
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward organic electronics with properties inspired by biological tissue
Timothy F.
O'Connor
,
Kirtana M.
Rajan
,
Adam D.
Printz
and
Darren J.
Lipomi
*
Department of NanoEngineering, University of California, San Diego, 9500 Gilman Drive, Mail Code 0448, La Jolla, CA 92093-0448, USA. E-mail: dlipomi@ucsd.edu
First published on 26th February 2015
Abstract
The carbon framework common to both organic semiconductors and biological structures suggests that these two classes of materials should be easily integrated. Substantial work, however, will be required to endow synthetic electroactive materials with properties resembling those of biological tissue, which exhibits extreme elasticity, biodegradability, and the capacity for self-repair. This Highlight reviews successful integration of organic semiconductor devices with biological systems, for example, in wearable and implantable health monitors and prosthetic devices. It then points to recent work in the areas of molecularly stretchable electronics, whole devices that can degrade under physiological conditions, and conjugated polymers capable of self-healing, which together suggest the possibility of a future in which organic electronics and biological tissue can interact seamlessly.
Tissue-like properties from organic semiconductors
The vision of organic electronics includes inexpensive,1 flexible and stretchable2 devices that can be fabricated by roll-to-roll printing3 and integrated with moving and non-planar parts of machines,4 buildings and windows,5,6 and the human body.7,8 Biomedical devices are regarded as an especially promising application of organic semiconductors—i.e., “organic bioelectronics”9,10—because of the perceived similarity of “soft” conjugated polymers and biomaterials. Despite the carbon framework common to both organic semiconductors and biological macromolecules, π-conjugated molecules have few properties in common with biological tissue.2,11 Conjugated polymers can be stiff (tensile modulus ca. 1 GPa)12–14 and small-molecule semiconductors have a brittleness characteristic of all van der Waals solids (crack-onset strain ca. 1%).12 In contrast, biological tissue is elastic (tensile modulus ∼10–100 kPa) and tough.15 Moreover, two additional properties of biological tissue—biodegradability16 and the capacity for self-repair17—would be desirable for biologically integrated electronics, but are not generally possessed by organic semiconductors.The best performance so far achieved for wearable and implantable electronic devices has been—despite the inherent flexibility of organic thin films—based on composite structures of high-performance inorganic devices embedded in (or upon) elastomeric supports.18–21 This approach, exemplified by the work of Rogers and others,18–23 uses (as one strategy) serpentine interconnects that absorb tensile strain by local bending and twisting.18 The active components can be located at areas of increased thickness20 so that strain is channelled into the interconnects. These devices can also form the basis of “transient electronics”,24 in which the active components, the interconnects, and the matrices can dissolve in programmable timescales under physiological conditions into harmless byproducts.25
Despite the success of systems comprising inorganic components embedded in organic matrices, further capabilities would be accessed by a complementary approach that used solely organic components.26–28 That is, organics have characteristics not easily replicated in inorganic semiconductors.2 These characteristics include solution-based printing,29 facile tunability of optical, electronic, and mechanical properties by synthesis,30 multimodal signal transduction, chemical selectivity,31 programmable degradation,32 and low embodied energy.2 The challenges of making organic electronics softer and more responsive to chemical and mechanical stimuli are not addressed in a research strategy whose principal goal is to replicate the high semiconducting performance of inorganic materials, but are addressed in a strategy in which the advantages unique to organics are exploited to the maximum extent possible.
This paper highlights recent progress in areas of research within organic electronics that are designed to make the materials more like living tissue (interpreted broadly). The work covered may have been motivated by purposes distinct from the present goal of increasing the tissue-like behaviour of organic semiconductors (for example, making organic solar cells more resistant to mechanical failure is tantamount to making them tougher and more elastic11) but the knowledge gained is independent of the motivation. We first highlight a few applications of stretchable organic electronics in electronic skin or biomedical applications, and then cover work whose goal is to increase the (1) elasticity, (2) biodegradability, and (3) self-repair of organic electronic materials and devices. Because of the shortness of this Highlight, and because of our expertise, we restrict our focus to the above properties 1–3 of thin-film organic semiconductors and their devices. We therefore must leave discussion of composite materials such as conductive hydrogels,33 important issues of polymer–cell interactions,34 and electrodes and electrochemical transistors based on poly(3,4-ethylenedioxythiophene) (PEDOT),10 to the outstanding work of experts in those fields.34–37
Stretchable and skin-like organic devices
The first requirement for devices to integrate with soft, biological structures is “stretchability”. This term implies biaxial elasticity, plasticity, or both. Systems exhibiting only flexibility—i.e., a thin film of nearly any material—can conform to cylindrical or conical shapes, while systems exhibiting stretchability are capable of reversible tensile deformations and bonding to complex three-dimensional surfaces.41,42 Most approaches to stretchable systems fall into one of two categories: random or deterministic composites.18 The first type of approach takes randomly dispersed structures and couples them to an elastic support or matrix.38,43,44 Strain applied to the bulk structure is thus accommodated by the elastic matrix, and the conducting or semiconducting properties remain intact so long as the embedded structures remain percolated. This approach has been used successfully for transparent conductive electrodes based on silver nanowires,45 carbon nanotubes (Fig. 1a and b),38,44 and poly(3-hexylthiophene) (P3HT) nanowires43 and scaffolds for tissue engineering.46 The second approach, as mentioned previously, uses deterministic patterns of rigid active components connected by wavy or otherwise stretchable interconnects.18 This “island-bridge” approach is amenable to both organic47 and inorganic21 circuits.48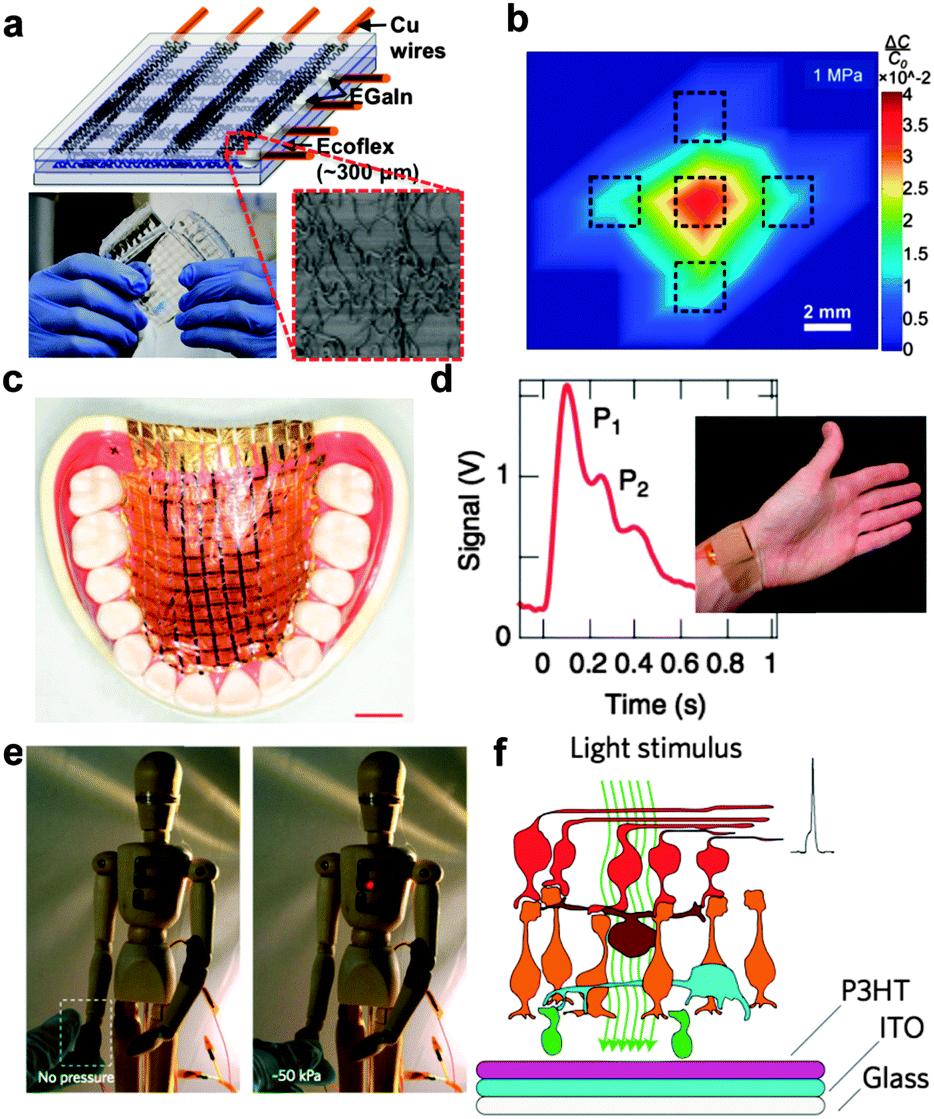 | ||
| Fig. 1 Overview of biological and biomedical applications of organic electronics. (a) Random composites, such as bundles of buckled carbon nanotubes, can be used to form stretchable, transparent, skin-like sensors of pressure and strain. By virtue of the compressibility, these devices exhibit low cross-talk between adjacent pixels (b). Reproduced with permission from ref. 38 Copyright 2011 Nature Publishing Group. (c) “Imperceptible” organic electronic devices built on ultrathin (<2 μm) polyester foils can easily conform to biological structures exhibiting complex topography. Reproduced with permission from ref. 26 Copyright 2013 Nature Publishing Group. (d) An organic thin-film transistor bearing a compressible dielectric layer can transduce the mechanical deformation of the radial artery into the pulse waveform. Reproduced with permission from ref. 39 Copyright 2013 Nature Publishing Group. (e) Self-healing elastic materials comprising metallic nanoparticles can exhibit a piezoresistive effect. Reproduced with permission from ref. 40 Copyright 2012 Nature Publishing Group. (f) Organic photodiodes placed in the vertebrate eye could form the basis of an artificial retina. Reproduced with permission from ref. 7 Copyright 2011 Nature Publishing Group. | ||
Conformability to complex biological surfaces can be achieved to some extent by using extremely thin substrates, even if they are not stretchable. For example, by building organic electronic devices on polyester foils less than 2 μm thick, Someya, Bauer, and coworkers have produced wearable biomedical devices (Fig. 1c)26 and solar cells with the highest ratio of power to mass reported (organic or of any type).49 The Bao laboratory has used thin substrates and microstructured compressible dielectric layers to produce organic transistors capable of measuring the pressure waveform from the radial artery in the wrist (Fig. 1d).39 A hybrid inorganic–organic material consisting of nickel nanoparticles embedded in a stretchable, self-healing polymer can sense pressure (Fig. 1e) as a piezoresistor.40 Organic photovoltaic cells are also thin and lightweight enough to be implanted into the vertebrate eye (in this case, a rat, Fig. 1f) toward the long-term goal of restoring vision in the blind.7
Toward intrinsically—“molecularly”—stretchable electronics
An emerging, third approach for generating stretchable electronic systems is perhaps the one which is least explored. That is, designing and synthesizing organic conductors and semiconductors that—by virtue of their molecular structure or solid-state microstructure—are also highly elastic and ductile.2 This approach, which we have nicknamed “molecularly stretchable electronics”, does not in principle rely on elastic matrices or substrates to supply the restoring force.2 This strategy has been used by our laboratory13,42,50–53 as well as those of Pei44,45,54–56 and Bao,57–59 for example, to generate stretchable piezoresistors capable of detecting human motion (Fig. 2a–c),51 stretchable light-emitting devices (Fig. 2d),45 and biaxially deformable solar cells that can be bonded conformally to hemispherical surfaces.42 Our group has attempted to characterize the mechanical properties of π-conjugated polymers to understand the molecular and microstructural parameters that allow mechanical compliance and semiconducting behavior to coexist.50 In particular, we have employed a technique60 based on the strain-induced instability (surface wrinkling)61 to measure the tensile moduli of thin films that are difficult to assay using conventional means.12 We have supported our measurements using a semi-empirical theory proposed by Seitz62 that predicts the tensile moduli through analysis of the thermal properties and the chemical structure.13 Among our conclusions was that the length of the alkyl pendant group (installed for increased solubility) is a key determinant of the tensile modulus.13 Lengthening of the aliphatic side chain decreases the number of load-bearing, main-chain carbon atoms per cross sectional area and reduces the ability of the main chains to associate. Both effects reduce the tensile modulus and the mobility of charge carriers, and thus polythiophene bearing octyl side chains is not only ten times more compliant—but also a poorer semiconductor—than polythiophene bearing hexyl side chains.2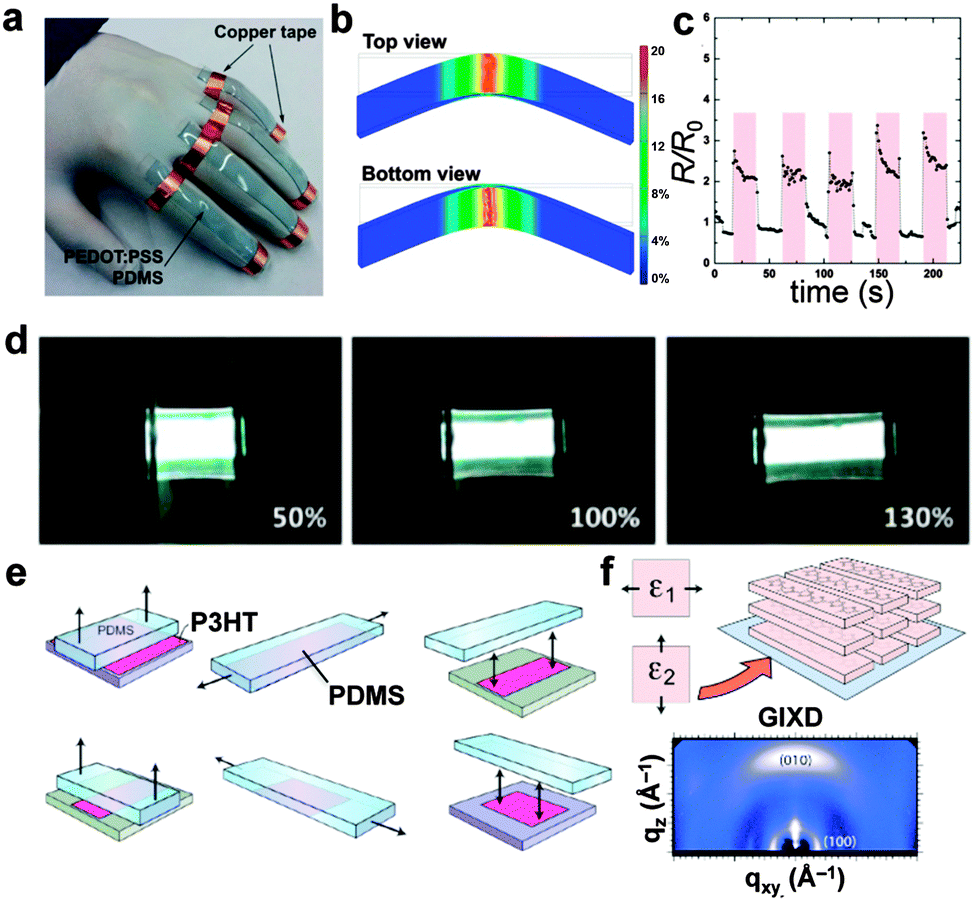 | ||
| Fig. 2 Molecularly stretchable organic electronics and mechanical properties of organic semiconductors. (a–c) Highly plasticized films of PEDOT:PSS can behave as piezoresistive signal transducers for detecting human motion. Reproduced with permission from ref. 51 Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA. (d) Stretchable organic light-emitting devices maintain their luminescence at strains ≥130%. Reproduced with permission from ref. 54 Copyright 2014 American Chemical Society. (e) Biaxial stretching of P3HT produces a texture (f) in which the axis of π-stacking is oriented normal to the substrate (“face-on”). Reproduced with permission from ref. 63 Copyright 2013 American Chemical Society. | ||
The apparent competition between mechanical and charge-transport properties led us to synthesize block copolymers and “segmented” polymers whose blocks and segments permitted co-engineering of mechanical compliance and charge-carrier mobility.50 We concluded that a highly elastic semiconducting polymer should exhibit three structural features: long alkyl side chains, a low percentage of fused rings in the main chain, and structural randomness (e.g., branched, racemic side chains and a small degree of regioirregularity) to reduce the crystallinity.2 It is not, however, sufficient that a semiconducting polymer simply accommodate strain without fracture; strain produces microstructural rearrangements in semiconducting polymers that inevitably affect the charge-transport properties of the materials in ways that must be mitigated (or at least predicted).58 While strain-induced alignment of chains has been known since the early days of conjugated polymers,64,65 mechanical deformation can also produce changes in the degree of crystallinity and texture.11 For example, O'Connor and coworkers have shown an interesting reorientation in texture from edge-on to face-on in biaxially oriented films of poly(3-hexylthiophene) (P3HT, Fig. 2e and f).63,66 Such strain-evolved microstructure must be understood and predicted in any circumstance in which the active material undergoes deformation.
Biodegradability
Biodegradability refers to the potential of a material to be broken down in the outdoor environment or in the body without the release of toxic byproducts.67–69 While environmentally benign disposal of electronics is increasingly mandated by regulation, physiologically benign disintegration of implanted devices imposes even stricter constraints on the design and selection of materials. (Though a molecule whose metabolic fate is benign is unlikely to be deleterious to the environment.) Rogers and coworkers have reported electronic devices based on silicon nanomembranes that are stretchable and also soluble under physiological conditions.24 Producing all-organic circuits from components that are all biodegradable is an exciting prospect,32 because the semiconductors, electrodes, and substrates could be tailored by synthesis to degrade. In an early example of a biodegradable organic electronic device, Bettinger et al. fabricated a thin-film transistor on a resorbable poly(lactic-co-glycolic acid) (PLGA) substrate (Fig. 3a),16 and poly(lactic acid) can also serve as a substrate for biodegradable organic devices.70 Silk fibroin is a biologically derived material that is both highly elastic and biodegradable.71 In a recent demonstration, Liu et al. produced an organic solar cell (Fig. 3b–d) for which at least the substrate was biodegradable.72 While the fate of organic semiconductors in the body has not been widely studied, researchers may be inspired by the fact that melanin—a naturally occurring semiconducting polymer—is degraded into harmless byproducts. Melanin, however, has a low mobility of charge carriers, and despite its biological origin, is mechanically brittle.16,67 New synthetic systems will be required that combine favorable charge-transport properties with programmable degradation in a single material.73–75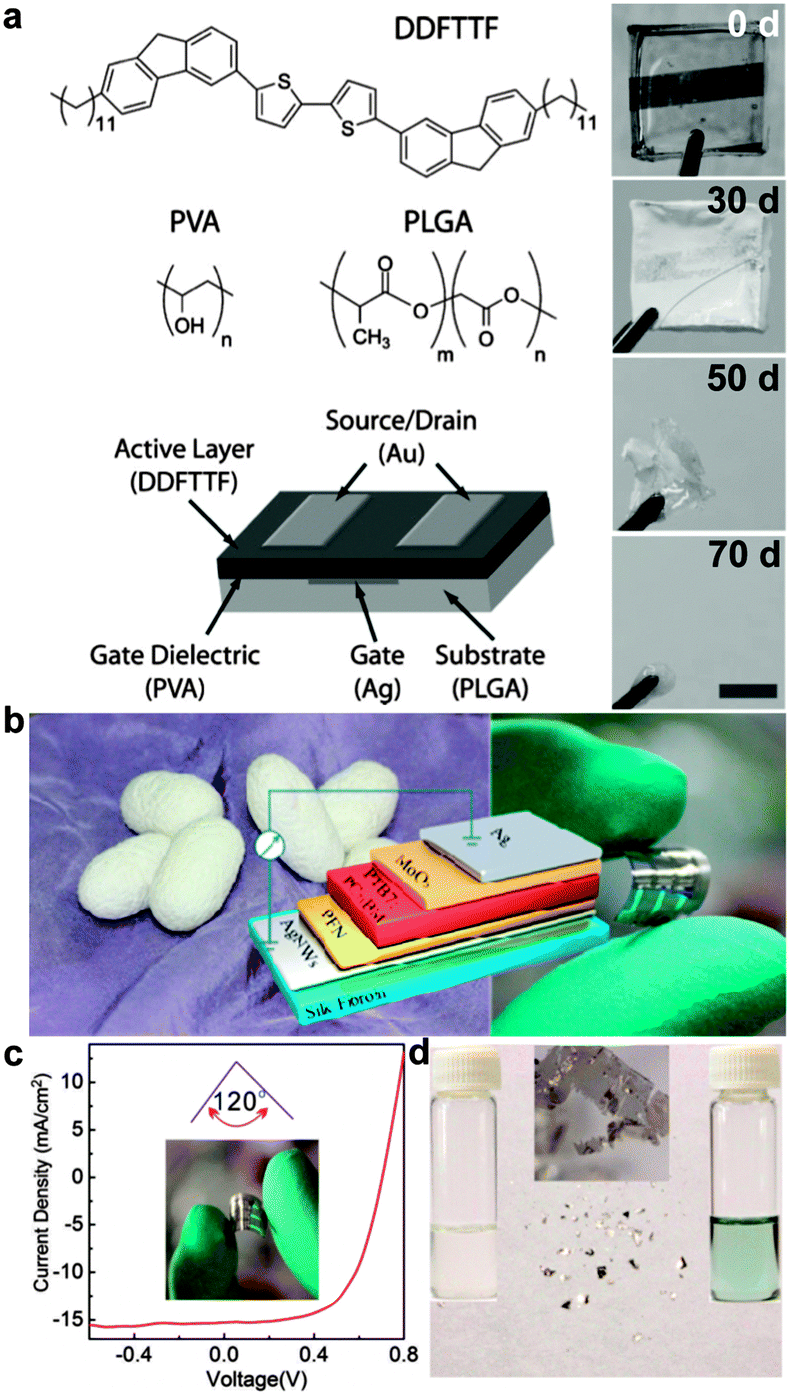 | ||
| Fig. 3 Biodegradable organic electronics. (a) Organic thin-film transistors fabricated on a biodegradable substrate decompose into byproducts that are not believed to be harmful over a period of 70 d in a solution mimicking the physiological environment. Reproduced with permission from ref. 16 Copyright 2010, Wiley-VCH Verlag, GmbH & Co. KGaA. (b) Organic solar cells fabricated on silk fibroin substrates exhibit reversible flexibility (c) and facile degradation (d). Reproduced with permission from ref. 72 Copyright 2015 American Chemical Society. | ||
Capacity for self-repair
Self-healing behavior in non-conjugated polymers is the subject of a large and expanding literature.17,76 However, the vast majority of systems for self-repair of bulk polymers—e.g., encapsulation of healing agents in hollow fibers and micron-sized particles—are not amenable to very thin films (≤100 nm) characteristic of organic optoelectronic devices.76 We are thus interested in intrinsically healing materials that repair simply upon contact of the fractured surfaces. Materials can be self-healed by non-covalent interactions and by covalent bonds. Hydrogen bonding77,78 and Diels–Alder cycloadditions55,79 have been explored for these purposes. A prerequisite to both types of repair is a sufficient level of mobility of polymer chains to allow intermixing of polymer chains across the fractured interface.80,81 This condition is generally satisfied in poly(3-alkylthiophenes) when the alkyl chain is heptyl or longer, for which the glass transition is significantly below room temperature.50 We hypothesize that unsaturated van der Waals bonds and increased mobility of polymer chains at fractured interfaces, however, may produce skin-depth effects82 that facilitate self-healing even in glassy samples.The self-healing efficiency for polymers intended for structural applications is typically expressed as a fraction of a mechanical property (e.g., strength) of the healed material relative to that of the pristine material.76 For self-healing electronic materials, however, an electronic self-healing efficiency must be used alongside a conventional one based on mechanical properties.40 Recent research from the laboratory of Kahn and coworkers has shown that conventional conjugated polymers may already exhibit the capacity for self-repair without any specialized chemical functionality.83,84 In particular, laminated interfaces of P3HT showed charge-transport properties that were similar to those of pristine layers of the same thickness.83,84
Conclusions
The design of biologically integrated electronics seems to be a problem for which organic semiconductors are an ideal solution. Despite the potential amenability of organic electronic materials to biological systems, significant work remains to be done to integrate materials from the electronic and biological realms in a way that is seamless. The strategy advocated in this Highlight is the molecular engineering of π-conjugated materials to have properties inspired by biological tissue: extreme elasticity, biodegradability, and the capacity for self-repair. While substantial progress toward this goal has already been made, a concerted, deliberate effort from chemists, materials scientists, engineers, biologists, and the medical community will be required to extract the maximum possible benefit from the versatility of organic semiconducting materials.Acknowledgements
This work was supported by startup funds from the University of California, San Diego. Additional support was provided by Air Force Office of Scientific Research (AFOSR) Young Investigator Program, Grant FA9550-13-1-0156. T. F. O. acknowledges support from the National Defense Science and Engineering Graduate Fellowship Program (NDSEG) sponsored by the Office of Naval Research (ONR).Notes and references
- N. Espinosa, M. Hosel, D. Angmo and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117–5132 CAS.
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski and D. J. Lipomi, Chem. Mater., 2014, 26, 3028–3041 CrossRef CAS.
- K. Fukuda, Y. Takeda, Y. Yoshimura, R. Shiwaku, L. T. Tran, T. Sekine, M. Mizukami, D. Kumaki and S. Tokito, Nat. Commun., 2014, 5, 4147 CAS.
- T. Someya, Y. Kato, T. Sekitani, S. Iba, Y. Noguchi, Y. Murase, H. Kawaguchi and T. Sakurai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12321–12325 CrossRef CAS PubMed.
- J. Y. Lee, S. T. Connor, Y. Cui and P. Peumans, Nano Lett., 2010, 10, 1276 CrossRef CAS PubMed.
- J. Jensen, M. Hosel, I. Kim, J. S. Yu, J. D. Jo and F. C. Krebs, Adv. Funct. Mater., 2013, 24, 1228–1233 CrossRef PubMed.
- D. Ghezzi, M. R. Antognazza, R. Maccarone, S. Bellani, E. Lanzarini, N. Martino, M. Mete, G. Pertile, S. Bisti, G. Lanzani and F. Benfanati, Nat. Photonics, 2013, 7, 400–406 CrossRef CAS PubMed.
- J. Reeder, M. Kaltenbrunner, T. Ware, D. Arreaga-Salas, A. Avendano-Bolivar, T. Yokota, Y. Inoue, M. Sekino, W. Voit, T. Sekitani and T. Someya, Adv. Mater., 2014, 26, 4967–4973 CrossRef CAS PubMed.
- C. M. Lochner, Y. Khan, A. Pierre and A. C. Aria, Nat. Commun., 2014, 5, 5745 CrossRef CAS PubMed.
- J. Rivnay, R. M. Owens and G. C. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Energy Environ. Sci., 2015, 8, 55–80 CAS.
- D. Tahk, H. H. Lee and D. Y. Khang, Macromolecules, 2009, 42, 7079–7083 CrossRef CAS.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS PubMed.
- B. O'Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef PubMed.
- A. N. Annaidh, M. Ottenio, K. Bruyer, M. Destrade and M. D. Gilchrist, IFMBE Proc., 2010, 31, 1000–1003 Search PubMed.
- C. J. Bettinger and Z. N. Bao, Adv. Mater., 2010, 22, 651–655 CrossRef CAS PubMed.
- F. Herbst, D. Dohler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 24, 203–220 CrossRef PubMed.
- J. A. Fan, W. H. Yeo, Y. W. Su, Y. Hattori, W. Lee, S. Y. Jung, Y. H. Zhang, Z. J. Liu, H. Y. Cheng, L. Falgout, M. Bajema, T. Coleman, D. Gregoire, R. J. Larsen, Y. G. Huang and J. A. Rogers, Nat. Commun., 2014, 5, 3266 Search PubMed.
- D. H. Kim, N. Lu, Y. G. Huang and J. A. Rogers, MRS Bull., 2012, 37, 226–235 CrossRef CAS.
- J. Lee, J. A. Wu, M. X. Shi, J. Yoon, S. I. Park, M. Li, Z. J. Liu, Y. G. Huang and J. A. Rogers, Adv. Mater., 2011, 23, 986–991 CrossRef CAS PubMed.
- D. H. Kim, N. S. Lu, R. Ma, Y. S. Kim, R. H. Kim, S. D. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T. I. Kim, R. Chowdhury, M. Ying, L. H. Xu, M. Li, H. J. Chung, H. Keum, M. McCormick, P. Liu, Y. W. Zhang, F. G. Omenetto, Y. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838–843 CrossRef CAS PubMed.
- S. Wagner and S. Bauer, MRS Bull., 2012, 37, 207–213 CrossRef.
- S. P. Lacour, D. Chan, S. Wagner, T. Li and Z. Suo, Appl. Phys. Lett., 2006, 88, 204103 CrossRef PubMed.
- S. W. Hwang, H. Tao, D. H. Kim, H. Y. Cheng, J. K. Song, E. Rill, M. A. Brenckle, B. Panilaitis, S. M. Won, Y. S. Kim, Y. M. Song, K. J. Yu, A. Ameen, R. Li, Y. W. Su, M. M. Yang, D. L. Kaplan, M. R. Zakin, M. J. Slepian, Y. G. Huang, F. G. Omenetto and J. A. Rogers, Science, 2012, 337, 1640–1644 CrossRef CAS PubMed.
- H. L. Hernandez, S.-K. Kang, O. P. Lee, S. W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. J. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef CAS PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwodiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- T. Sekitani and T. Someya, MRS Bull., 2012, 37, 236–245 CrossRef.
- T. Sekitani, U. Zschieschang, H. Klauk and T. Someya, Nat. Mater., 2010, 9, 1015–1022 CrossRef CAS PubMed.
- F. C. Krebs, N. Espinosa, M. Hosel, R. R. Sondergaard and M. Jorgensen, Adv. Mater., 2014, 26, 29–39 CrossRef CAS PubMed.
- V. Benfenati, N. Martino, M. R. Antognazza, A. Pistone, S. Toffanin, S. Ferroni, G. Lanzani and M. Muccini, Adv. Healthcare Mater., 2014, 3, 392–399 CrossRef CAS PubMed.
- W. G. Huang, K. Besar, R. LeCover, P. Dulloor, J. Sinha, J. F. M. Hardigree, C. Pick, J. Swavola, A. D. Everett, J. Frechette, M. Bevan and H. E. Katz, Chem. Sci., 2014, 5, 416–426 RSC.
- A. Campana, T. Cramer, D. T. Simon, M. Berggren and F. Biscarini, Adv. Mater., 2014, 26, 3874–3878 CrossRef CAS PubMed.
- C. Keplinger, J. Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- M. R. Abidian, J. M. Corey, D. R. Kipke and D. C. Martin, Small, 2010, 6, 421–429 CrossRef CAS PubMed.
- P. Leleux, J. Rivnay, T. Lonjaret, J. M. Badier, C. Benar, T. Herve, P. Chauvel and G. C. Malliaras, Adv. Healthcare Mater., 2015, 4, 142–147 CrossRef CAS PubMed.
- E. Stavrindou, O. Winther-Jensen, B. S. Shekibi, V. Armel, J. Rivnay, E. Ismailova, S. Sanaur, G. C. Malliaras and B. Winther-Jensen, Phys. Chem. Chem. Phys., 2014, 16, 2275–2279 RSC.
- N. Bhagwat, K. L. Kiick and D. C. Martin, J. Mater. Res., 2014, 29, 2835–2844 CrossRef CAS.
- D. J. Lipomi, M. Vosgueritchian, B. C.-K. Tee, C. H. Fox, J. A. Lee and Z. N. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS PubMed.
- G. Schwartz, B. C.-K. Tee, J. G. Mei, A. L. Appleton, D. H. Kim, H. L. Wang and Z. N. Bao, Nat. Commun., 2013, 4, 1859 CrossRef PubMed.
- B. C.-K. Tee, C. Wang, R. Allen and Z. N. Bao, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS PubMed.
- H. C. Ko, M. P. Stoykovich, J. Z. Song, V. Malyarchuk, W. M. Choi, C. J. Yu, J. B. Geddes, J. L. Xiao, S. D. Wang, Y. G. Huang and J. A. Rogers, Nature, 2008, 454, 748–753 CrossRef CAS PubMed.
- T. F. O'Connor, A. V. Zaretski, B. A. Shiravi, S. Savagatrup, A. D. Printz, M. I. Diaz and D. J. Lipomi, Energy Environ. Sci., 2014, 7, 370–378 Search PubMed.
- M. Shin, J. H. Song, G. H. Lim, B. Lim, J. J. Park and U. Jeong, Adv. Mater., 2014, 26, 3706–3711 CrossRef CAS PubMed.
- Z. B. Yu, X. F. Niu, Z. Liu and Q. B. Pei, Adv. Mater., 2011, 23, 3989–3994 CrossRef CAS PubMed.
- J. J. Liang, L. Li, X. F. Niu, Z. B. Yu and Q. B. Pei, Nat. Photonics, 2013, 7, 817–824 CrossRef CAS PubMed.
- M. Simonet, N. Stingelin, J. G. F. Wismans, C. W. J. Oomens, A. Driessen-Mol and F. P. T. Baaijens, J. Mater. Chem. B, 2014, 2, 305–313 RSC.
- T. Sekitani, H. Nakajima, H. Maeda, T. Fukushima, T. Aida, K. Hata and T. Someya, Nat. Mater., 2009, 8, 494–499 CrossRef CAS PubMed.
- S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger and R. Schwodiauer, Adv. Mater., 2014, 26, 149–162 CrossRef CAS PubMed.
- M. Kaltenbrunner, M. S. White, E. D. Glowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Macromolecules, 2014, 47, 1981–1992 CrossRef CAS.
- S. Savagatrup, E. Chan, S. Renteria-Garcia, A. D. Printz, A. V. Zaretski, T. F. O'Connor, D. Rodriquez, E. Valle and D. J. Lipomi, Adv. Funct. Mater., 2015, 25, 427–436 CrossRef CAS PubMed.
- A. D. Printz, S. Savagatrup, D. Rodriquez, T. F. O'Connor and D. J. Lipomi, Sol. Energy Mater. Sol. Cells, 2015, 134, 64–72 CrossRef CAS PubMed.
- A. D. Printz, S. Savagatrup, D. J. Burke, T. Purdy and D. J. Lipomi, RSC Adv., 2014, 4, 13635–13643 RSC.
- J. Liang, L. Li, K. Tong, Z. Ren, W. Hu, X. F. Niu, Y. S. Chen and Q. B. Pei, ACS Nano, 2014, 8, 1590–1600 CrossRef CAS PubMed.
- J. P. Li, J. Liang, L. Li, F. B. Ren, W. Hu, J. Li, S. H. Qi and Q. B. Pei, ACS Nano, 2014, 8, 12874–12882 CrossRef CAS PubMed.
- Z. B. Yu, Q. W. Zhang, L. Li, Q. Chen, X. F. Niu, J. Liu and Q. B. Pei, Adv. Mater., 2011, 23, 664–668 CrossRef CAS PubMed.
- H. C. Wu, S. J. Benight, A. Chortos, W. Y. Lee, J. G. Mei, J. W. F. To, C. Lu, M. Q. He, J. B.-H. Tok, W. C. Chen and Z. N. Bao, Chem. Mater., 2014, 26, 4544–4551 CrossRef CAS.
- A. Chortos, J. Lim, J. W. F. To, M. Vosgueritchian, T. J. Dusseault, T. H. Kim, S. W. Hwang and Z. N. Bao, Adv. Mater., 2014, 26, 4253–4259 CrossRef CAS PubMed.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. G. Mei and Z. N. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS PubMed.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. Vanlandingham, H. C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545–550 CrossRef CAS PubMed.
- N. Bowden, S. Brittain, A. G. Evans, J. W. Hutchinson and G. M. Whitesides, Nature, 1998, 393, 146–149 CrossRef CAS.
- J. T. Seitz, J. Appl. Polym. Sci., 1993, 49, 1331–1351 CrossRef CAS PubMed.
- D. Gargi, R. J. Kline, D. M. DeLongchamp, D. A. Fischer, M. F. Toney and B. T. O'Connor, J. Phys. Chem. C, 2013, 117, 17421–17428 CAS.
- Y. Cao, P. A. Smith and A. J. Heeger, Synth. Met., 1991, 41–43, 181–184 CrossRef.
- Y. Cao, P. Smith and A. J. Heeger, Polymer, 1991, 32, 1210–1218 CrossRef CAS.
- B. O'Connor, R. J. Kline, B. R. Conrad, L. J. Richter, D. Gundlach, M. F. Toney and D. M. DeLongchamp, Adv. Funct. Mater., 2011, 21, 3697–3705 CrossRef PubMed.
- C. J. Bettinger, J. P. Bruggeman, A. Misra, J. T. Borenstein and R. Langer, Biomaterials, 2009, 30, 3050–3057 CrossRef CAS PubMed.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl, C. J. Bettinger and R. Langer, Biomacromolecules, 2007, 8, 3067–3073 CrossRef CAS PubMed.
- M. Strange, D. Plackett, M. Kaasgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 805–813 CrossRef CAS PubMed.
- G. Mattana, D. Briand, A. Marette, A. V. Quintero and N. F. de Rooij, Org. Electron., 2015, 17, 77–86 CrossRef CAS PubMed.
- D. H. Kim, J. Viventi, J. J. Amsden, J. L. Xiao, L. Vigeland, Y. S. Kim, J. A. Blanco, B. Panilaitis, E. S. Frechette, D. Contreras, D. L. Kaplan, F. G. Omenetto, Y. G. Huang, K. C. Hwang, M. R. Zakin, B. Litt and J. A. Rogers, Nat. Mater., 2010, 9, 511–517 CrossRef CAS PubMed.
- Y. Q. Liu, N. Qi, T. Song, M. L. Jia, Z. H. Xia, Z. C. Yuan, W. Yuan, K.-Q. Zhang and B. Q. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20670–20675 CAS.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef CAS PubMed.
- M. Irimia-Vladu, N. S. Sariciftci and S. Bauer, J. Mater. Chem., 2011, 21, 1350–1361 RSC.
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, L. Shmygleva, Y. Kanbur, G. Schwabegger, M. Bodea, R. Schwödiauer, A. Mumyatov, J. W. Fergus, V. F. Razumov, H. Sitter, N. S. Sariciftci and S. Bauer, Adv. Funct. Mater., 2010, 20, 4069–4076 CrossRef CAS PubMed.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS PubMed.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. B. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS PubMed.
- N. B. Pramanik, G. B. Nando and N. K. Singha, Polymer, 2015 DOI:10.1016/j.polymer.2015.01.023.
- R. P. Wool and K. M. O'Connor, J. Appl. Phys., 1981, 52, 5953–5963 CrossRef CAS PubMed.
- S. J. Garcia, Eur. Polym. J., 2014, 53, 118–125 CrossRef CAS PubMed.
- C. M. Stafford, B. D. Vogt, C. Harrison, D. Julthongpiput and R. Huang, Macromolecules, 2006, 39, 5095–5099 CrossRef CAS.
- A. L. Shu, A. Dai, H. Wang, Y. L. Loo and A. Kahn, Org. Electron., 2013, 14, 149–155 CrossRef CAS PubMed.
- A. Dai, Y. H. Zhou, A. L. Shu, S. K. Mohapatra, H. Wang, C. Fuentes-Hernandez, Y. D. Zhang, S. Barlow, Y. L. Loo, S. R. Marder, B. Kippelen and A. Kahn, Adv. Funct. Mater., 2013, 24, 2197–2204 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |