
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directional Plk1 inhibition-driven cell cycle interruption using amphiphilic thin-coated peptide-lanthanide upconversion nanomaterials as in vivo tumor suppressors†
Chi-Fai
Chan
a,
Rongfeng
Lan
a,
Ming-Kiu
Tsang
e,
Di
Zhou
d,
Sam
Lear
c,
Wai-Lun
Chan
a,
Steven L.
Cobb
*c,
Wai-Kwok
Wong
a,
Jianhua
Hao
e,
Wing-Tak
Wong
*bg and
Ka-Leung
Wong
*af
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: klwong@hkbu.edu.hk
bDepartment of Applied Biological and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong. E-mail: bcwtwong@polyu.edu.hk
cDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: s.l.cobb@durham.ac.uk
dSchool of Chemistry and Material Engineering Jiangsu, Key Laboratory of Advanced Functional Materials, Changshu Institute of Technology, Changshu, China
eDepartment of Applied Physics, The Hong Kong Polytechnic University, Hung Hom, Hong Kong
fChangshu Research Institute of Hong Kong Baptist University, Changshu Economic and Technological Development Zone, Jiangsu 215500, China
gState Key Laboratory for Chirosciences from The Ministry of Science and Technology of the People's Republic of China, The Hong Kong Polytechnic University, Hung Hom, Hong Kong
First published on 5th February 2015
Abstract
Polo-like kinase 1 (Plk1) is a major serine/threonine protein kinase which regulates key mitotic events such as centrosome duplication, spindle assembly and chromosome separation. Overexpression and aberrant activities of Plk1 can be detected in different types of cancer. Given that the unique polo box domain (PBD) pocket provides an excellent drug target for Plk1 binding and inhibition, we have rationally designed multifunctional lanthanide-doped upconversion nanomaterials. NaGdF4:Yb3+, Er3+ (NaGdF4) and BaGdF5:Yb3+, Er3+ (BaGdF5) nanoparticles of two different sizes (60 nm and 10 nm, respectively) have been thin-coated with Plk1 specific peptides (–P1 = PLHSpT, –P2 = PLHSD, and –P3 = GGPLHSpT) to prepare novel nanomaterials. Comparative studies on cellular uptake, anti-cancer activity and imaging properties were then carried out. The experimental data obtained support our original hypothesis that the designed nanomaterials can successfully deliver Plk1 specific peptides into cancer cells causing Plk1 inhibition while simultaneously allowing direct NIR imaging and monitoring. Among the NaGdF4–Pn and BaGdF5–Pn nanoparticle series prepared in this study, NaGdF4–P1 emerged as the best candidate for Plk1 binding and imaging. NaGdF4–P1 can effectively exert cell cycle G2/M arrest and thus selective tumor inhibition both in vitro and in vivo and as such it offers a potentially interesting system for the development of new cancer therapies.
1. Introduction
The human polo-like kinase family (Plk1-5) is responsible for a wide variety of cell regulatory activities such as the DNA damage response, bipolar spindle formation, mitotic entry as well as cytokinesis.1–5 However, the actual role of Plk1 in carcinogenesis is still yet to be fully elucidated. Some pre-clinical evidence suggests that overexpression of Plk1 is linked to an increased survival rate of cells,6 and this is supported by the fact that Plk1 is normally highly expressed in actively proliferating tissue.7 Strategic inactivation of Plk1 results in cell cycle arrest and subsequently apoptosis, thus Plk1 has emerged as a favorable therapeutic target for cancer treatment.8Within Plk1, there are two distinct domains—an amino-terminal kinase domain and a unique carboxyl terminal polo box domain (PBD). Several organic small molecules which can inhibit the Plk1 kinase domain and the PBD have been identified. Molecules such as BI2536, CBB2001 and MLN0905 target the kinase domain of Plk1 causing inhibition as well as pronounced dose-limiting toxicity (DLT),9–11 however unspecific activities and unwanted side-effects have really limited the further development of Plk1 kinase domain targeting molecules. On the other hand, peptides and their analogues that can bind to the PBD present a novel and promising avenue for Plk1 targeting and inhibition.12,13 Peptides can be employed as tailor-made inhibitors and potential drugs but there are issues in working with this class of biological molecules. Peptides often display physical and chemical instabilities,14 poor cellular uptake, and localization issues.15 Real-time assessments, including MTT, in vitro imaging and tumor inhibition, are also crucial elements in cancer prognosis and treatment. Conventionally, for imaging purposes complex methods of analysis such as immunostaining of Plk1 using antibodies or fusion fluorescent proteins have been employed.16,17 Recently, E. K. Ryu et al. demonstrated that a Plk1-specific peptide (PLHSpT) conjugated with the αvβ3 integrin receptor inhibitor peptide (RGD) and a radioligand can achieve the much sought-after theranostic effect.14 However, this is a rather indirect method of analysis as it only tests cancer cells which involves multiple steps to interrupt the cell cycle, inhibit the tumor size, and diagnose the cancer tissues with PET. Most recently, we have reported a porphyrin conjugated to a Plk1-specific peptide (e.g. PLHSpT) as a multi-modal bio-probe, with the molecular system serving as the vector.18 Its overall tumor growth inhibition efficiency through the singlet oxygen generation has, indeed, still room for further improvement.
In the present study we have prepared lanthanide-doped upconversion nanoparticles (UCNPs) which can act as delivery vehicles for Plk1-specific peptides. The nano-particle–peptide conjugates were shown to inhibit Plk1 activity, while cell-cycle arrest and visualization can also be achieved simultaneously. Owing to the excellent photo-physical properties of lanthanide ions, the dopant ions (ytterbium/erbium) can be excited and emit in the near-infrared (NIR) region. The nano-systems developed also offer additional benefits for bio-imaging such as improved light penetration depth, lower cytotoxicity and, above all, elimination of autofluorescence due to the long emission lifetime (μs to ms) of lanthanides compared with that of organic biomarkers.19–28 By conjugating the PBD binding peptide onto UCNPs, the cellular uptake problem of the individual peptides can also be addressed because the resulting organic–inorganic nanomaterial is highly biocompatible,29,30 and the peptides are protected from enzymatic degradation.31 Although, in the case of cyclin D, the inhibitory effects of our previously established UCNPs (NaGdF4:Yb3+, Er3+) conjugated with cyclin D-specific peptides were not as satisfactory as expected,32 we have rationalized that this could be ascribed to unwanted cross-binding among various cyclins and their cyclin-dependent kinases (CDKs),33 in addition, the in vitro emission signal was also quenched after bio-conjugation when compared to the unmodified NaGdF4:Yb3+, Er3+. With these factors in mind, we have now developed a new type of novel peptide-functionalized UCNP which can achieve termination and visualization of cancer cells concurrently through binding and inhibition of Plk1.
Herein, we report the synthesis of eight UCNPs, using two solid systems of different sizes—60 nm NaGdF4:Yb3+, Er3+ (NaGdF4) and 10 nm BaGdF5:Yb3+, Er3+ (BaGdF5). These two types of UCNPs are square, doped with Yb3+ and Er3+ at molar ratio 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and coated with polyethyleneimine (PEI), PLHSpT (P1),13,14 PLHSD (P2) and GGPLHSpT (P3) respectively (Fig. 1). All three peptides have already shown strong binding with PBD in Plk1.13P3, which contained an additional Gly–Gly motif, was used primarily to check whether any significant impact on the binding affinity between the peptide (i.e. PLHSpT) and Plk1, as well as the anti-tumor activity was observed when the linking distance between the UNCPs and the peptides was increased in length and flexibility. Comprehensive in situ (photophysical properties, protein pull down assay), in vitro (emission spectra, imaging, co-staining with GFP-Plk1, ICP-MS, cellular toxicity) and in vivo studies (imaging and solid tumor inhibition) in both cancer and normal cell lines have been conducted and the data are presented. Experimental results revealed that our 60 nm UNCPs have a better performance than the 10 nm UCNPs in terms of uptake efficiency and imaging quantity. In particular for NaGdF4–P1 coated with the P1 peptide; control experiments have illustrated that NaGdF4–P1 is selective in its mode of action and it was demonstrated to have no anti-growth effect in normal cells.
:2 and coated with polyethyleneimine (PEI), PLHSpT (P1),13,14 PLHSD (P2) and GGPLHSpT (P3) respectively (Fig. 1). All three peptides have already shown strong binding with PBD in Plk1.13P3, which contained an additional Gly–Gly motif, was used primarily to check whether any significant impact on the binding affinity between the peptide (i.e. PLHSpT) and Plk1, as well as the anti-tumor activity was observed when the linking distance between the UNCPs and the peptides was increased in length and flexibility. Comprehensive in situ (photophysical properties, protein pull down assay), in vitro (emission spectra, imaging, co-staining with GFP-Plk1, ICP-MS, cellular toxicity) and in vivo studies (imaging and solid tumor inhibition) in both cancer and normal cell lines have been conducted and the data are presented. Experimental results revealed that our 60 nm UNCPs have a better performance than the 10 nm UCNPs in terms of uptake efficiency and imaging quantity. In particular for NaGdF4–P1 coated with the P1 peptide; control experiments have illustrated that NaGdF4–P1 is selective in its mode of action and it was demonstrated to have no anti-growth effect in normal cells.
 | ||
| Fig. 1 Schematic diagram of conjugating Plk1 specific peptides onto NaGdF4 and BaGdF5. | ||
2. Results and discussion
Through conjugation to various inorganic molecules such as nanoparticles, peptides can be effectively shielded from enzymatic degradation. In addition peptide coated UNCPs can be excited at 980 nm enabling their real-time imaging. In this study, we intended to exploit both these aforementioned properties coupled with Plk1 targeting to design and prepare nanosystems capable of simultaneous Plk1 inactivation and real time imaging, something that is not currently easily achievable. Two sets of UNCPs of different sizes have been conjugated with three kinds of Plk1 specific peptides and photophysical properties, cellular uptake efficiency, in vitro imaging and selectivity for Plk1 of these systems have been evaluated. The UCNPs have been characterized and verified by a series of experiments including spectroscopic measurements, microscopic analyses and competitive assays through protein binding and western blotting. In addition to investigating the selection of Plk1 specific peptides and different UCNPs, we also wished to probe the effects of the size and shape of UCNPs themselves. Thus we used 60 nm and 10 nm UCNPs as both of them have a similar crystal lattice and they are square in shape. Through this comprehensive study, we aimed to satisfy our hypothesis that Plk1 specific coated nanomaterials can act as promising systems in the future development of multi-modal anti-cancer upconversion nano-drugs.2.1 Synthesis and characterization of nanoparticles conjugated with Plk1 (Pn) specific peptides
The surface groups of our NaGdF4 and BaGdF5 were examined by FTIR. The corresponding IR transmission spectra of amine-functionalized NaGdF4 and BaGdF5 are shown in Fig. S1.† The broad absorption band centered at 3459 cm−1 is due to the O–H/N–H stretching vibration. The IR absorption peaks at 1637 and 1436 cm−1 are attributable to the N–H bending of the amino group (NH2) and the C–N bond stretching vibration. Therefore, the IR transmission spectrum fully supports the capping of poly(ethyleneimine) (PEI) on the surfaces of NaGdF4 and BaGdF5. The amine-functionalized NaGdF4 and BaGdF5 were then modified into amide-functionalized NaGdF4-ene and BaGdF5-ene. The conversion of amine to amide was verified by the IR spectrum in Fig. S1(b).† The overall spectrum is similar to Fig. S1(a).† In fact, the absorption peaks at 1648 cm−1 newly appear under the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.34 Fig. S1(c)† shows the IR spectrum recorded after addition of the target specific peptide Pn on the surfaces of NaGdF4 and BaGdF5. The small absorption peaks around 3022 to 3417 cm−1 represent the N–H bond stretching while the broad peak at 3307 cm−1 stands for carboxylic acid. Hence, we had confirmed the successful coating of target specific peptide Pn onto our NaGdF4 and BaGdF5.
O bond.34 Fig. S1(c)† shows the IR spectrum recorded after addition of the target specific peptide Pn on the surfaces of NaGdF4 and BaGdF5. The small absorption peaks around 3022 to 3417 cm−1 represent the N–H bond stretching while the broad peak at 3307 cm−1 stands for carboxylic acid. Hence, we had confirmed the successful coating of target specific peptide Pn onto our NaGdF4 and BaGdF5.
The shape and morphology of NaGdF4 or BaGdF5 and peptide coated NaGdF4–Pn or BaGdF5–Pn, on the other hand, were investigated by TEM. In Fig. 2a and b, the morphology of NaGdF4 and BaGdF5 was square in shape which corresponds to the cubic phase of NaGdF4 and BaGdF5. This observation is similar to the previously reported synthesis of the cubic phase NaGdF4 (ref. 30) and BaGdF5 (ref. 22) which was verified by the SAED pattern of NaGdF4 and BaGdF5 as shown in Fig. S2.† From the high resolution TEM (HRTEM), a thin layer is observed (as indicated by red arrows) and the size distribution in TEM images reveals also that the overall sizes of Plk1 specific peptide coated NaGdF4–Pn and BaGdF5–Pn increase by 1–2 nm compared with the bare NaGdF4 and BaGdF5, thereby justifying the conjugation of NaGdF4 and BaGdF5 with peptide Pn. As demonstrated by the dynamic light scattering analysis, the size of the nanomaterials is also increased by 1–3 nm after the peptide conjugation (Fig. S5†). Besides, the surface charges of NaGdF4 and BaGdF5 should be different before and after the peptide conjugation. In Fig. 3, there is clearly a drop of zeta potential which again qualifies the successful peptide conjugation onto our nanomaterials.
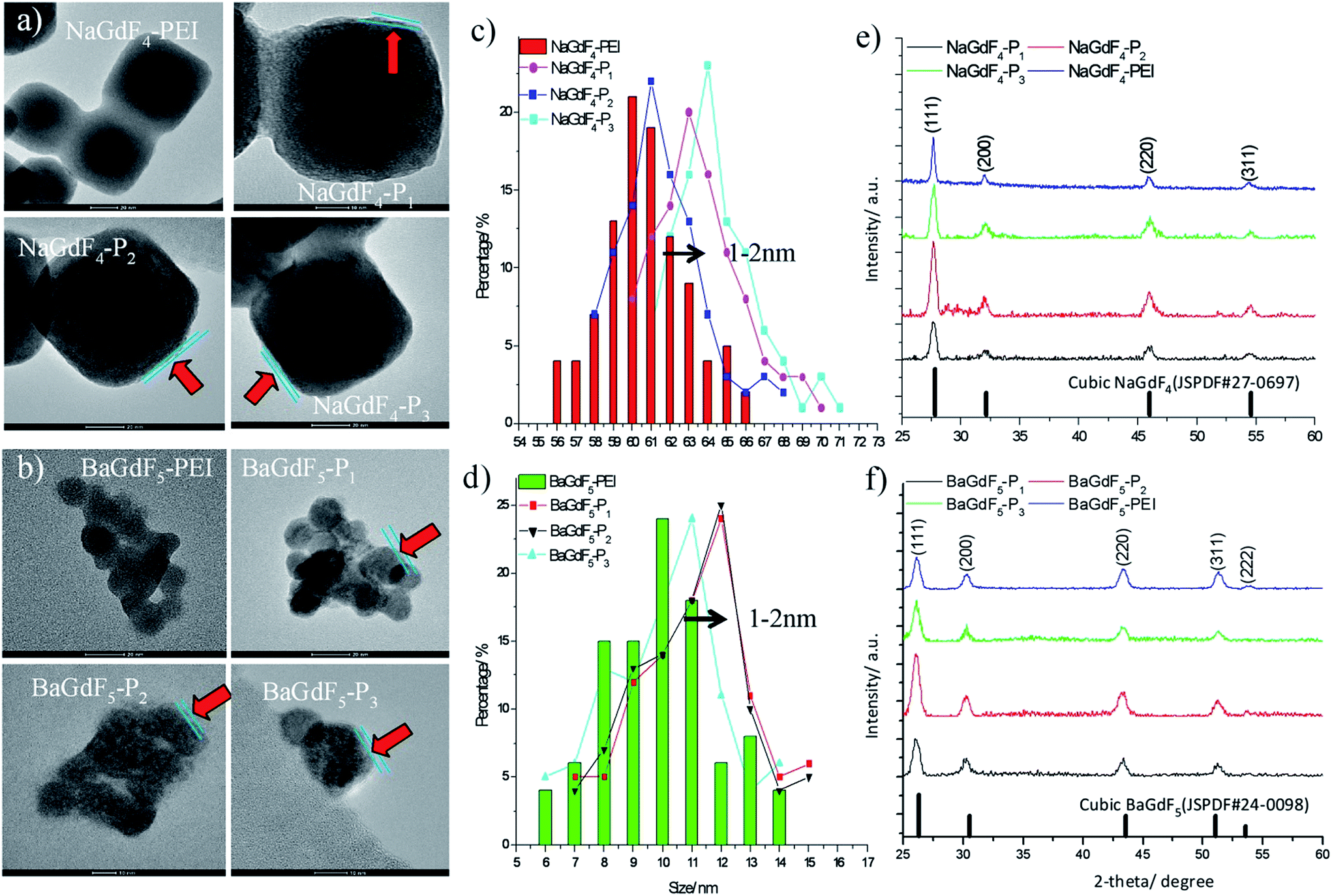 | ||
| Fig. 2 Surface characterization by (a and b) TEM analysis; (c and d) size distribution and (e and f) XRD pattern analysis. | ||
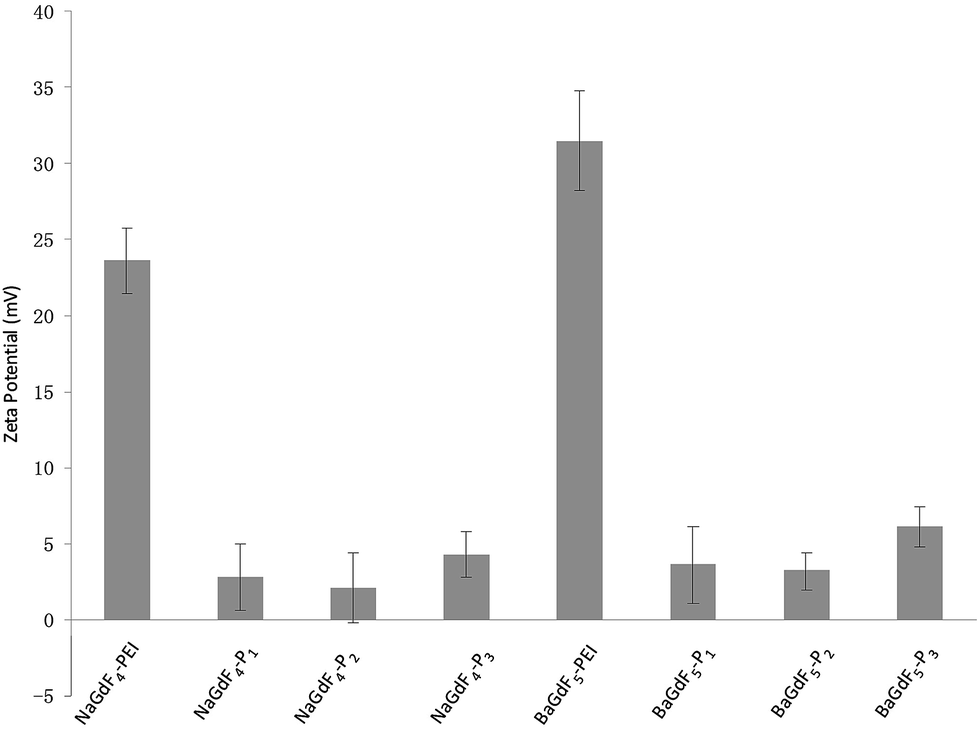 | ||
| Fig. 3 Peptide conjugation leads to a drop of zeta potential since peptides are relatively more negative compared with the PEI on NaGdF4 or BaGdF5. | ||
The phase composition of NaGdF4 or BaGdF5 and peptide coated NaGdF4–Pn or BaGdF5–Pn was revealed by powder X-ray diffraction (XRD). In Fig. 2e and f, the NaGdF4 and BaGdF5 were indexed to the standard cubic phase NaGdF4 (JSPDF#27-0697) and BaGdF5 (JSPDF#24-0098) respectively. The diffraction peaks agree well with the standard pattern, with the matching results indicating that our NaGdF4 and BaGdF5 are present in the pure cubic phase with no other phase impurities. After the coating of Plk1 specific peptides on the surface of NaGdF4–Pn or BaGdF5–Pn, the XRD patterns show no changes in the 2-theta angles and this implies that the coating of peptides did not induce changes in the crystal structures of both NaGdF4 and BaGdF5. Also, this observation was consistent with the non-crystalline nature of peptides.
2.2 Photophysical properties of upconversion nanoparticles
In Yb3+ and Er3+ co-doped nanoparticles, under 980 nm laser diode excitation, Yb3+ ions possessing a large absorption cross-section, much longer excited state lifetime and the energy (2F5/2 → 2F7/2) well resonant with higher-energy states of other lanthanide ions can transfer energy continuously and efficiently to Er3+ ions, leading to bright upconversion luminescence in the visible region. Fig. 4 shows the emission spectra of the peptide coated NaGdF4 and BaGdF5 (NPs), as well as the spectra of all the coated NPs indicating the same structures. The three emission peaks at 521 nm, 541 nm, and 650 nm arise from 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 transitions respectively, which are ascribed to the two-photon upconversion process under 980 nm excitation with 340 mW and commonly present in nanomaterials doped with Yb3+ and Er3+ ions. The nearby peaks originated from transitions between other Stark levels of the corresponding excited energy levels and the ground state. In order to understand the upconversion mechanism well, we measured the decay time curves for the peptide coated UCNPs. The insets of Fig. 4a and b display the luminescence decay curves of the 4S3/2 → 4I15/2 transition of Er3+ ions in NaGdF4–Pn and BaGdF5–Pn. Each of the down edges of the decay curves can be fitted well into a single exponential function I(t) = I0exp(−t/τ), where I0 is the initial emission intensity at t = 0, and τ is the decay time. The lifetime of the 541 nm emission peak is ∼30 μs. Moreover, all the decay curves consist of rising edges, indicating that the population of 4S3/2 originates from the energy transfer of the sensitizer Yb3+ ions. The upper level 4F7/2 was populated by two-step upconversion energy transfer of Yb3+ ions and then nonradiatively relaxed to 4S3/2, which resulted in the 541 nm emission. Thus the population of the 4S3/2 level is closely related to the phonon state density and the emission would be sensitive to temperature. From Fig. 4a, the nanoparticles of NaGdF4 coated with different peptides show remarkably different emission intensities, thereby implying the obvious influence of the surface coating on the upconversion properties of the nanoparticles. In short, the peptide coated nanoparticle emitting long lifetime bright visible light under near infrared excitation could be an ideal material to achieve termination and visualization of cancer cell simultaneously.
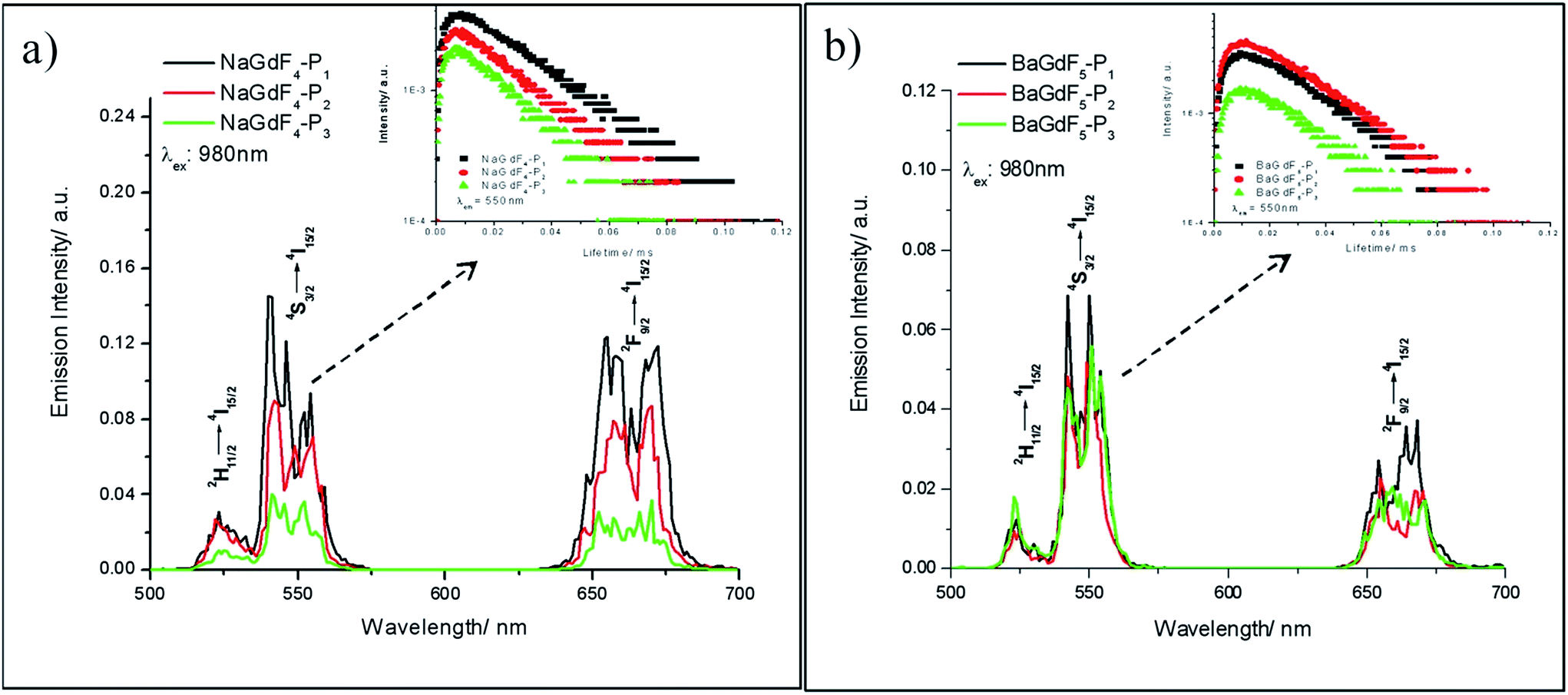 | ||
| Fig. 4 Upconversion emission spectra of (a) NaGdF4–Pn and (b) BaGdF5–Pn. The insets are their corresponding lifetimes at λem: 541 nm. | ||
2.3 In vitro cell based assay of UCNPs
The application of peptides as potential anti-cancer agents is not a new concept; but, there are very limited methods for probing directly the uptake of such peptides in vitro. In our system, the lanthanide content in peptide-functionalized UCNPs offers an in-built and direct monitoring opportunity for ICP-MS evaluation of peptide uptake efficiency in vitro (Fig. 5). From the analysis of the ICP-MS results (Fig. 5), it can be seen that the uptake of two unfunctionalized nanoparticles is similar in both the cancer (HeLa) and the normal (liver derived QSG-7701) cells lines. A variation in the uptake rate occurs when the UNCPs are coated with peptides, especially peptides P1 and P3 (see arrows in Fig. 5). In fact, in general, there are three main differences between peptide coated and unfunctionalized UCNPs. First, coating with peptides facilitates cell permeability in both cancer (HeLa) and normal (liver derived QSG-7701) cells. Secondly, the 60 nm NaGdF4–Pn shows slightly higher cellular uptake efficiency in comparison with 10 nm BaGdF5–Pn in both cancer and normal cell lines. The underlying reason for this could simply be that in the 60 nm systems a larger surface area is available, and thus more peptides are coated on the nanoparticle. Lastly, peptides P1 and P3 helped to improve the selectivity of the uptake, most possibly due to the fact that hydrophilic peptides enhanced the distribution of nanoparticles. In addition, cellular Plk1 may also increase nanoparticle accumulation through specific peptide–protein binding (i.e. greater difference between uptake in cancer versus normal cells). From this later point it also appears that the specific sequence of the peptides, but not their actual length, is important in the binding interactions with Plk1.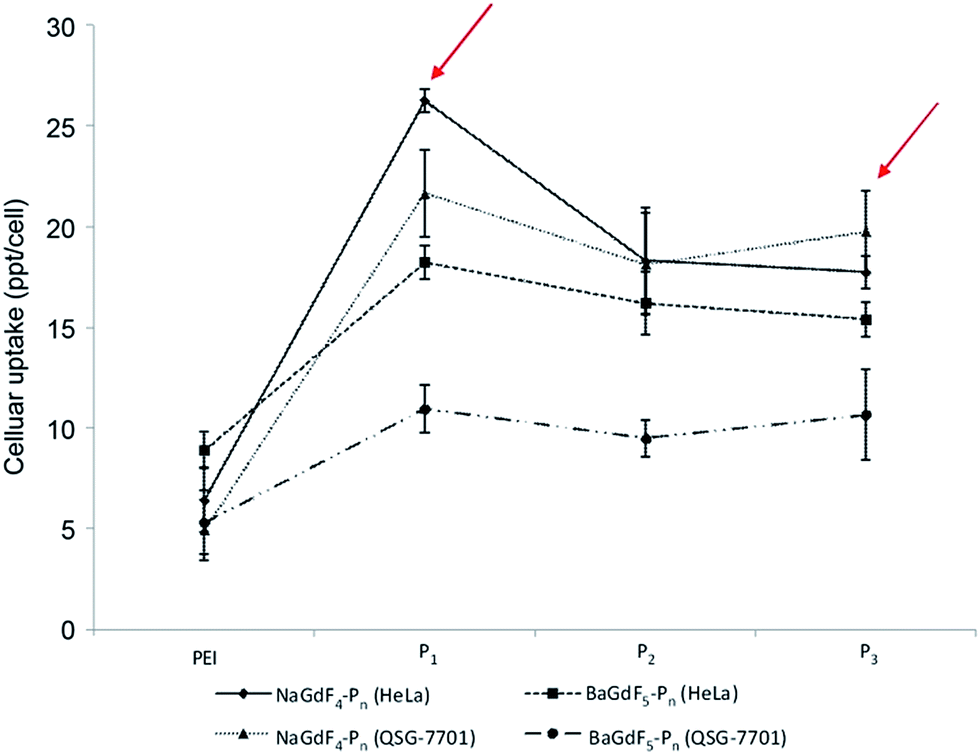 | ||
| Fig. 5 HeLa and QSG-7701 cellular uptake profile of NaGdF4–Pn and BaGdF5–Pn by ICP-MS. The y-axis is the cellular uptake concentration at the ppt level per cell. | ||
Given that our nanomaterials demonstrated interesting photo-physical properties within the biological window, high selectivity for Plk1 (by western blotting), and that they are more readily taken up by cancer cells, the potential of NaGdF4–Pn or BaGdF5–Pn to act as in vitro imaging bio-probes for Plk1 appeared very promising. To investigate this further, in vitro experiments were conducted in cervical carcinoma HeLa cells with two-photon microscopes (Fig. S3†). NaGdF4–Pn or BaGdF5–Pn have, as expected, detectable emissions within the biological window (500 nm to 700 nm) via the near-infrared excitation at 980 nm. Such upconversion inside the cells can also help us to confirm the cell permeability of our UCNPs. The near-infrared (excitation) to near-infrared (emission) in vitro imaging of NaGdF4–Pn or BaGdF5–Pn can be correlated with the uptake results in Fig. S3.† Under the NIR excitation by the multi-photon confocal microscope, impressive green (4S3/2 → 4I15/2) and red (Fig. 6, 2F9/2 → 4I15/2) f–f emissions can be observed in HeLa cells due to the presence of NaGdF4–Pn or BaGdF5–Pn after 24 hours incubation. The in vitro emission spectra were recorded with the lambda scan system on a confocal microscope (resolution = 3 nm), the erbium emission bands 4S3/2 → 4I15/2 and 2F9/2 → 4I15/2 are found which is the same as our measurements with these UCNP–Pn in aqueous solution. Obviously, the in vitro emission of 60 nm NaGdF4–Pn is far more intense than the 10 nm BaGdF5–Pnin vitro. This observation can be found from the in vitro imaging and its emission spectra. NaGdF4–P1 and NaGdF4–P3 show impressive red emission inside the cell.
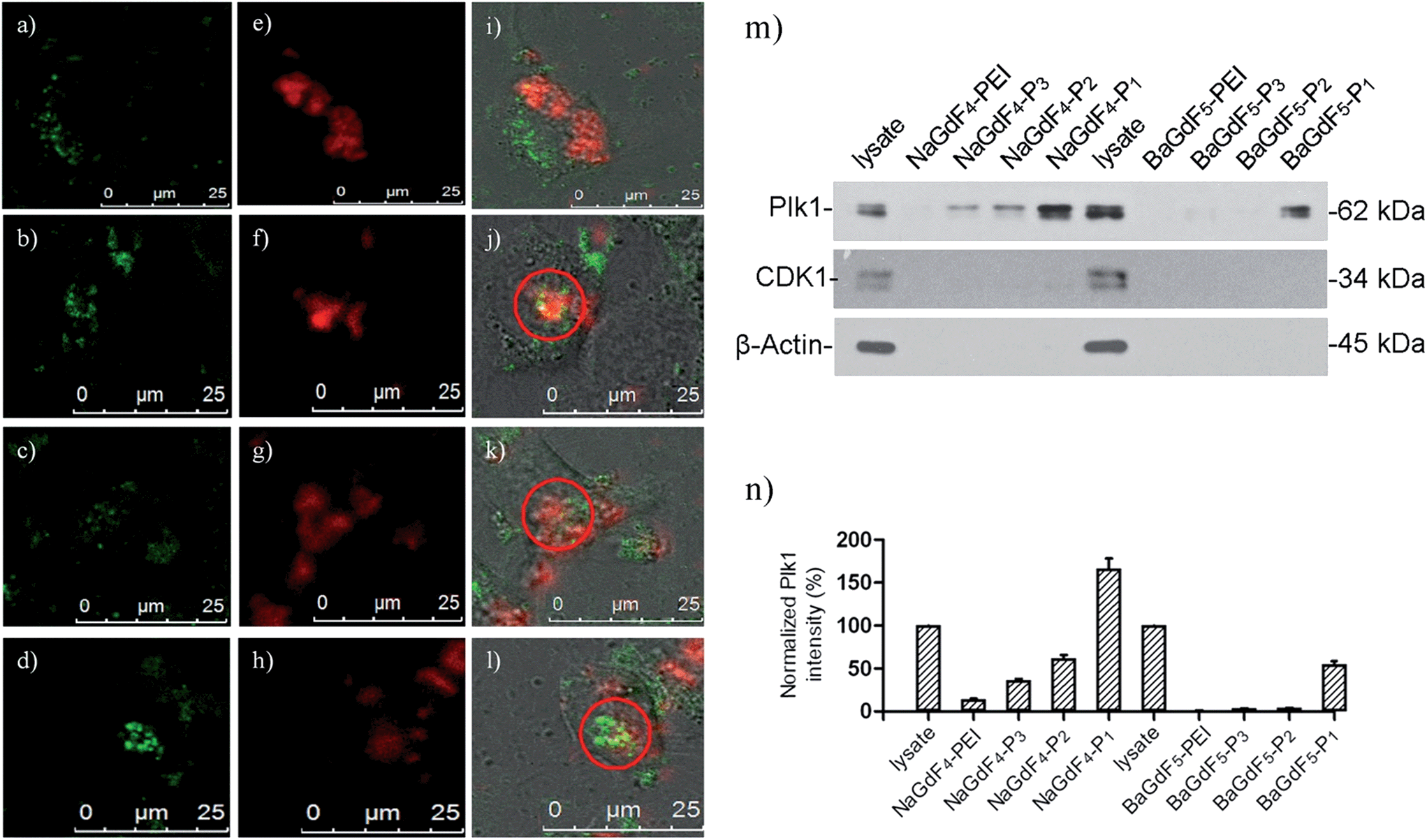 | ||
| Fig. 6 In vitro images of (a–d) expressed GFP-Plk1; (e–h) upconversion emission of NaGdF4-PEI, –P1, –P2, –P3 respectively and (i–l) their overlay images in HeLa cells. (m and n) The pull down assay compared the binding affinity among NaGdF4–Pn or BaGdF5–Pn towards Plk1 and their relative Plk1 intensity respectively. | ||
The selectivity of the prepared UCNPs toward Plk1 binding was confirmed by co-staining experiments with GFP-Plk1 specific fluorescent protein. (GFP-Plk1 protein is not available with the excitation at 980 nm). The co-staining experiments were carried out with the transient expression of the GFP-Plk1 protein in the cancer cells through plasmid transfection, followed by incubation with the UCNPs for another 24 hours. In Fig. 6a, the red emission from bare NaGdF4 (λex = 980 nm) is not overlapped with the green emission from the GFP-Plk1 protein (λex = 488 nm). However, in Fig. 6b and d, the red emission from NaGdF4–P1 and NaGdF4–P3 overlapped with the green emission from GFP-Plk1 well (red circle and white arrow in Fig. 6b and d). From in vitro microscopy, the up-conversion spectra showing the uptake behaviours of those nanomaterials are similar to what we found in ICP-MS uptake assays. The up-conversion red emission from NaGdF4–P1 and NaGdF4–P3 can be overlayed with the green GFP emission, further supporting the nano-material selectivity towards Plk1. In order to further verify the binding selectivity of NaGdF4–Pn or BaGdF5–Pn towards Plk1, the nanoparticle–peptide bound Plk1 proteins in the total cell lysate were examined using anti-Plk1 antibodies and western blotting by pull down assay. Among the various NaGdF4–Pn or BaGdF5–Pn tested, the NaGdF4–P1 shows the highest binding affinity towards Plk1 (about 10-fold higher) as revealed by the graph of relative Plk1 intensity (Fig. 6m and n). Therefore, we propose that by conjugating the Plk1 specific peptide P1 onto the surface, the cellular uptake of our UCNPs should be much higher rather than conjugating other peptides (P2 and P3). Hence, NaGdF4–P1 is the best candidate for in vivo imaging nanomaterials compared with the other Plk1 specific peptide analogues.
2.4 Inhibition of cell viability and cancer cell cycle interruption
Plk1 expression in different kinds of tumor cells is higher than that in normal healthy cells. Therefore, before further studies and investigation of applications, the inhibitory effect of NaGdF4–Pn or BaGdF5–Pn on cancer cells had to be investigated. Firstly, the cell viability was examined using an MTT assay in various cell lines (details are given in the Experimental section). Cell inhibition rates were calculated via viability related absorbance. Fig. 7 shows that NaGdF4–Pn or BaGdF5–Pn exerted a higher inhibitory effect on cell viability, in comparison with the results using NaGdF4-PEI and BaGdF5-PEI in tested cell lines. Among the different Plk1 specific peptides conjugated to the NaGdF4–Pn or BaGdF5–Pn systems, NaGdF4–Pn, showed a better inhibitory effect on cancer (HeLa and HK-1) cells in contrast to BaGdF5–Pn as highlighted in Fig. 7. The results from the MTT assays carried out in normal cell lines, namely MRC-5 and QSG-7701, demonstrated the selectivity of NaGdF4–Pn or BaGdF5–Pn towards cancer cell lines (which over-express Plk1). In fact, there is a comparable amount of as-prepared nanoparticles taken up by HeLa cells and QSG-7701 cells but the inhibitory effect on HeLa is much higher than that on QSG-7701, thereby proving that our NaGdF4–Pn or BaGdF5–Pn can selectively kill cancer cells but remained relatively non-toxic to the normal cells. The IC50 values of NaGdF4–Pn or BaGdF5–Pn are provided in ESI Table S1.† Furthermore, cell cycle interruption was evaluated by flow cytometry. All of the Plk1 specific peptide coated NaGdF4–Pn or BaGdF5–Pn (0–100 μg mL−1) and control (NaGdF4-PEI or BaGdF5-PEI) had been tested for the inhibitory activity and the data are shown in Fig. 7e. Normally, the cell phase distribution of usual cell division is ∼67%, ∼10% and ∼23% in G1, G2 and S phases respectively. However, when treated with various NaGdF4–Pn or BaGdF5–Pn, only the NaGdF4–P1 showed a significant G2 phase arrest at a dosage concentration of 100 μg mL−1 (indicated with statistical significance) while other NaGdF4–Pn or BaGdF5–Pn showed milder cell cycle interruption.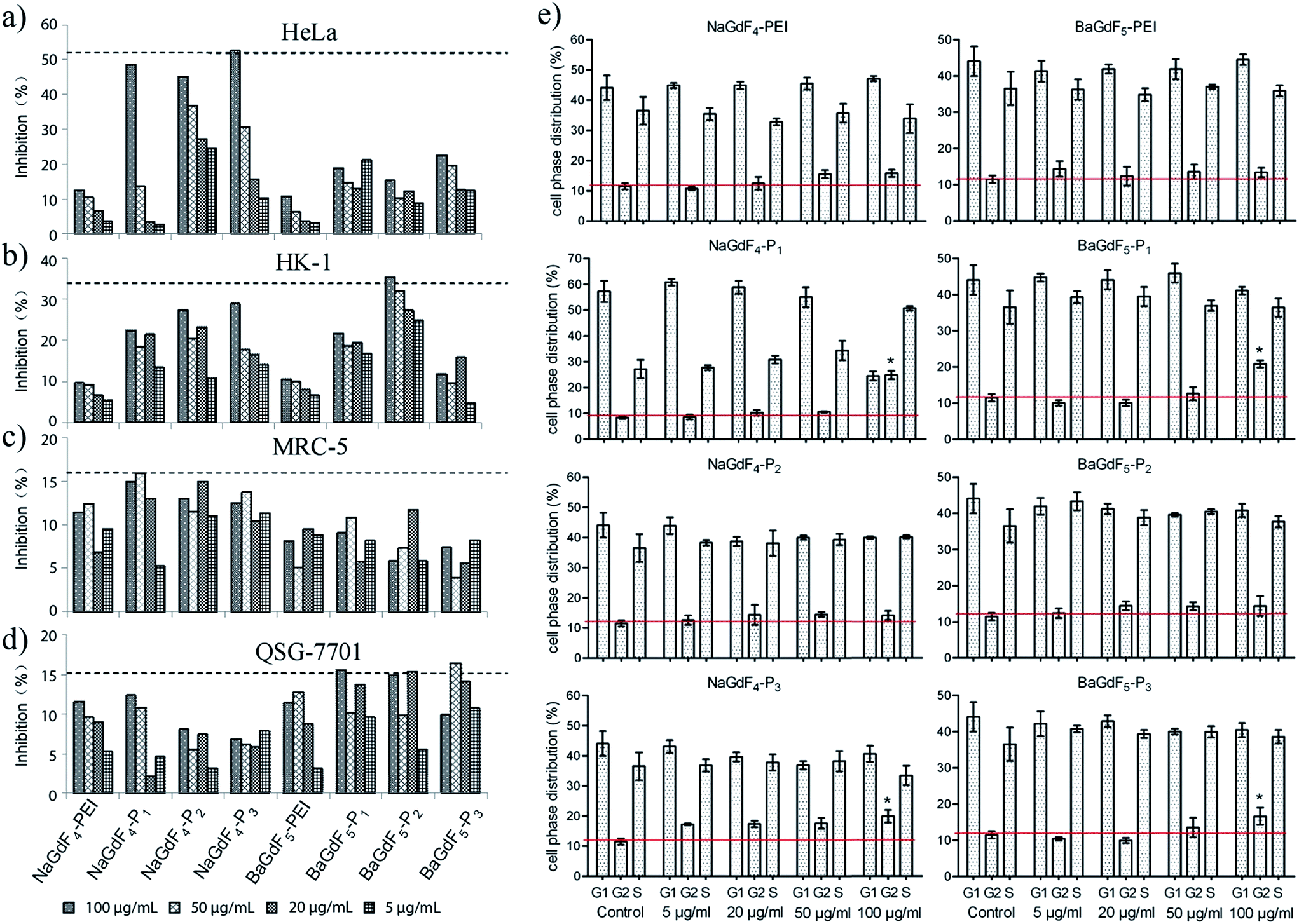 | ||
| Fig. 7 MTT inhibition assays of NaGdF4–Pn and BaGdF5–Pn in (a) human cervical cancer HeLa; (b) human nasopharyngeal carcinoma HK-1; (c) human normal lung fibroblast MRC-5 cell; (d) human liver-derived QSG-7701 cell, and (e) cell cycle interruption in HeLa with flow cytometric analysis. * represents the statistical significance of P < 0.01. | ||
2.5 In vivo bio-distribution and mouse xenograft tumor inhibition
In order to study the metabolism of NaGdF4–Pn or BaGdF5–Pn in an animal model, gadolinium concentration in different mouse tissues was analyzed with ICP-MS. Fig. 8a–c show that NaGdF4-PEI and NaGdF4–P1 accumulate in mice spleen and liver 6 hours after caudal vein injection, while BaGdF5-PEI shows low accumulation in these organs. The reason for this is that BaGdF5-PEI was quickly metabolized and excluded. Since liver and spleen are the primary organs responsible for detoxification, our nano-materials were captured and precipitated in these organs. Interestingly, compared with NaGdF4-PEI, NaGdF4–P1 not only accumulates in the spleen and liver, but it is also excreted in high concentration in the urine which suggests a longer retention time in the body (Fig. 8a and c). In addition, mouse xenograft tumor inhibition was also investigated using our nano-materials which were subcutaneously injected around the tumor. Fig. 8d clearly shows that treatment with NaGdF4–Pn (especially NaGdF4–P1) results in tumor shrinkage while BaGdF5–Pn shows only a slight effect on the tumor. Therefore, the tumor inhibition performance of the NaGdF4–Pn systems was better than that of the BaGdF4–Pn systems which is completely in line with the results from the in vitro cell line based inhibition studies.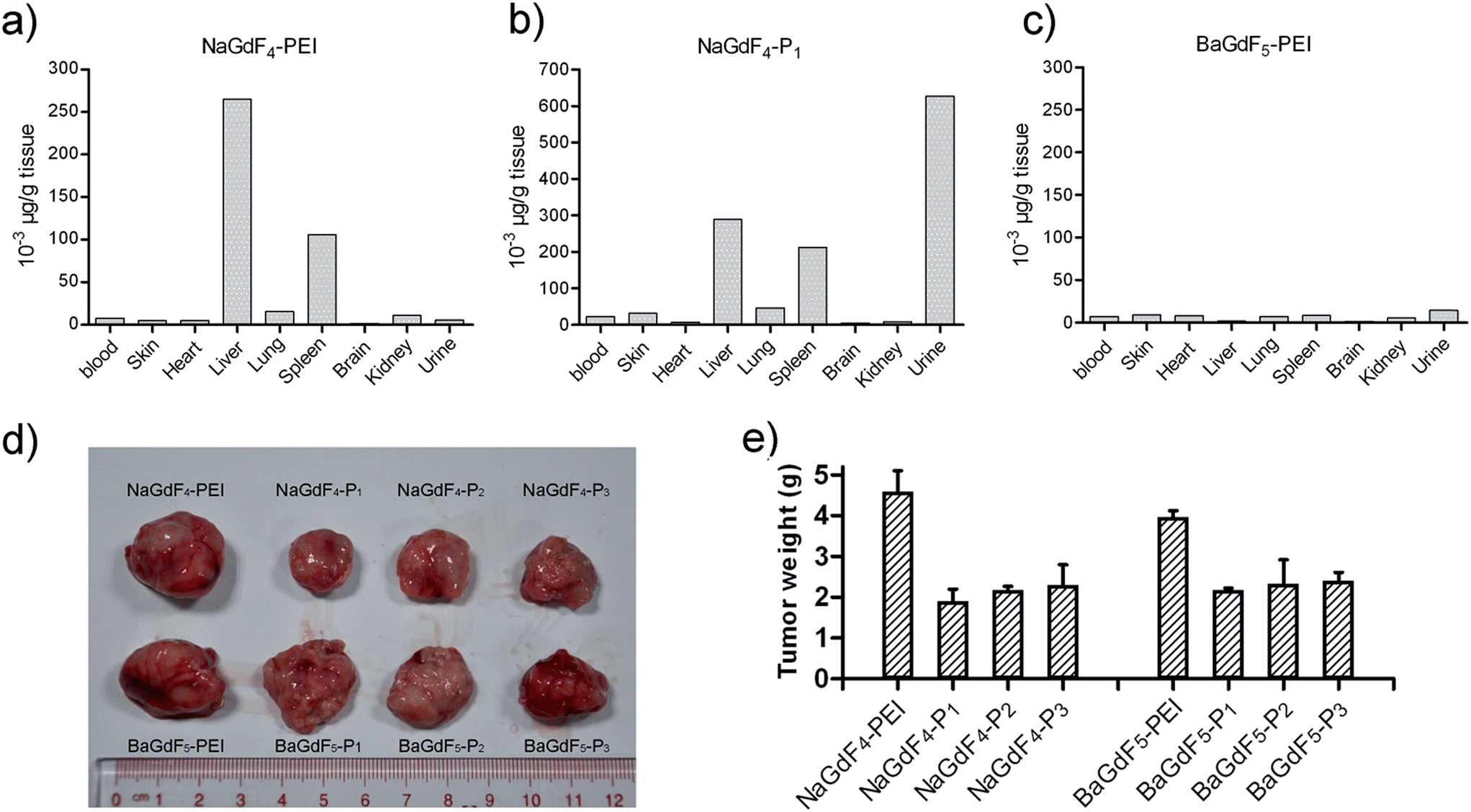 | ||
| Fig. 8 (a–c) In vivo uptake profiles of NaGdF4-PEI, NaGdF4–P1 and BaGdF5-PEI analyzed by ICP-MS respectively. (d) Tumor inhibition effect of NaGdF4–Pn and BaGdF5–Pnvia subcutaneous injection around the tumor. (e) Xenograft tumor weight after the treatment of NaGdF4–Pn and BaGdF5–Pn. | ||
3. Experimental
3.1 Chemicals and materials
Ln(NO3)3·6H2O (Ln = Gd, Yb, Er) were purchased from Aldrich and dissolved in de-ionized water (DI-water) to form solutions with concentrations of 0.5 M and 0.1 M. Acrylic acid, triisopropylsilane (TIS) and N-hydroxy-succinimide (NHS, 98%) were purchased from Aldrich. Ethylene glycol (EG, 99%), branched polyethylenimine (PEI, 25 kDa), BaCl2 (99.99%) and trifluoroacetic acid (TFA, 99%) were purchased from Sigma-Aldrich. NH4F (99.99%) and NaCl (99.99%) were obtained from Sinopharm Chemical Reagent Co., China., and N,N′-dicyclohexylcarbodiimide (DCC, 99%) was purchased from Meyer. 3-(Tritylthio)propanoic acid (>99%) was purchased from Dieckmann. All of these chemicals were used as received without further purification.3.2 Synthesis of NaGdF4 and BaGdF5 upconversion nanoparticles
Water-dispersed NaGdF4 was prepared by the one-step hydrothermal method. Typically, 1 mmol of NaCl and 1 mmol of Gd(NO3)3 (0.5 M), Yb(NO3)3 (0.5 M) and Er(NO3)3 (0.1 M) with the molar ratio of 78:20:2 were dissolved in 20 mL of ethylene glycol (EG) and stirred for 30 min. After capping with polyethyleneimine (PEI, branched 25 kDa), approximately 10 mL of EG containing 5.5 mmol of NH4F was then added to the above solution and stirred for another 30 min. The obtained mixture was then transferred into a 50 mL stainless Teflon-lined autoclave, which was sealed and kept at 190 °C for 24 h. After that, the autoclave was cooled to room temperature naturally and the reaction mixture was separated through centrifugation. Ethanol and DI-water were used to wash the precipitate several times and dried at 70 °C for 12 h to obtain the as-prepared samples. As a result, NaGdF4 with PEI as the surface modifier was prepared by the hydrothermal method. The synthesis of BaGdF5 is similar to the above procedure except for replacing NaCl with BaCl2. The zeta-potential for the NaGdF4 or BaGdF5 colloidal solution was measured to be around +23.6 mV and +31.5 mV respectively, indicating the successful conjugation of positively charged PEI on the surface of NPs.
3.3 Synthesis of Plk1 specific peptides
The three peptides were obtained by stepwise elongation of the peptide chain by the method outlined below. For peptide P1, 500 mg of the rink amide resin (0.45 mmol g−1 loading) was suspended in a 20% solution of piperidine in DMF, stirred for 20 min at room temperature and washed with DMF prior to subsequent steps. The first amino acid to be coupled, Fmoc-(Bn-p)-Thr-OH (420 mg, 4 equiv.), was dissolved in DMF and coupled to the resin in the presence of PyBOP (645 mg, 4 equiv.) and NMM (135 μL, 4 equiv.) with the use of microwave chemistry as described previously.15,32 Other Fmoc amino acid derivatives, Fmoc-Ser(tBu)-OH (420 mg, 4 equiv.), Fmoc-His(Trt)-OH (438 mg, 4 equiv.), Fmoc-Leu-OH (350 mg, 4 equiv.), and Fmoc-Pro-OH (320 mg, 4 equiv.), were connected to the resin using an analogous synthetic strategy. Following the final removal of the Nα-Fmoc group, the peptide-resin was washed with DMF. The free peptide was obtained by cleavage through the use of 9 mL of TFA in the presence of 750 μL of water and 750 μL of TIPS according to the standard procedure. After cleavage, the peptide was purified by preparative reverse phase HPLC. The main peptide-containing fractions were collected and lyophilized. The purity of all final products was confirmed by analytical reverse phase HPLC. P2 and P3 were obtained and purified by the same methodology as the preparation of P1. P1: PLHSpT MALDI-TOF HRMS ([M + 1]+, m/z): calcd for C24H42N8O10P, 633.27; found for [M + H]+, 633.26; P2: PLHSD MALDI-TOF HRMS ([M + 1]+, m/z): calcd for C23H37N8O8, 568.37; found for [M + H]+, 568.45; P3: GGPLHSpT MALDI-TOF HRMS ([M + 1]+, m/z): calcd for C28H47N10O12P, 746.31; found for [M + H]+ 747.330.3.4 Bio-conjugation of Plk1 specific peptides onto NaGdF4 or BaGdF5
50 mg PEINaGdF4 nanocrystals were added to 744 mg DCC, 200 mg acrylic acid and 266 NHS in DMF to react at room temperature overnight. The excess DCC, acrylic acid and NHS were removed by washing with DMF and methanol. The precipitate was then vacuum dried. 30.8 mg DCC, 15.9 mg NHS and 40 mg 3-(tritylthio)propanoic acid were added to 50 mg Plk1 peptide in DMF and stirred at room temperature overnight. Excess DCC, NHS and 3-(tritylthio)propanoic acid were removed by washing with DMF and methanol. The resin of the modified Plk1 peptide was removed by reacting with TFA, TIS and de-ionized water in the ratio of 90:5:5 for 4 hours at room temperature. Finally, the modified NaGdF4 nanocrystal and Plk1 peptide reacted with each other in PBS buffer at pH 7.4 at room temperature overnight. The product was washed with water and methanol three times and vacuum dried overnight. The bioconjugation of Plk1 specific peptides (Pn) onto BaGdF5 is similar to that onto NaGdF4 except for replacing NaGdF4 with BaGdF5. The characterization of the NaGdF4–Pn or BaGdF5–Pn was performed with FTIR transmittance and zeta potential analysis.
3.5 Characterization
Powder X-ray diffraction (XRD) patterns of the as-prepared NaGdF4 or BaGdF5 and NaGdF4–Pn or BaGdF5–Pn were recorded using a Rigaku smart lab 9 kW (Rigaku Japan) with Cu-Kα radiation (λ = 1.5406 Å). The step scan covered the angular range from 25° to 60° in steps of 0.02°. The morphology of the as-prepared samples was characterized by using a Tecnai G2 20 S-TWIN Transmission Electron Microscope operated at 200 kV. A fourier transform infrared (FT-IR) spectrometer (Nicolet Avator 630) was used to record the infrared spectra of the samples by using a KBr pellet technique. The powder materials were pressed into a tungsten mesh grid and installed in situ FT-IR transmittance cell, and the samples were degassed in a vacuum system with a residual pressure of less than 3 × 10−4 Torr at room temperature. UC spectra were recorded using an FLS920P Edinburgh analytical instrument apparatus equipped with a 980 nm diode laser as an UC pump source.3.6 Cell culture
Human nasopharyngeal carcinoma (HK-1) cells and cervical carcinoma (HeLa) cells were cultured in DMEM medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and antibiotics (penicillin 50 μg mL−1; streptomycin 50 μg mL−1). Human liver-derived (QSG-7701) cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Human normal lung fibroblast (MRC-5) cells were grown in MEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. All the cells were incubated at 37 °C in a humidified environment with 5% CO2.3.7 Study of cellular uptake of NaGdF4–Pn or BaGdF5–Pn by ICP-MS
To measure the intracellular concentration of the NaGdF4–Pn or BaGdF5–Pn. 1 × 105 cells were plated in each well and incubated with the complex for at 100 μg mL−1, which was the same as in vitro imaging. After co-incubation, the cell culture medium containing NaGdF4–Pn or BaGdF5–Pn was removed and the exposed cells were further washed with 1 mL PBS 3 times to remove NaGdF4–Pn or BaGdF5–Pn adhering to the outer cell membrane and the bottom of the confocal dishes. Then the cells were trypsinized and dispersed into 1 mL of culture medium. The exposed cells were collected by centrifugation at 4000 rpm and the cell pellet was digested in 100 μL of concentrated HNO3 (69%) at 70 °C for 3 hours. The cellular uptake of NaGdF4–Pn or BaGdF5–Pn was determined using an Agilent 7500 series of inductively coupled plasma mass spectroscopy (ICP-MS). All ICP experiments were performed in triplicate and the values obtained were averaged. The concentration of Gd per cell was calculated by determining the concentration of Gd in the cell lysate by ICP-MS and then dividing it by the number of cells which counted using a haematocytometer.3.8 In vitro bio-imaging
To investigate the suitability of the obtained and Plk1 specific peptides coated UCNPs as bio-probes, imaging of HeLa cells (1 × 105) was conducted. After the incubation with NaGdF4–Pn or BaGdF5–Pn for 24 hours, the cells were washed by PBS 3 times before imaging. The in vitro imaging was performed on a confocal laser scanning microscope, Leica TCS SP5, equipped with a Ti:sapphire laser (Libra II, coherent). The samples were excited by a 980 nm wavelength laser.3.9 Plk1 GFP co-staining in vitro bio-imaging
To validate the binding between NaGdF4–Pn or BaGdF5–Pn and Plk1, GFP co-staining experiment was carried out. Plk1 full length cDNA was PCR amplified from pOTB7-Plk1 and inserted into multiple cloning sites (MCS) of pEGFP-C3 (CLONETECH, #6082-1) using restriction digestion sites of XhoI/EcoRI for GFP-Plk1 expression. The positive recombinant was selected through kanamycin proved by sequencing. Plasmids were purified using a StarPrep Plasmid Miniprep Kit (Genstar). For transfection, a 3.5 cm dish of HeLa cells (with 70–80% confluence) was transfected with 8 μg DNA using lipofectamine2000 according to the manufacturer instructions. Then, 500 μL of the lipofectamine 2000 and plasmid were added to the confocal dish with 500 μL cell medium. After incubation for 8 hours, 100 μg mL−1 of NaGdF4–Pn or BaGdF5–Pn was added and incubated for 24 hours. The imaging procedures and parameters were the same as given in the previous in vitro bio-imaging section.3.10 MTT inhibition rate assay
1 × 105 of HK-1, HeLa, QSG-7701 and MRC-5 were treated with NaGdF4-PEI or BaGdF5-PEI and NaGdF4–Pn or BaGdF5–Pn for 24 hours. Then the cells were treated with nanoparticles at 4 concentrations (100 μg mL−1, 50 μg mL−1, 20 μg mL−1 and 5 μg mL−1). The cell monolayers were rinsed with phosphate buffer saline (PBS) once and incubated with 0.5 mg mL−1 MTT solution. The cellular inhibitory potency of the nanoparticles was examined by the formation of formazan after addition of MTT (3-(4,5-dimethylthiazol-2-yl)-2 and 5-diphenyltetrazolium bromide) for 4 hours to allow formazan production during cell metabolism. After that, formazan was fully dissolved in DMSO through oscillating. Finally, the absorbance of solution was measured with a Biotek Power wave xsMicroplate Reader at a wavelength of 570 nm and 690 nm.3.11 Flow cytometry
106 cells were harvested by trypsine digestion, washed twice in PBS, and then fixed in 70% ethanol for 2 hours at 4 °C. After removal of the ethanol, cells were resuspended in PBS and then subjected to PI staining (1% triton-100, 50 μg mL−1 RNase A, 20 μg mL−1 propidium iodide) for 40 minutes at 37 °C. Cell phase distributions were analyzed in BD FACS Calibur and the results were processed by Flowjo 7.6.1, shown as the offset histogram and column.3.12 Antibodies and western blotting
Anti-β-actin (#66009-1-Ig) antibodies were purchased from Proteintech Group, Inc. Anti-CDK1 (#9116) and anti-Plk1 (#4535) antibodies were from Cell Signaling Technology. HeLa cells in a 12 well-plate were treated with nanoparticles (1, 10, 50, 100 μg mL−1 each) for 24 hours, and then rinsed in PBS twice after the culture medium was discarded. Direct lysis in 50 mM Tris–HCl, pH 6.8, 1% SDS, 10% glycerol and thorough denaturation in a boiling water bath were followed. The supernatant was collected as a clear lysate after centrifugation steps. Equal amounts of total proteins were loaded onto the 10% nitrocellular membranes, which was followed by western blotting. All the results finally were analyzed using a Gel-Pro analyzer and GraphPad Prism 5 software.3.13 Pull down assay
Five 10 cm dishes of cells were lysed in RIPA buffer (50 mM Tris pH = 7.4, 150 mM NaCl, 1% NP-40, phenylmethanesulfonyl fluoride (PMSF) 1 mM, cocktail protease inhibitors) for 15 minutes on ice, and then centrifuged at 13000 rpm for 20 minutes. The clear supernatant was collected and the total 5 mL cellular lysate was divided into eight samples that were subjected to incubation with the NaGdF4 or BaGdF5 nanoparticle series (100 μg each) at 4 °C for 2 hours. Nanoparticle precipitated proteins were collected by short centrifugation and washed with RIPA buffer three times to remove unbound proteins. Finally, the pull-down proteins underwent SDS-PAGE separation followed by antibody detection.
3.14 Animals and ethics
BALB/c nude mice were provided by Guangdong Medical Laboratory Animal Center, with the licensing number of SCXK-2008-0002 approved by Guangdong Science and Technology Department. Mice were kept in the isolated bio-safety facility for specific pathogen-free animals. All animal feeding and experiments were performed according to the Procedures for the Care of Laboratory Animals approved by the Shenzhen Institute of The Hong Kong Polytechnic University (with Use Permit of Experiment Animal license SCXK-2010-0062).3.15 Mouse xenograft tumor inhibition and ICP-MS bio-distribution analysis
For tumor inhibition assays, human HeLa cells were trypsinized, harvested and resuspended in DMEM. 1 × 106 cells in 100 μL DMEM were injected s.c. into the flanks of the mice (6 weeks old). After 14 days when the tumors were about 150 mm3, the animals were randomized into groups for experiments. NaGdF4 and BaGdF5 series of nanoparticles of Plk1 (500 μg for a tumor) were injected around the tumors and the mice were kept for another 7 days. Finally, the tumors were extracted and imaged. For ICP-MS bio-distribution analysis, 200 μg nanoparticles were i.p. injected into the mice, and the tissues as well as urine after 24 hour post-injection were collected. Tissues were digested with nitric acid and subjected to ICP-MS for measuring the concentrations of Gd.4. Conclusions
Plk1 has emerged as a promising target for the development of new anti-cancer agents. The polo box domain (PBD) of Plk1 provides an excellent target for the development of molecules to inhibit Plk1 activity and as such we have rationally designed multifunctional lanthanide-doped upconversion nanomaterials to exploit this. Specifically our aim was to design novel nanomaterials that could deliver Plk1 specific peptides into cancer cells causing Plk1 inhibition while simultaneously allowing direct NIR imaging and monitoring. NaGdF4:Yb3+, Er3+ (NaGdF4) and BaGdF5:Yb3+, Er3+ (BaGdF5) nanoparticles of two different sizes (60 nm and 10 nm, respectively) were prepared and then conjugated with Plk1 specific peptides (–P1 = PLHSpT, –P2 = PLHSD, and –P3 = GGPLHSpT) to generate novel nanomaterials. Systematic and comparative studies of the nanomaterials prepared were carried out, and structural morphology, phase composition, upconversion photophysical properties, cellular uptake efficiency, cytotoxicity, tumor selectivity, and Plk1 binding affinity and inhibitory potency were also investigated in detail. The experimental results obtained showed that several of the nanomaterials prepared could achieve our original dual-function aim of anti-cancer activity and NIR imaging. Of the novel bio-conjugated nanomaterials investigated in our study, NaGdF4–P1, demonstrated considerable promise as a system for further development. NaGdF4–P1 emerged as the best candidate for Plk1 binding and imaging and it could effectively exert cell cycle G2/M arrest and thus tumor inhibition both in vitro and in vivo. It is also worth noting that NaGdF4–P1 is selective in its mode of action and that no anti-growth effects on the normal cells lines used in this study were observed. In summary, this study represents one of few examples where systems have been developed that are capable of both Plk1 inhibition and simultaneously direct NIR imaging and monitoring of cancer cells.Acknowledgements
This work was funded by grants from The Hong Kong Research Grants Council (HKBU 203013), ESPRC, Hong Kong Baptist University, The Hong Kong Polytechnic University and Durham University.Notes and references
- B. T. Martin and K. Strebhardt, Cell Cycle, 2006, 5, 2881 CrossRef CAS.
- M. A. Van Vugt and R. H. Medema, Oncogene, 2005, 24, 2844 CrossRef CAS PubMed.
- X. Liu, M. Lei and R. L. Erikson, Mol. Cell. Biol., 2006, 26, 2093 CrossRef CAS PubMed.
- B. Vogelstein, N. Papadopoulos, V. E. Velculescu, S. Zhou, L. A. Diaz Jr and K. W. Kinzler, Science, 2013, 339, 1546 CrossRef CAS PubMed.
- S. M. Lens, E. E. Voest and R. H. Medema, Nat. Rev. Cancer, 2010, 10, 825 CrossRef CAS PubMed.
- K. Strebhardt, L. Kneisel, C. Linhart, A. Bernd and R. Kaufmann, J. Am. Med. Assoc., 2000, 283, 479 CrossRef CAS.
- V. Maire, F. Némati, M. Richardson, A. Vincent-Salomon, B. Tesson, G. Rigaill, E. Gravier, B. Marty-Prouvost, L. De Koning, G. Lang, D. Gentien, A. Dumont, E. Barillot, E. Marangoni, D. Decaudin, S. Roman-Roman, A. Pierré, F. Cruzalegui, S. Depil, G. C. Tucker and T. Dubois, Cancer Res., 2013, 73, 813 CrossRef CAS PubMed.
- X. Liu and R. L. Erikson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5789 CrossRef CAS PubMed.
- R. Lan, G. Lin, F. Yin, J. Xu, X. Zhang, J. Wang, Y. Wang, J. Gong, Y. Ding, Z. Yang, F. Lu and H. Zhang, Lab. Invest., 2012, 92, 1503 CrossRef CAS PubMed.
- K. V. Gleixner, V. Ferenc, B. Peter, A. Gruze, R. A. Meyer, E. Hadzirjusufovic, S. Cerny-Reiterer, M. Mayerhofer, W. F. Pickl, C. Sillaber and P. Valent, Cancer Res., 2010, 70, 1513 CrossRef CAS PubMed.
- J. Q. Shi, K. Lasky, V. Shinde, B. Stringer, M. G. Qian, D. Liao, R. Liu, D. Driscoll, M. T. Nestor, B. S. Amidon, Y. Rao, M. O. Duffey, M. G. Manfredi, T. J. Vos, N. D'Amore and M. L. Hyer, Mol. Cancer Ther., 2012, 11, 2045 CrossRef CAS PubMed.
- F. Liu, J. E. Park, W. J. Qian, D. Lim, M. Gräber, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr, Nat. Chem. Biol., 2011, 9, 595 CrossRef PubMed.
- S. M. Yun, T. Moulaei, D. Lim, J. K. Bang, J. E. Park, S. R. Shenoy, F. Liu, Y. H. Kang, C. Liao, N. K. Soung, S. Lee, D. Y. Yoon, Y. Lim, D. H. Lee, A. Otaka, E. Appella, J. B. McMahon, M. C. Nicklaus, T. R. Burke Jr, M. B. Yaffe, A. Wlodawer and K. S. Lee, Nat. Struct. Mol. Biol., 2009, 16, 876 CAS.
- S. M. Kim, S. Yoon, N. Choi, K. S. Hong, R. N. Murugan, G. Cho and E. K. Ryu, Biomaterials, 2012, 33, 6915 CrossRef CAS PubMed.
- I. R. Ruttekolk, F. Duchardt, R. Fischer, K. H. Wiesmuller, J. Rademann and R. Brock, Bioconjugate Chem., 2008, 10, 2081 CrossRef PubMed.
- J. Yuan, A. Hoerlin, B. Hock, H. J. Stutte, H. W. Ruebsamen and K. Strebhardt, Am. J. Pathol., 1997, 4, 1165 Search PubMed.
- Z. H. Chen, R. J. Zhao, R. H. Li, C. P. Guo and G. J. Zhang, PLoS One, 2013, 8, e53291 CAS.
- H. Li, C. F. Chan, W. L. Chan, S. Lear, S. L. Cobb, N. Mak, T. C. Lau, R. Lan, W. K. Wong and K. L. Wong, Org. Biomol. Chem., 2014, 12, 5876 CAS.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 34, 12608 CrossRef PubMed.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 8, 7200 CrossRef PubMed.
- P. Escribano, B. Julian-Lopez, J. Planelles-Arago, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23 RSC.
- S. Zeng, M. K. Tsang, C. F. Chan, K. L. Wong and J. Hao, Biomaterials, 2012, 33, 9232 CrossRef CAS PubMed.
- R. Naccache, P. Chevallier, J. Lagueux, Y. Gossuin, S. Laurent, L. Vander Elst, C. Chilian, J. A. Capobianco and M. A. Fortin, Adv. Healthcare Mater., 2013, 2, 1478 CrossRef CAS PubMed.
- N. Bogdan, E. Martín Rodríguez, F. Sanz-Rodríguez, M. C. Iglesias-de la Cruz, A. Juarranz de la Fuente, D. Jaque, J. GarcíaSolé and J. A. Capobianco, Nanoscale, 2012, 4, 3647 RSC.
- Q. Liu, W. Feng, T. Yang, T. Yi and F. Li, Nat. Protoc., 2013, 8, 2033 CrossRef CAS PubMed.
- W. Feng, C. Han and F. Li, Adv. Mater., 2013, 25, 5287 CrossRef CAS PubMed.
- L. D. Carlos, R. A. S. Ferreira, V. Z. Bermudez, B. Julian-Lopez and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC.
- M. L. Debasu, D. Ananias, S. L. C. Pinho, C. F. G. C. Geraldes, L. D. Carlos and J. Rocha, Nanoscale, 2012, 4, 5154 RSC.
- J. Wu, Q. Tian, H. Hu, Q. Xia, Y. Zou, F. Li, T. Yi and C. Huang, Chem. Commun., 2009, 27, 4100 RSC.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 13, 5029 CrossRef PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652 RSC.
- C. F. Chan, M. K. Tsang, H. Li, R. Lan, F. L. Chadbourne, W. L. Chan, G. L. Law, S. L. Cobb, J. Hao, W. T. Wong and K. L. Wong, J. Mater. Chem. B, 2014, 2, 84 RSC.
- P. Loyera, J. H. Trembleya, R. Katonac, V. J. Kidda and J. M. Lahti, Cell. Signalling, 2005, 17, 1033 CrossRef PubMed.
- E. Pretsch, P. Bühlmann and C. Affolter, Structure Determination of Organic Compounds, Springer, Berlin Heidelberg, 2000, p. 250 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb02104e |
| This journal is © The Royal Society of Chemistry 2015 |