotonated thiol group), a neutral cysteine is able to serve as a ligand in ferrous heme iron while non-deprotonable methionine can bind through its sulphur lone pair.20 Thus, it is conceivable that these two peptides are able to form complexes with Zn2+ thereby retaining more Zn2+ in solution than is possible in their absence. Since cysteine is known to be a stronger Zn2+ binder to Zn2+ compared to methionine,20 EC-12 was expected to retain more Zn2+ in solution compared to EM-12.| EM-12 Sequence | E1 | A2 | H3 | V4 | M5 | H6 | K7 | V8 | A9 | P10 | R11 | P12 | Net charge |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Side chain charge | − | + | + | + | |||||||||
| Terminal charge | + | − |
| EC-12 sequence | E1 | A2 | H3 | V4 | C5 | H6 | K7 | V8 | A9 | P10 | R11 | P12 | Net charge |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Side chain charge | − | + | + | + | |||||||||
| Terminal charge | + | − |
As anticipated, the addition of EC-12 to the ZnAc2–NH3 reaction under identical conditions retained more Zn2+ in solution throughout the reaction compared to EM-12 and the increase was proportional to the concentration of the EC-12 added (ESI 5†). In the presence of EM-12 (0.1–0.5 mM), ∼36–42% and ∼46–50% of Zn2+ was retained in solution at t = 1 hour and t = 96 hours, respectively; compared to 34% and 45% for the Blank. In the presence of EC-12 at comparable concentrations, ∼37–47% of Zn2+ was retained in solution at t = 1 hour while 48–57% was retained at t = 96 hours. The higher Zn2+-complexation capability by EC-12 may be caused either by cysteine being a stronger binder compared to methionine and/or by the difference in the conformation of the peptides in solution. The comparison of structures simulated in water at 300 K (GROMACS, force field: GROMOS95, dt: 0.002 ps, total time: 2 ns)45 using Visual Molecular Dynamics (VMD)46 in Fig. 8 showed that EM-12 (red structure) and EC-12 (blue structure) have a highly superimposable C-termini segment (same peptide sequence). However, the structure of EM-12 containing methionine at position 5 is more compact than EC-12 having a cysteine at position 5, even though their side chain functionalities are highly similar. It is also possible that the more extended glutamic acid (marked by a black arrow in Fig. 8) at the N-terminal having an anionic side chain may contribute to the higher complexation of Zn2+ in solution with EC-12 compared to EM-12.
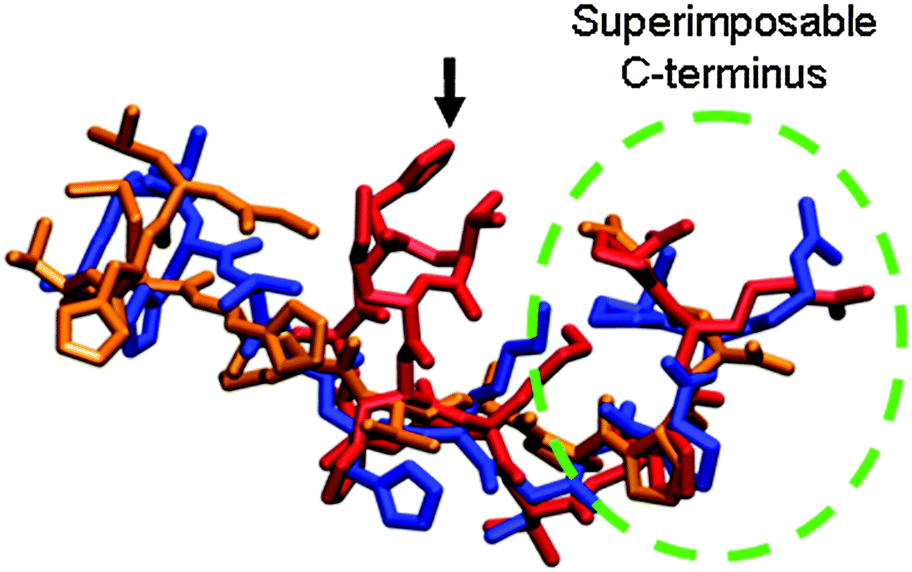 | ||
| Fig. 8 Visualization of the superimposition of molecular structures (simulated in water at 300 K) for EM-12 (EAHVMHKVAPRP) in red, EC-12 (EAHVCHKVAPRP) in blue, and G-12 (GLHVMHKVAPPR) in orange; images generated using VMD.46 | ||
In contrast to complete retention of EM-12 in solution throughout the reaction (Fig. 7a), 40–75% of the added EC-12 was absent from the solution at the beginning of the reaction (Fig. 9a). The complete retention of EC-12 in solution occurred within ∼7 hours of reaction for the 0.1 mM EC-12 precipitate, ∼24 hours for 0.2–0.3 mM EC-12 and ∼30 hours for 0.5 mM EC-12. In agreement with the different starting time points for complete retention, the amide-I band (FTIR) could be detected for the 24 hour precipitates when higher concentration of EC-12 were added (Fig. 9b), but not when EM-12 was added (Fig. 7b).
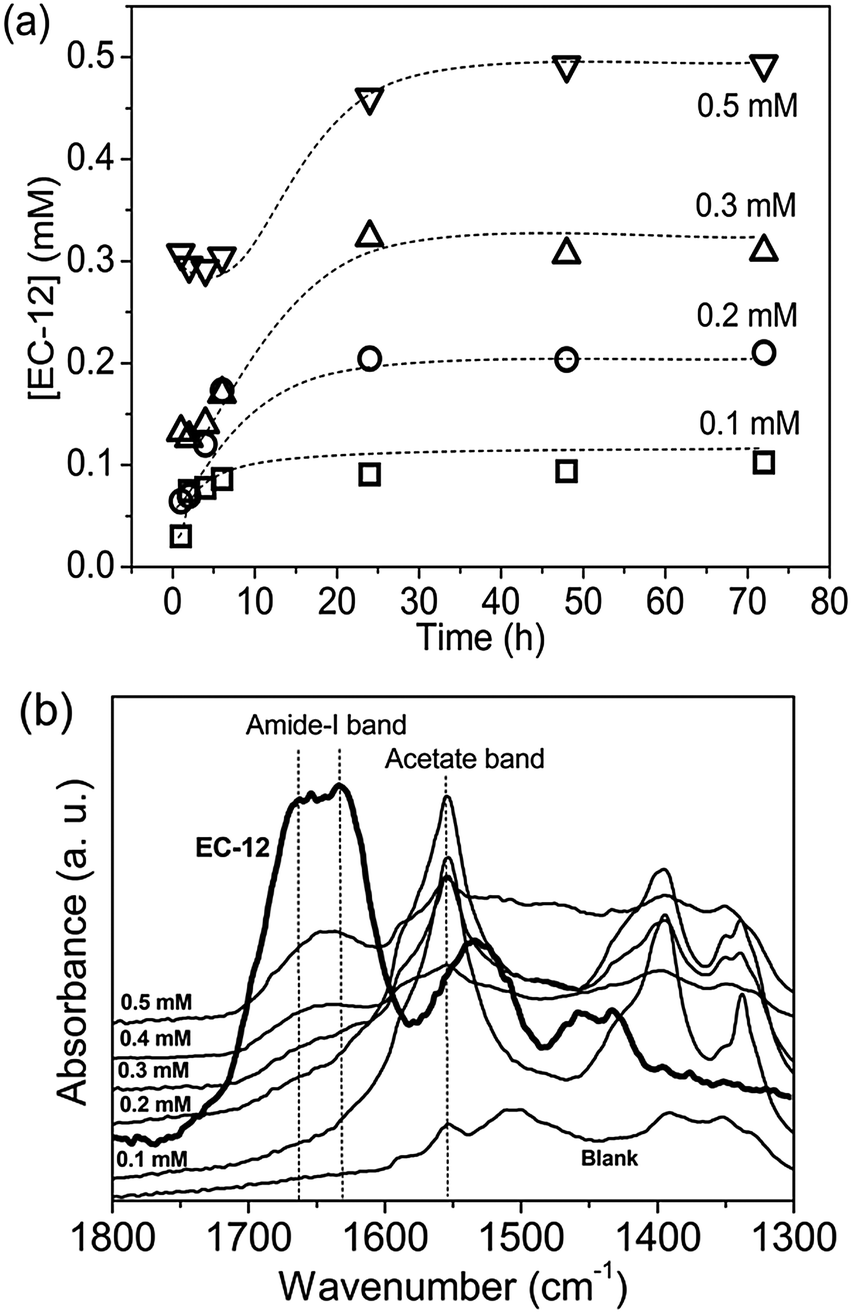 | ||
| Fig. 9 (a) EC-12 concentration, [EC-12], in supernatants collected at different reaction time from reaction with initial [EC-12] of 0.1 mM, 0.2 mM, 0.3 mM and 0.5 mM from UV-vis measured (b) FTIR spectra of precipitates collected at 24 hours. | ||
The detection of EC-12 in the precipitate was also shown by XPS. The sample with 0.4 mM EC-12 was chosen for analysis due to the prior detection of peptide by FTIR. The XPS survey spectrum, Fig. 10a, was similar to that observed when studying EM-12 (Fig. 3a); however, it not only showed the presence of Zn, O and C, but also N. The high resolution core level spectrum of the N 1s region gave a peak at 399.6 eV (Fig. 10b), attributed to the presence of amino groups in the peptide (399.0 ± 0.3 eV).22 The C 1s core level region also showed a peak at 286.5 eV, Fig. 10c, corresponding to the tertiary carbon connected with the nitrogen atom of the amino group,47–49 which was not detected in precipitates containing EM-12 (Fig. 4c).
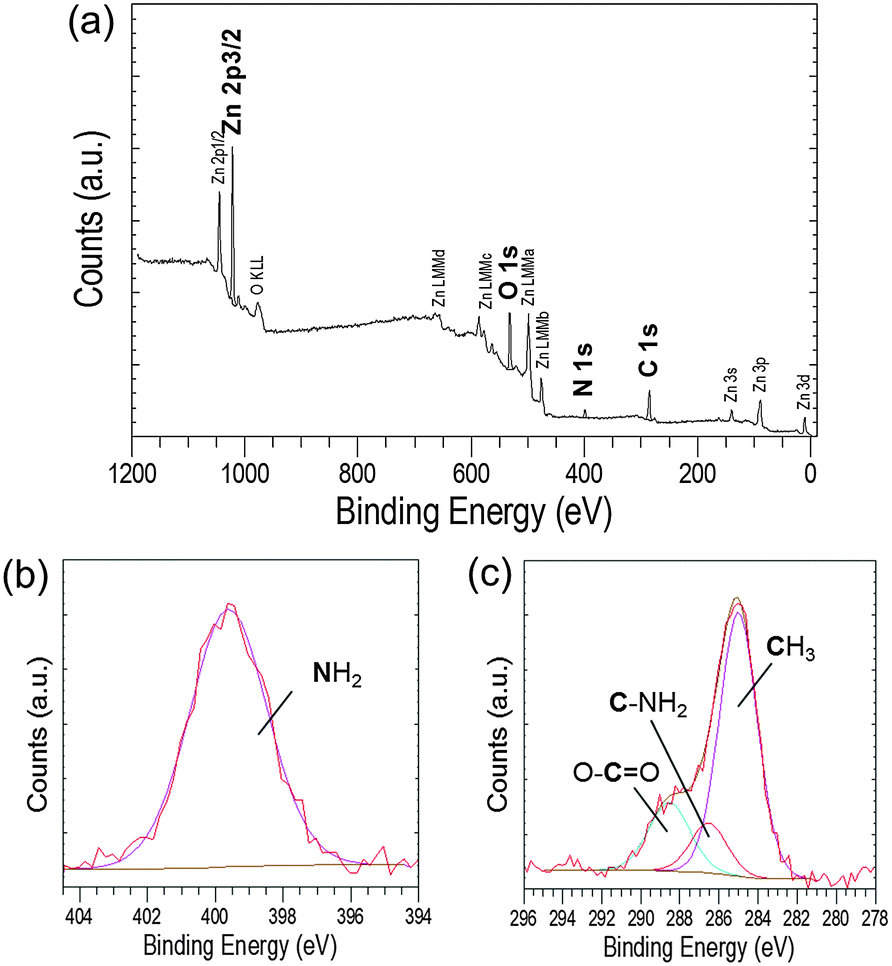 | ||
| Fig. 10 (a) XPS survey spectrum recorded from an as-deposited precipitate collected at 24 hours ([EC-12] at 0.4 mM) and the corresponding high resolution core level spectra of (b) N 1s and (c) C 1s regions. | ||
Further, a broad peak was observed in the S 2p region, which could arise from the cysteine thiol group50 present in EC-12. The broad peak at around 162.2 eV (Fig. 11) was deconvoluted into two peaks, 2p3/2 and 2p1/2 at 161.8 and 163.0 eV, respectively. The S 2p doublet separation could be fitted using a peak area ratio of 2:1 and a separation of 1.18 eV. The overlap of the two peaks was most likely due to the small amount of sulphur in the samples. Previous studies have showed that the binding energy of the S 2p3/2 signal determines the presence of sulphur as a thiol (unbound) or thiolate (bound to the surface), the BE values being at ∼163 eV and ∼162 eV, respectively.47,48,50–52 Our data suggests that the interaction of EC-12 with the solid-phase was via Zn2+–sulphur (from cysteine). Despite the presence of a sulphur atom in both peptides, methionine for EM-12 and cysteine for EC-12, the higher complexation capability of EC-12 seems to be at least in part due to the presence of the thiol group in cysteine.
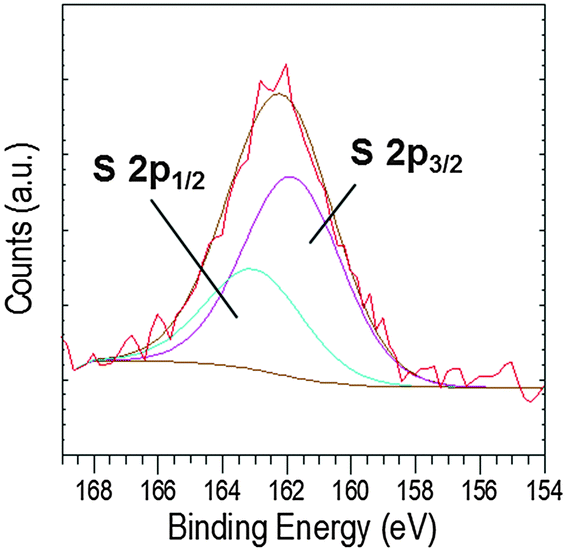 | ||
| Fig. 11 XPS high resolution core level spectra of the S 2p region. | ||
From the characterization of the 24 hour precipitate formed in the presence of EC-12, ZnO was the main crystalline phase detected by XRD (ESI 6†). However, as the weight loss of this precipitate by TGA was 25%, the experimental data suggests that LBZA was also present in the solid phase. Thus, the precise solid phase(s), LBZA and/or ZnO, with which EC-12 interacted, cannot be uniquely identified. Based on XRD and TGA results (ESI 6†), the higher complexation capability of EC-12 did not lead to a stronger delay/suppression effect than EM-12 on ZnO formation when higher concentrations of peptide were added. From comparison of the % thermal decomposition weight loss, generally all 24 hour precipitates obtained in the presence of EC-12 contained a higher fraction of ZnO (weight loss ∼25%) compared to those obtained in the presence of EM-12 (weight loss ∼34%), and no ZnO suppression effect was observed at the highest EC-12 concentration employed (Fig. 12). However, a weaker but still delayed-formation of ZnO due to the presence of EC-12 was remarkably caused when lower concentrations of EC-12 were used (ESI 4†). That EC-12 is able to interact with species in solution and also with species in the solid state is the key to this behavior. According to Umetsu et al., ET-17 could catalyze ZnO formation from a stable Zn(OH)2 sol.9 Although EC-12 did not demonstrate any catalytic capability as shown by ET-17, its weaker delay/suppression effect on ZnO formation (compared to EM-12) suggests the possibility of there being a balance between a catalytic effect and a delay/suppression effect by the modification of EM-12 through the incorporation of a cysteine residue in the peptide. This (effects tuning) possibility cannot be discounted because (i) EM-12, EC-12 and ET-17 have the same/highly similar combination of amino acid side chain functionalities and (ii) any structural differences arising from peptide sequence may be used to tune the strength of these effects. In terms of crystal morphology (Fig. 12), ZnO crystals formed at 24 hours were mostly twinned but asymmetrical. Crystals formed in the presence of 0.1–0.2 mM EC-12 have more defined facets (Fig. 12a and b) while those formed at higher EC-12 concentrations assumed the shape of mushrooms (Fig. 12c and d). When the reaction was extended to 48 hours, the asymmetrical crystals developed into more symmetrical crystals with smaller diameter at their twinned planes, having either a dumbbell shape (Fig. 12e and f) or an hourglass-like shape (Fig. 12g and h). It is noticeable that these ZnO crystals were morphologically different from those obtained from the Blank reaction that had uniform diameters along their c-axes.
 | ||
| Fig. 12 SEM images of precipitates collected at (a) 24 hours (scale bars = 5 μm, inset scale bars = 1 μm) and (b) 48 hours (scale bar = 2 μm). EC-12 concentration for SEM images are (i) 0.1 mM for (ii) 0.2 mM, (iii) 0.4 mM and (iv) 0.5 mM. | ||
Effects of EM-12 and EC-12 on ZnO morphology
In the presence of EM-12, ZnO crystals generally transformed from symmetrical twinned ZnO crystals (Blank) to asymmetric twinned crystals (in the majority) followed by un-twinned crystals as EM-12 concentration was increased from 0.1 mM to 0.4 mM, Fig. 13(a)–(c) and Scheme 1a. The aspect ratio was studied for all ZnO containing precipitates and showed the reduction of L/D with the addition of peptide (Fig. 14). For the Blank (0 mM of EM-12) the points are overlapped due to the symmetrical hexagonal rods and for [EM-12] ≥ 0.3 mM only one value is given due to the crystal growth equilibrated before/at the end of growth of the first rod. Further, EM-12 not only caused the delayed formation of ZnO (conversion from LBZA to ZnO) but also caused the inhibition of crystal growth as seen in Fig. 14 (inset), where the Blank increased its aspect ratio with time and for samples containing EM-12 the aspect ratio was maintained over time. In the presence of EC-12, a similar trend to the Blank was observed, which suggested that addition of EC-12 did not lead to an inhibition of crystal growth as seen for EM-12. | ||
| Fig. 13 SEM images of precipitates collected at 48 hours for: (a) 0.1 mM, (b) 0.2 mM, and (c) 0.4 mM of EM-12. All scale bars are 5 μm and inset scale bars at 0.5 μm. | ||
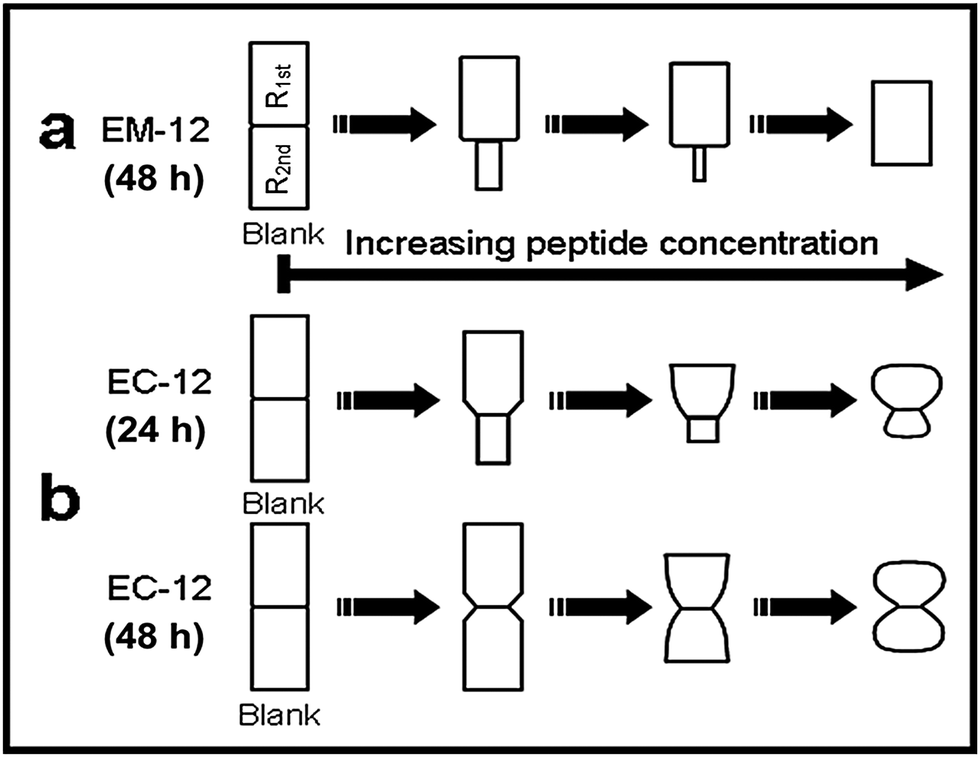 | ||
| Scheme 1 The illustration of ZnO morphology (principal) in the presence of (a) EM-12 at different concentrations (based on 48 hour-precipitates) and, (b) EC-12 at different concentrations collected after 24 hours and 48 hours. Non-scaled images are for morphology comparisons only. | ||
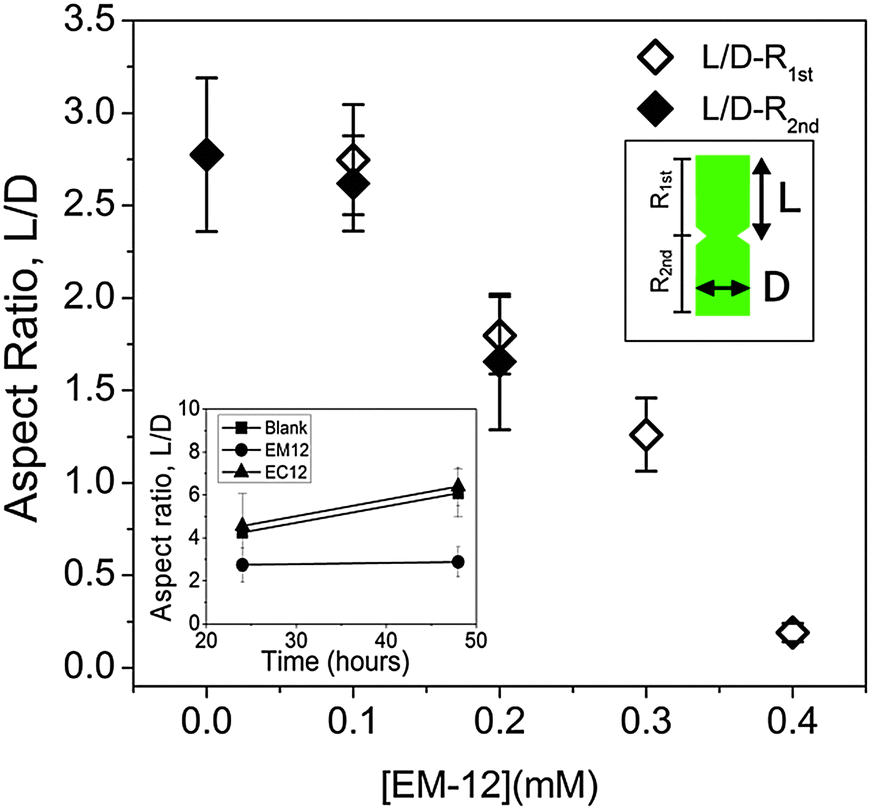 | ||
| Fig. 14 Average aspect ratio (L/D) of ZnO crystal rods (n ≥ 30) as a function of [EM-12] where large and small indicated on the legend represent the larger or smaller crystal on each side of the twin plane. The inset shows the L/D of entire ZnO crystals (n ≥ 30) as a function of time for Blank, EM-12 and EC-12 samples. | ||
ZnO crystals generated in the presence of EC-12, Scheme 1b, were different from those obtained in the presence of EM-12 in three aspects. Firstly, all crystals remained twinned as the EC-12 concentration was increased. Secondly, the diameter of the crystals was not uniform along their c-axis. Thirdly, the surface planes of the crystals became less well-defined with increasing EC-12 concentration with mushroom-like ZnO crystals being obtained in the presence of higher concentrations of EC-12.
It has been proposed that twinned hexagonal rods are formed by the aggregation of two approximately spherical particles.53 However, the observed morphology changes (with time) in the presence of EC-12 suggest that crystallization occurred by the formation of the first hexagonal rod (R1st) followed by the growth of the second rod (R2nd) on the first one.15 Note that with the increase of EC-12 the hexagonal rods became spheres (S1st and S2nd). It is believed that EM-12 added reactions follow the same mechanism. It has been reported that the reduction of the aspect ratio on crystals can be due by specific polypeptides playing a role in the competitive adsorption-nucleation process by interacting with specific crystal planes15 and for ZnO, theoretical and experimental studies,12,15 have suggested that polypeptides can interact with the (0002) plane corresponding to the c-axis with a reduction in the aspect ratio being observed; however, in this current work, the changes in twinning behavior with increasing amount of EM-12 can be explained as follows. Zn2+-EM-12 complexation reduced the amount of Zn2+ available for crystal growth. At lower EM-12 concentrations (0.1–0.2 mM), the growth of the second rod (R2nd) was not complete resulting in the formation of asymmetrically twinned ZnO crystals. In the presence of 0.3 and 0.4 mM EM-12, crystal growth equilibrated before/at the end of growth of the first rod (R1st), thus, untwinned ZnO crystals were obtained, which was supported by the observation of untwinned crystals being shorter but having comparable diameters with those (larger side of asymmetrical twinned crystals) formed at lower EM-12 concentrations (Fig. 14). Addition of R2nd can be considered as a secondary nucleation process15 which in this case was inhibited by the presence of EM-12.
The ZnO morphology transformation (from dumbbell to mushroom-like) by increasing EC-12 concentration is believed to be due to interaction with the solid-phase. This peptide may have modified the ZnO morphology via reversible adsorption on ZnO crystals.54 The adsorption of G-12 (GLHVMHKVAPPR),8 a highly similar ZnO-BP (67% and 58% similar to EM-12 and EC-12, respectively), has been shown to reduce the aspect ratio of ZnO crystals via an adsorption-growth inhibition mechanism by interacting with Zn2+ and/or O2− on ZnO surfaces.12 The differences and similarities between the effects of EM-12, EC-12 and G-12 on ZnO crystal morphology and formation may have arisen (i) at least partly from the presence of glutamic acid (E) in EM-12 and EC-12 but not in G-12; (ii) their superimposable C-termini, (iii) superimposable N-termini between EC-12 and G-12, and (iv) the extended structure of EC-12 in solution as opposed to the more compact structure of EM-12, Fig. 8.
The modification of ZnO morphology via the reversible adsorption of EC-12 on ZnO crystals cannot be confirmed due to the presence of other solid phase(s), LBZs. The interlayer spacing of LBZA formed in the presence of EC-12 did not change significantly compared to the Blank (ESI 6†) which implied that either EC-12 was not intercalated or it was oriented parallel to the zinc hydroxide layer while intercalated. Thus, the possibility of EC-12 intercalating into LBZA formed and then released back into the solution during LBZA conversion to ZnO cannot be discounted.
A recent study has shown that some common amino acids can be incorporated into the ZnO lattice,55 particularly, amino acids containing S or Se, as is the case of cysteine and selenocysteine, respectively. Subsequently, it was found that incorporation of the amino acids led to a change in the optical and paramagnetic properties of the material as well as the crystal morphology. The possibility that EC-12 could have been incorporated into the lattice of ZnO was studied, ESI.† ZnO lattice strain modification was observed when EC-12 was added into the system further suggesting the possibility of this peptide being incorporated into the lattice of ZnO.
Experimental
Materials and methods
000 rpm for 3 minutes to separate precipitates from the supernatants. The precipitates were washed three times with ddH2O. The supernatant obtained was recentrifuged using the same conditions. This supernatant was used for the determination of the concentration of Zn2+ and peptide in the solution. Cleaned precipitates and selected supernatants were lyophilized at −70 °C using a Virtis-110 freeze-dryer.
Conclusions
The effect of the peptides EM-12 and EC-12 during ZnO formation via the ZnAc2–NH3 reaction, showed that the presence of the peptide EM-12 causes a delay and/or suppression of ZnO formation when the peptide concentration was increased, with LBZA being the major compound present at the highest concentration of EM-12 added. Further, the presence of EM-12 not only caused the delayed formation of ZnO (conversion from LBZA to ZnO) but it also caused the inhibition of crystal growth (wurtzite structured hexagonal rods) along the c-axis ((0002) plane). The presence of EC-12; however, caused a more drastic change in the mineral morphology with mushroom-like ZnO crystals being formed, and remarkably, no ZnO suppression was observed for the highest EC-12 concentrations added.The data presented appears to show that the mechanism of ZnO crystallisation occurs by the formation of a first hexagonal rod (R1st) followed by the growth of a second rod (R2nd) on the first one, considering the addition of R2nd as a secondary nucleation process.
Experimental data showed that EM-12 interacts with Zn2+ species in solution, whereas EC-12 interacts either with Zn2+ species in solution and in the solid phase(s). Peptide interaction with the solid-phase cannot be observed when using EM-12 although weak interactions cannot be discounted. The EC-12 interaction mechanism is proposed to include Zn2+–sulphur (from cysteine) interactions. Further, the reduction of the amount of Zn2+ in solution in the presence of EM-12 was not the only cause for the delayed/suppression of ZnO formation in the ZnAc2–NH3 reaction, since a higher Zn2+-EC-12 complexation compared with EM-12 was observed. ZnO lattice strain modification was observed when EC-12 was added into the system and it is suggested that this peptide may be incorporated into the lattice of ZnO.
Acknowledgements
We are very grateful to the U.S. Air Force Office of Scientific Research (AFOSR) for funding (FA9550-13-1-0040 and FA9550-06-1-0154). We thank Dr G. Hickman for the help with Mass spec analysis, Dr Olivier Deschaume for computational studies and Dr Marion Limo for her knowledge in this field.Notes and references
- S. Mann, Biomimetic Materials Chemistry, VCH, New York, 1996 Search PubMed.
- S. V. Patwardhan, S. J. Clarson and C. C. Perry, Chem. Commun., 2005, 1113–1121 RSC.
- A. Xu, Y. Ma and H. Coelfen, J. Mater. Chem., 2007, 17, 415–449 RSC.
- C. Sanchez, H. Arribart and M. M. Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS.
- Z. R. Tian, J. A. Voigt, J. Liu, B. McKenzie and M. J. McDermott, J. Am. Chem. Soc., 2002, 124, 12954–12955 CrossRef CAS PubMed.
- C. K. Thai, H. X. Dai, M. S. R. Sastry, M. Sarikaya, D. T. Schwartz and F. Baneyx, Biotechnol. Bioeng., 2004, 87, 129–137 CrossRef CAS PubMed.
- K. Kjaergaard, J. K. Sorensen, M. A. Schembri and P. Klemm, Appl. Environ. Microbiol., 2000, 66, 10–14 CrossRef CAS PubMed.
- M. M. Tomczak, M. K. Gupta, L. F. Drummy, S. M. Rozenzhak and R. R. Naik, Acta Biomater., 2009, 5, 876–882 CrossRef CAS PubMed.
- M. Umetsu, M. Mizuta, K. Tsumoto, S. Ohara, S. Takami, H. Watanabe, I. Kumagai and T. Adschiri, Adv. Mater., 2005, 17, 2571–2575 CrossRef CAS PubMed.
- D. Rothenstein, B. Claasen, B. Omiecienski, P. Lammel and J. Bill, J. Am. Chem. Soc., 2012, 134, 12547–12556 CrossRef CAS PubMed.
- P. Golec, J. Karczewska-Golec, M. Łoś and G. Wgrzyn, J. Nanopart. Res., 2012, 14, 1–6 CrossRef PubMed.
- M. Liang, O. Deschaume, S. V. Patwardhan and C. C. Perry, J. Mater. Chem., 2011, 21, 80–89 RSC.
- M. J. Limo, R. Ramasamy and C. C. Perry, Chem. Mater., 2015, 27, 1950–1960 CrossRef CAS.
- T. Togashi, N. Yokoo, M. Umetsu, S. Ohara, T. Naka, S. Takami, H. Abe, I. Kumagai and T. Adschiri, J. Biosci. Bioeng., 2011, 111, 140–145 CrossRef CAS PubMed.
- M. Muthukumar, J. Chem. Phys., 2009, 130, 161101 CrossRef CAS PubMed.
- H. Bai, F. Xu, L. Anjia and H. Matsui, Soft Matter, 2009, 5, 966–969 RSC.
- N. Yokoo, T. Togashi, M. Umetsu, K. Tsumoto, T. Hattori, T. Nakanishi, S. Ohara, S. Takami, T. Naka, H. Abe, I. Kumagai and T. Adschiri, J. Phys. Chem. B, 2010, 114, 480–486 CrossRef CAS PubMed.
- S. V. Patwardhan, G. Patwardhan and C. C. Perry, J. Mater. Chem., 2007, 17, 2875–2884 RSC.
- M. Okochi, T. Sugita, S. Furusawa, M. Umetsu, T. Adschiri and H. Honda, Biotechnol. Bioeng., 2010, 106, 845–851 CrossRef CAS PubMed.
- I. Bertini, Biological inorganic chemistry: structure and reactivity, University Science Books, Sausalito, California, 2007 Search PubMed.
- P. Gerstel, P. Lipowsky, O. Durupthy, R. C. Hoffmann, P. Bellina, J. Bill and F. Aldinger, J. Ceram. Soc. Jpn., 2006, 114, 911–917 CrossRef CAS.
- J. Baier, T. Naumburg, N. Blumenstein, L. Jeurgens, U. Welzel and J. Pleiss, Biointerface Res. Appl. Chem., 2012, 2, 380–391 CAS.
- H. H. Wang, C. S. Xie, D. W. Zeng and Z. H. Yang, J. Colloid Interface Sci., 2006, 297, 570–577 CrossRef CAS PubMed.
- Y. Wang, Y. Li, Z. Zhou, X. Zu and Y. Deng, J. Nanopart. Res., 2011, 13, 5193–5202 CrossRef CAS.
- L. Poul, N. Jouini and F. Fievet, Chem. Mater., 2000, 12, 3123–3132 CrossRef CAS.
- M. S. Tokumoto, S. H. Pulcinelli, C. V. Santilli and V. Briois, J. Phys. Chem. B, 2003, 107, 568–574 CrossRef CAS.
- E. Jang, J. Won, Y. Kim, Z. Cheng and J. Choy, J. Solid State Chem., 2010, 183, 1835–1840 CrossRef CAS PubMed.
- G. Patrinoiu, J. M. Calderon-Moreno, D. C. Culita, R. Birjega, R. Ene and O. Carp, Solid State Sci., 2013, 23, 58–64 CrossRef CAS PubMed.
- E. Hosono, S. Fujihara, T. Kimura and H. Imai, J. Colloid Interface Sci., 2004, 272, 391–398 CrossRef CAS PubMed.
- J. J. Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef.
- M. Liang, M. J. Limo, A. Sola-Rabada, M. J. Roe and C. C. Perry, Chem. Mater., 2014, 26, 4119–4129 CrossRef CAS.
- Y. Masuda, N. Kinoshita and K. Koumoto, Electrochim. Acta, 2007, 53, 171–174 CrossRef CAS PubMed.
- Q. Cui, K. Yu, N. Zhang and Z. Zhu, Appl. Surf. Sci., 2008, 254, 3517–3521 CrossRef CAS PubMed.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829, DOI:10.1088/0953-8984/16/25/R01.
- H. Morioka, H. Tagaya, J. Kadokawa and K. Chiba, J. Mater. Sci. Lett., 1999, 18, 995–998 CrossRef CAS.
- A. Kasai and S. Fujihara, Inorg. Chem., 2006, 45, 415–418 CrossRef CAS PubMed.
- Z. Xia, Y. Wang, Y. Fang, Y. Wan, W. Xia and J. Sha, J. Phys. Chem. C, 2011, 115, 14576–14582 CAS.
- H. Amekura, Y. Sakuma, M. Yoshitake, Y. Takeda, N. Kishimoto and Ch. Buchal, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 257, 64–67 CrossRef CAS PubMed.
- S. Inoue and S. Fujihara, Inorg. Chem., 2011, 50, 3605–3612 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons Inc, New York, 1986 Search PubMed.
- T. Schmidt, G. Muller, L. Spanhel, K. Kerkel and A. Forchel, Chem. Mater., 1998, 10, 65–71 CrossRef CAS.
- M. Rombach, M. Gelinsky and H. Vahrenkamp, Inorg. Chim. Acta, 2002, 334, 25–33 CrossRef CAS.
- R. Vogler, M. Gelinsky, L. F. Guo and H. Vahrenkamp, Inorg. Chim. Acta, 2002, 339, 1–8 CrossRef CAS.
- ChemAxon, Marvin 5.0.0 was used for drawing and characterizing chemical structures and substructures, http://www.chemaxon.com/.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- D. T. Clark, J. Peeling and L. Colling, Biochim. Biophys. Acta, 1976, 453, 533–545 CrossRef CAS.
- O. Cavalleri, G. Gonella, S. Terreni, M. Vignolo, L. Floreano, A. Morgante, M. Canepa and R. Rolandi, Phys. Chem. Chem. Phys., 2004, 6, 4042–4046 RSC.
- Y. K. Gao, F. Traeger, O. Shekhah, H. Idriss and C. Woell, J. Colloid Interface Sci., 2009, 338, 16–21 CrossRef CAS PubMed.
- G. Dodero, L. De Michieli, O. Cavalleri, R. Rolandi, L. Oliveri, A. Dacca and R. Parodi, Colloids Surf., A, 2000, 175, 121–128 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- M. Volmer, M. Stratmann and H. Viefhaus, Surf. Interface Anal., 1990, 16, 278–282 CrossRef CAS PubMed.
- M. A. Verges, A. Mifsud and C. J. Serna, Surf. Interface Anal., 1990, 86, 959–963 CAS.
- J. Zhang, H. Liu, Z. Wang and N. Ming, J. Cryst. Growth, 2008, 310, 2848–2853 CrossRef CAS PubMed.
- A. Brif, G. Ankonina, C. Drathen and B. Pokroy, Adv. Mater., 2014, 26, 477–481 CrossRef CAS PubMed.
- W. Chan and P. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York, 2000 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS analysis for the study of the ubiquitous carbonaceous contamination, and XPS data including the relative ratio of Zn/O and BE (eV) for O 1s, C 1s and Zn 2p3/2 for samples analyzed; calibration curves used for peptide quantification in solution; calculated pKa of peptide side chains using Marvin calculator; Zn2+ in solution for EC-12-added reactions; XRD and TGA results for 24 hour-precipitates produced in the presence of EC-12; dimension analysis of ZnO crystals obtained from EM-12-added reactions (48 hours); lattice strain modification for ZnO planes in the presence of EC-12; and mass spectra for peptides. See DOI: 10.1039/c5tb00253b |
| ‡ EM-12, 12-mer peptide containing methionine; EC-12, 12-mer peptide containing cysteine. |
| This journal is © The Royal Society of Chemistry 2015 |