
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrating silver compounds and nanoparticles into ceria nanocontainers for antimicrobial applications†
J.
Gagnon
*a,
M. J. D.
Clift
b,
D.
Vanhecke
b,
D. A.
Kuhn
b,
P.
Weber
a,
A.
Petri-Fink
b,
B.
Rothen-Rutishauser
b and
K. M.
Fromm
*a
aDepartment of Chemistry and Fribourg Center for Nanomaterials, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: Katharina.Fromm@unifr.ch; Jacinthe.Gagnon@iaf.inrs.ca; Fax: +41 26 3009738; Tel: +41 26 3008732
bAdolphe Merkle Institute and Fribourg Center for Nanomaterials, University of Fribourg, Route de l'Ancienne Papeterie, P.O. Box 209, 1723 Marly, Switzerland
First published on 9th January 2015
Abstract
Silver compounds and nanoparticles (NPs) are gaining increasing interest in medical applications, specifically in the treatment and prevention of biomaterial-related infections. However, the silver release from these materials, resulting in a limited antimicrobial activity, is often difficult to control. In this paper, ceria nanocontainers were synthesized by a template-assisted method and were then used to encapsulate silver nitrate (AgNO3/CeO2 nanocontainers). Over the first 30 days, a significant level of silver was released, as determined using inductively coupled plasma optical emission spectroscopy (ICP-OES). A novel type of ceria container containing silver NPs (AgNP/CeO2 containers) was also developed using two different template removal methods. The presence of AgNPs was confirmed both on the surface and in the interior of the ceria containers by X-ray diffraction (XRD), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). Upon removal of the template by calcination, the silver was released over a period exceeding three months (>90 days). However, when the template was removed by dissolution, the silver release was shortened to ≤14 days. The antimicrobial activity of the silver-containing CeO2 containers was observed and the minimum bactericidal concentration (MBC) was determined using the broth dilution method. Investigation on human cells, using a model epithelial barrier cell type (A549 cells), highlighted that all three samples induced a heightened cytotoxicity leading to cell death when exposed to all containers in their raw form. This was attributed to the surface roughness of the CeO2 nanocontainers and the kinetics of the silver release from the AgNO3/CeO2 and AgNP/CeO2 nanocontainers. In conclusion, despite the need for further emphasis on their biocompatibility, the concept of the AgNP/CeO2 nanocontainers offers a potentially alternative long-term antibactericidal strategy for implant materials.
1. Introduction
Implant-associated bacterial infections remain a significant issue. In the U.S.A., 4.3% of implants inserted into humans become infected.1,2 Bacteria that adhere at the site of implantation can create a biofilm,3 competing with the host tissues for integration with the implant as well as proper host cell integration, both essential aspects for the biocompatibility of any implant. Once bacteria adhere to a biomaterial surface, they are very difficult to be removed by the host immune system or displaced by the host cells.4 Thus preventing bacterial contamination during and after surgery improves the host tissue integration of the implant. It has been demonstrated that the decisive period for preventing bacterial invasion relates to only 6 hours after surgery.5 Infections can lead to additional pain for the patient as well as malfunction of the implant, which in turn leads to a need for its replacement, or in extreme cases to amputation or even death of the patient. From an economical point of view, bacterial infections also create considerable additional medical costs which could be avoided if bacterial invasion is prevented.2 Moreover, implants are being increasingly used and at the same time bacteria are becoming more resistant to conventional antibiotics.6 Therefore, there is an essential need for new developments in preventing and curing implant-related bacterial infections.In this context, silver compounds and nanoparticles (NPs) are gaining more interest from the scientific community as a possible replacement for commonly used antibiotics, such as penicillin. Silver-containing materials may be used to coat implants in order to prevent bacterial biofilm formation. However, these compounds may be too soluble and even promote adverse effects towards the host if the silver release is not controlled.7 Ideally, the release of silver ions should be able to kill bacteria without affecting biocompatibility. The release of silver ions, however, largely depends on the environment (i.e. pH, ionic strength, organic matter) and is therefore difficult to control in the human body. As we have shown in previous reports, it is possible to design the light stability and solubility of silver coordination polymers as efficient antimicrobial coatings for implant materials.8,9 However, with these compounds the silver release occurs permanently, irrespective of the presence of bacteria.
Another strategy to control the release of silver is the integration of silver NPs (AgNPs) into a biocompatible coating,10 or directly into the biomaterials, such as in bone cements.11 In addition to such outlooks, encapsulation of AgNPs can be advantageous in order to control and prolong the release of different compounds,12,13 in turn increasing the stability and biocompatibility of silver drugs. Encapsulation is also of great interest for many medical applications, such as targeted drug delivery.14 For example, it has been shown that encapsulated drugs are thermally more resistant.13,15
Ceria (CeO2) is an excellent material for encapsulating drugs since it is highly stable for long time periods in aqueous solutions and elicits limited adverse biological effects.16,17 It has been demonstrated that CeO2-NPs can elicit certain antioxidant properties that protect cells against the onset of oxidative stress.16,18 The aim of the present study, therefore, was to determine if it were possible to specifically encapsulate silver compounds and/or NPs within ceria-based container systems in order to slow down the silver release and thus making silver coatings increase their active antimicrobial period. Two approaches were envisaged to fill nanocontainers with silver compounds. A first method was to make hollow spheres and then fill them with the desired compound by soaking, resulting in the AgNO3/CeO2 nanocontainers. Secondly, it is also possible to include the AgNPs during the synthesis of the nanocontainers. Both methods are hereby described with the resultant material's characterization. The silver release was measured over a period of 3 months. The antimicrobial activity and the cytotoxicity of the novel AgNO3/CeO2 and AgNP/CeO2 containers were evaluated in prokaryotic and eukaryotic systems respectively.
2. Experimental
2.1 Materials and reagents
All materials were obtained from Sigma-Aldrich, Switzerland. Cerium(III) acetylacetonate (Ce(acac)3), polyvinylpyrrolidone (PVP, average molecular weight: 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), potassium persulfate (KPS), silver(I) oxide (Ag2O), silver(I) nitrate (AgNO3), sodium dodecyl sulfate (SDS), urea and ethanol were of analytical grade and used without further purification. Styrene and water were doubly distilled prior to use. Lysogeny broth (LB) medium and agar plates were obtained “ready-for-use”.
000), potassium persulfate (KPS), silver(I) oxide (Ag2O), silver(I) nitrate (AgNO3), sodium dodecyl sulfate (SDS), urea and ethanol were of analytical grade and used without further purification. Styrene and water were doubly distilled prior to use. Lysogeny broth (LB) medium and agar plates were obtained “ready-for-use”.
2.2 Polystyrene nanosphere (PS NS) synthesis
Anionic PS NSs were synthesized via emulsion polymerization.19 The reaction mixture was prepared by mixing styrene (3.70 g, 35.5 mmol), KPS (0.30 g, 1.1 mmol), and SDS (0.09 g, 0.3 mmol) in water (250 mL). The solution was stirred at 80 °C under argon for 42 hours. The PS NSs were then washed three times by centrifugation (30 min at 10000 rpm). The supernatant was subsequently discarded and the pellets re-suspended in water.
2.3 CeO2 container synthesis
The CeO2 containers were formed via a sol–gel method:13 PS NSs (0.3 g), Ce(acac)3 (0.7 g, 1.6 mmol), PVP (0.3 g, 0.0075 mmol), urea (0.3 g, 5.0 mmol) and water (40 mL) were mixed for 5 minutes. The reaction was then allowed to proceed at 100 °C for 4 to 5 days without any agitation. Urea was used to prevent aggregation but had no effect on the reaction.13 The suspension was centrifuged for 30 min at 15000 rpm. The supernatant was discarded and the pellets were re-suspended in water using sonication. This washing step was repeated 3 times and the PS NSs coated with CeO2 were dried in an oven at 40 °C for 1 day. Hollow CeO2 containers were obtained by removing the PS core by calcination in air in an oven at 600 °C for 4 hours.
2.4 Encapsulation of AgNO3 in CeO2 containers
AgNO3 was encapsulated into CeO2 containers using a method adapted from Kartsonakis et al.13 Briefly, CeO2 containers were placed in a closed vial and submitted to a vacuum. They were then rapidly immersed in a saturated solution of AgNO3 in ethanol and the mixture was stirred at room temperature for 2 hours in the closed vial. The mixture was centrifuged and the supernatant was discarded. The AgNO3/CeO2 containers were washed three times with fresh ethanol and collected by centrifugation (10 min at 10000 rpm). They were then dried overnight at room temperature.
2.5 Silver nanoparticle (AgNP) synthesis
The method used to synthesize AgNPs was adapted from Evanoff and Chumanov.20 A 250 mL flask was filled with PS NSs (0.140 g) and silver(I) oxide (0.045 g, 0.2 mmol) in water (60 mL). The flask was then placed into an autoclave. The autoclave was filled with 10 bar H2 and the reaction was allowed to proceed at 70 °C for 4 hours with constant stirring. After cooling, the mixture was centrifuged (15 min at 15000 rpm). The supernatant was discarded and the pellets were re-suspended in water (5 mL).
2.6 Encapsulation of AgNPs in PS NSs
The method used to encapsulate the AgNPs into the PS beads was adapted from Kumbhar and Chumanov.21 After the AgNP synthesis, the mixture (2 mL) was added to acetone (5 mL) and sonicated for 4 hours at room temperature. The resulting mixture was centrifuged (15 min at 15000 rpm) and the pellets were resuspended in water (5 mL).
2.7 AgNP/CeO2 container synthesis
The PS NSs encapsulating AgNPs were coated with CeO2, similar to the synthesis of the CeO2 nanocontainers: the obtained AgNP/PS spheres (0.12 g), Ce(acac)3 (0.50 g, 1.1 mmol), PVP (0.300 g, 0.0075 mmol), urea (0.20 g, 3.3 mmol) and water (30 mL) were mixed for 5 min. Then the reaction was allowed to proceed at 100 °C for 4 to 5 days without any agitation. The suspension was centrifuged for 30 min at 15000 rpm. The supernatant was discarded and the pellets were re-suspended in water using sonication. The washing step was repeated three times and the AgNP/PS NSs coated with CeO2 were dried in an oven at 40 °C for at least one day. Hollow AgNP/CeO2 containers were obtained by calcination in air in an oven at 600 °C for 4 hours, or by many successive dissolution treatments in boiling xylene containing triethylamine for 12 hours.12 Formed containers were then allowed to dry in an oven at 40 °C.
2.8 Characterization
The synthesized particles were visualized by transmission electron microscopy (TEM) using a FEI/Philips CM-100 Biotwin microscope. The TEM grids used were 300 square mesh copper TEM grids covered by a carbon film, provided by Electron Microscopy Sciences, USA. The particles were also visualized by scanning electron microscopy (SEM) using a FEI XL 30 Sirion FEG. SEM samples were prepared by spreading them on a carbon tape glued on a SEM holder. All images were obtained without sputter coating pretreatment. Images were obtained at different voltages using the Everhart–Thornley detector (EDT) or solid state detector (SSD) either in contrast or back-scattering mode. X-ray diffraction (XRD) patterns were measured using a STOE transmission X-ray powder diffraction system. Fourier transform infrared (FTIR) spectra were obtained with a Bruker Tensor 27 FTIR and a MKII Golden Gate Single Attenuated Total Reflection System. Thermogravimetric analysis (TGA) was performed in a Mettler Toledo SDTA/TGA 851e ids. The silver concentration was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Perkin Elmer Optima 7000DV ICP-OES. To assess the morphology of the A549 cells following exposure to the different CeO2 nanocontainers, an inverted Zeiss laser scanning microscope (LSM) 710 Meta, Switzerland, was used.2.9 Silver release
In order to determine the silver release from AgNO3/CeO2 nanocontainers and AgNP/CeO2 containers, 4 duplicates (75 mg) of each sample were prepared. Each duplicate was placed in a well, covered with distilled water (1.2 mL) and left at room temperature in the dark. At certain time intervals over a 3 month period, a fraction of the supernatant (0.500 mL) was collected and added to 1% nitric acid solution (5 mL) and was then ready for silver concentration determination. Freshly distilled water (0.500 mL) was placed back into the well. The silver concentration in each aliquot was determined by ICP-OES.2.10 Antibacterial tests
The Escherichia coli (E. coli) K-12 bacterial strain was used for both zone of growth inhibition determination and minimum bactericidal concentration (MBC) determination.:1 ratio. A sterility control, composed of a fresh LB medium, was also prepared. A total of 100 μL of each new suspension was then placed on a LB agar plate and incubated overnight at 37 °C. The following day, the concentration at which total inhibition of bacterial growth occurred was recorded.
2.11 Cell culture and exposure
The A549 cell line, an adenocarcinomic human alveolar Type II epithelial cell culture (ATCC, USA), was cultured as previously described by Rothen-Rutishauser et al.26The experiments were performed on BD Falcon 2-chamber culture slides. Each well had a growth surface area of 4 cm2. The day before exposure, nanocontainer samples of 58 mg and 174 mg were suspended in a cell culture medium (2 mL) and added to a well of the chamber slide. The samples were then incubated overnight at 37 °C and 5% CO2. The following day, the supernatant was removed and replaced with 2 mL of culture medium containing 0.5 × 106 cells per mL. The samples were then incubated at 37 °C and 5% CO2 for 4 and 7 days. After each exposure period, the supernatant was removed and was used to determine the level of epithelial cell cytotoxicity.
2.12 Epithelial cell cytotoxicity
Cytotoxicity was determined by assessment of cell membrane damage (permeability) via measuring the release of the cytosolic enzyme lactate dehydrogenase (LDH) in the cell supernatant. LDH is the enzyme responsible for the conversion of pyruvate into lactate in the glycolysis pathway and is present in the cytosol of eukaryotic cells. Upon disruption to the cell membrane LDH can be released from the cytosol and subsequently measured in the cell culture medium as a measure of cell damage (i.e. cytotoxicity). Following exposure to 58 and 174 mg per well for 4 and 7 days at 37 °C, 5% CO2, LDH levels were determined using a diagnostic kit (Roche Applied Sciences, Switzerland). As a positive control, 0.2% TritonX-100 in PBS was used. A negative control of the cell culture medium only was also performed along with each sample series. All analysis was repeated in triplicate (n = 3).2.13 Epithelial cell morphology
Initially, following the exposure period, cells were fixed and labelled as previously described by Lehmann et al.27 Briefly, samples were labelled with a 250 μL mix of a 1:50 dilution of phalloidin-rhodamine (label for cell cytoskeleton) and 1:100 dilution of 4′,6-diamidino-2-phenylindole (DAPI stain for cell nuclei). Coverslips were then inverted and mounted onto microscope slides using Glycergel (Dako, Carpinteria, USA) and imaged by LSM.
2.14 Statistical and data analysis
The results from the silver release experiments and antibacterial tests are presented as the mean ± standard error of the mean. All cytotoxicity data are also presented as the mean ± standard error of the mean. This latter dataset was found to be normally distributed (data not shown) and so a parametric two-way ANOVA was conducted with subsequent Tukey's post-hoc test (SPSS, IBM, USA). Datasets were considered significant if p < 0.05. Cytotoxicity analysis was performed in triplicate (n = 3), while silver release experiments were performed in 4 duplicates (n = 4).3. Results and discussion
3.1 Encapsulation of AgNO3 into ceria nanocontainers
In a first attempt, CeO2 nanocontainers were prepared by a procedure similar to that described by Kartsonakis et al.,19 using PS NSs as a template. The slightly adapted synthesis route yields white PS NSs of 220 ± 15 nm diameter. After coating with CeO2 and removing the core by calcination at 600 °C, pale yellow nanocontainers with diameters between 175 and 200 nm and shell wall thicknesses of 12 to 20 nm were obtained. To introduce AgNO3 into the containers, the nanocontainers were exposed to a vacuum in order to remove the air within the sample and were then immersed in a saturated solution of AgNO3 in ethanol for 2 hours. In order to test if AgNO3 was integrated into the containers, thermogravimetric analysis (TGA) was performed on the obtained grey CeO2 nanocontainers previously dried in an oven at 60 °C under a vacuum and on free AgNO3 (Fig. 1). CeO2 containers alone (Fig. 1A) did not have any significant weight loss. Free AgNO3 (Fig. 1B) underwent thermal decomposition at 370–430 °C. The AgNO3-containing CeO2 nanocontainers (Fig. 1C) showed a weight loss not only in the same temperature range, which corresponds to free AgNO3, but also at 30–220 °C, 220–290 °C and 780–900 °C. This first weight loss corresponds to desorption of physically adsorbed water. The weight loss at 220–290 °C corresponds to the encapsulated ethanol that was used as a solvent during the encapsulation. Such a difference in thermal decomposition of ethanol is due to the protection of the encapsulated ethanol in the CeO2 shell. The weight loss at 780–900 °C still remains uncertain, although there is convincing evidence that this could be due to the formation of AgNPs in the CeO2 shell from the reduction of AgNO3.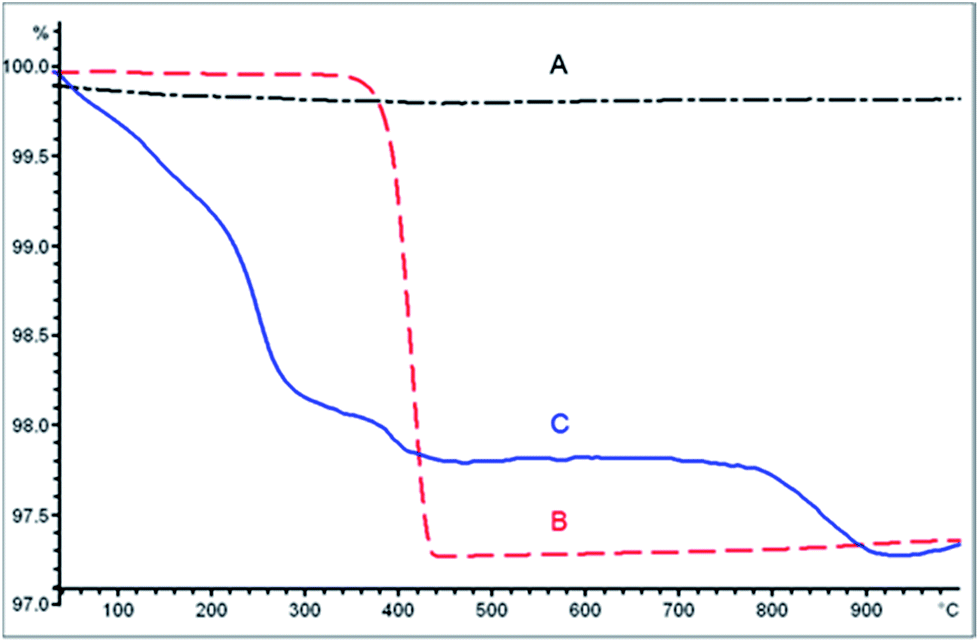 | ||
| Fig. 1 TGA of untreated CeO2 nanocontainers (A), free AgNO3 (B) and AgNO3/CeO2 nanocontainers (C). | ||
CeO2 can be photosensitized.28 It may thus be inferred that simple day light is responsible for the blackening of the CeO2 nanocontainers, reducing part of the Ag+ from AgNO3 to Ag(0). SEM images of these nanocontainers confirmed the presence of a few AgNPs (Fig. S1†). Silver release from these AgNO3/CeO2 nanocontainers showed an enhanced release during the first 5 days after immersion in water. Thereafter, the silver release considerably slowed and reached a threshold after approximately 30 days (Fig. 2). This release pattern is comparable to the silver release from many other materials.29,30
 | ||
| Fig. 2 Silver release as a function of immersion time in water for CeO2 containers containing AgNO3. Data are the % mean ± standard error of the mean compared to the positive control (n = 4). | ||
3.2 Synthesis of AgNP/CeO2 containers
In order to further control the release of silver ions from the containers, the potential to encapsulate AgNPs within a CeO2 nanocontainer shell was investigated. Depending on the pore size of the ceria container wall, it was expected that this process would be increasingly complex and so it was subsequently assumed that this encapsulation method would not be appropriate in this specific case. Thus, to include AgNPs in the nanocontainers during their synthesis an alternative method was used. Based on the work by Evanoff and Chumanov20 and Kumbhar and Chumanov,21 AgNPs of approximately 40 nm were synthesized via reduction with H2 in the presence of PS NSs before being embedded within them. The so-synthesized AgNPs easily adhered to the PS NSs (Fig. 3). After sonication in an acetone/water solution, they were all encapsulated inside the beads, as confirmed by TEM images (Fig. 4). Following encapsulation, the formed hybrid AgNP/PS spheres became polydispersed, and their diameters varied between 220 nm and >1 μm, as determined by TEM. This diverse size range was attributed to the incorporation of the AgNPs and the coalescence between the PS NSs. | ||
| Fig. 3 TEM images of AgNPs attached to PS NSs after synthesis. | ||
 | ||
| Fig. 4 TEM images of AgNPs encapsulated in the PS NSs after sonication demonstrating the full encapsulation of nanoparticles. | ||
The hybrid AgNP/PS spheres were then coated with CeO2 as described above. The X-ray diffraction (XRD) pattern (Fig. 5) showed that the particles were composed of ceria (JCPS no. 34-0394) and elemental silver (JCPS no. 04-0783), while polystyrene was not detected as a crystalline component. As determined by TEM and SEM, the diameter of the containers varied from 150 nm to 12 μm and the ceria shell thickness varied from 15 nm to 20 nm. Surprisingly, some AgNPs could also be seen on the outside surface of the CeO2 shells (Fig. 6B). In order to construct hollow spheres, the PS core was removed either by heating the spheres to 600 °C or via a dissolution process with xylene. After the PS removal, the AgNPs were still evidently present on the surface of the CeO2 nanocontainers (Fig. 6D and F). The difference of texture seen in these SEM images of the containers can be attributed to the treatment method used to remove the core material. When the polystyrene core was removed by calcination, the nanocontainer surface contained numerous pores and cracks, although the containers remained closed (Fig. 6C and D). Alternatively, when the PS core was removed by dissolution, the containers conserved their smooth surface (Fig. 6E and F). With both methods, the polystyrene core was removed, as shown by the loss of bands corresponding to the polystyrene on the IR spectra (Fig. S2†).
 | ||
| Fig. 5 XR diffractogram of AgNP/CeO2 containers. Red lines and green stars correspond to the peak positions of ceria and silver XRD patterns respectively. | ||
 | ||
| Fig. 6 TEM images (A, C and E) and SEM images (B, D and F) of silver nanoparticles encapsulated in the PS NSs and coated with CeO2 before the removal of the polystyrene core (A and B), and after the removal of the core via calcination (C and D) and via dissolution in xylene (E and F). Silver nanoparticles can be observed in all images. In SEM images, they clearly appear on the surface of the CeO2 coating, as well as in the cavity of the broken containers (F). | ||
Open containers could be observed in only a few samples. SEM analysis clearly showed that the AgNPs were also present inside the container cavity (Fig. 6F), suggesting that the AgNPs are indeed well integrated within the ceria shell. The mechanism by which the AgNPs move from the container cavity to the outside of the ceria shell is still not fully understood. However, it can be hypothesized that by the diffusion process during the coating with CeO2, or indeed the transport process, which involves the extraction of the polystyrene core out of the containers, be it either by heating or dissolution, may also entrain partial transport of the AgNPs to the outer side of the containers.
The silver release from these hybrid AgNP/CeO2 containers was determined by ICP-OES. In contrast to AgNO3/CeO2 nanocontainers (Fig. 2), the silver release from AgNP/CeO2 containers prepared by calcination was much slower (Fig. 7). The AgNP/CeO2 containers obtained by calcination therefore enabled a silver release over a period of at least 3 months. Even after this extended period of time, silver release was still found to be considerable. It is well known that silver ions, even at low concentrations, have an enhanced antimicrobial activity,30 which suggests that this newly designed silver-containing material could potentially prevent bacterial invasion during the most important period following implant surgery.
 | ||
| Fig. 7 Silver release as a function of immersion time for AgNP/CeO2 containers prepared by calcination. | ||
On the other hand, AgNP/CeO2 containers prepared by dissolution have a lower and faster silver release, i.e. over a period of less than a month (Fig. 8). This is explained by the dissolution steps in which a considerable amount of silver was washed away. The containers prepared by dissolution would therefore be less efficient in the prevention of infections compared to the ones prepared by calcination.
 | ||
| Fig. 8 Silver release as a function of immersion time for AgNP/CeO2 containers prepared by dissolution. Data are the % mean ± standard error of the mean (n = 4). | ||
To the best of our knowledge, this is the first time that AgNPs are encapsulated into inorganic containers using a method in which AgNPs are first encapsulated into a PS template. This resulted in well-dispersed AgNPs throughout the CeO2 shell and a silver release exceeding 3 months. In addition, despite the fact that orthopaedic implants are normally in place for ten to fifteen years,31 only a few studies described the long-term release of silver from implant materials.32 The AgNP/CeO2 containers obtained by calcination are therefore very promising for the development of long-term antibacterial implant coatings.
3.3 Antibacterial tests
Neither tests for the zone of growth inhibition or MBC determination showed any inhibition of E. coli growth for empty CeO2 nanocontainers (Fig. 9A). On the other hand, both silver-containing CeO2 containers demonstrated heightened antimicrobial activity (Fig. 9B and C), indicating the intrinsic antimicrobial properties of silver. The zone of inhibition of AgNO3/CeO2 nanocontainers was 4 mm in radius, while the inhibition zone of AgNP/CeO2 containers was 2 mm in radius. AgNP/CeO2 containers showed a slower release of silver, explaining why the AgNO3/CeO2 nanocontainers elicited a larger inhibitory zone in such a short time period (i.e. overnight incubation). AgNP/CeO2 containers prepared by dissolution also showed an inhibitory zone as for AgNP/CeO2 containers prepared by calcination. | ||
| Fig. 9 Photographs of disc diffusion tests (n = 3) of CeO2 nanocontainers (A), AgNO3/CeO2 nanocontainers (B) and AgNP/CeO2 containers (C). | ||
The MBC determination by the broth dilution method demonstrated that AgNO3/CeO2 nanocontainers have a MBC of 93 ± 5 mg mL−1 (0.54 ± 0.03 mol L−1) and for AgNP/CeO2 containers, the MBC is 117 ± 6 mg mL−1 (0.68 ± 0.03 mol L−1). These findings further demonstrate the heightened antibacterial activity of silver-containing CeO2 containers. The enhanced antibacterial activity of AgNO3/CeO2 nanocontainers is consistent with their higher silver load being released after a one-day period (Fig. 2) compared to the silver release of AgNP/CeO2 containers (Fig. 7).
3.4 Epithelial cell cytotoxicity and morphology
Fig. 10 shows that CeO2 nanocontainers elicit no significant increase (p > 0.05) in LDH release from the A549 cells at either time point or concentration tested. However, further analysis via LSM highlighted that these nanocontainers induced signs of apoptosis (e.g. shrinkage of the cell size and membrane as well as condensed nuclear regions (Fig. 10B)). In addition, the monolayer, as clearly shown in the sample treated with cell culture media only (negative control) (Fig. 10A), was completely disrupted by the CeO2 nanocontainers. Thus, the lack of any detectable LDH release can be attributed to the fact that the cell membrane remained intact. Although CeO2 NPs have been shown to elicit a heightened antioxidant effect,33 the potential for this nanomaterial to cause apoptosis in A549 cells is also well known, as recently reported by Mittal and Pandey.34 While Mittal and Pandey34 highlighted the mechanistic pathways associated with the onset of cell death following interactions (i.e. uptake) of CeO2 NPs with A549 cells, in the present study the apoptosis observed can be strongly attributed to the rough surface of the CeO2 nanocontainers that the cells were cultured upon. Although an imperative understanding of the raw form of the CeO2 nanocontainers has been gained here, it is hypothesized that the effect of CeO2 nanocontainers on cell morphology will be significantly reduced following the impregnation of the CeO2 nanocontainers into a polymer-based biomaterial for their eventual application. | ||
| Fig. 10 Cytotoxicity (bar graph) of 58 mg per well (light grey) and 174 mg per well (black) of CeO2 nanocontainers, AgNO3/CeO2 nanocontainers and AgNP/CeO2 containers after 4 and 7 days of exposure to A549 epithelial cells. Cell culture media only served as the negative control in both occasions (white bars). Data are the % mean ± standard error of the mean compared to the positive control (0.2% TritonX 100) (n = 3). *Represents p < 0.05 compared to the negative control at 4 days. #Represents p < 0.05 compared to the negative control at 7 days. LSM images showing the XY panel and the XZ panel (below XY image) of the negative control (cell culture media only) (A), CeO2 nanocontainers (B), AgNO3/CeO2 nanocontainers (C) and AgNP/CeO2 containers (D). The scale bar corresponds to 50 μm. The nuclei are stained with DAPI (yellow) and the F-actin with rhodamine-phalloidin (cyan). | ||
Following exposure to AgNO3/CeO2 and AgNP/CeO2 containers respectively, the epithelial cell monolayer was also significantly disrupted, as evident in Fig. 10C and D. With respect to the AgNO3/CeO2 nanocontainer exposure, a significantly lower (p < 0.05) LDH release compared to control levels was observed for both time points and concentrations tested. This effect, augmented further by the noticeable unspecific rhodamine-phalloidin staining throughout the sample, can be associated with the nanocontainers inducing immediate necrosis following exposure to the A549 cells. This explains the low LDH concentration in the culture medium for AgNO3/CeO2. Since cell death occurred rapidly after exposure, cells could not build up a LDH load over the incubation periods, as was the case for the other samples and the control. This effect is most likely due to a rapid silver ion release from these nanocontainers that causes membrane disruption and actin depolymerisation, as previously reported by Cheng and colleagues.35
With regard to the AgNP/CeO2 nanocontainers, similar to the CeO2 nanocontainers, a limited, non-significant (p > 0.05) cytotoxicity was observed after a 4 day exposure. Following a 7 day exposure, however, the AgNP/CeO2 nanocontainers elicited a significant increase (p < 0.05) in LDH activity at both concentrations tested. This effect can be attributed to a higher concentration of silver in the medium after 7 days' exposure that constantly interacts with the cells during this prolonged period. Subsequent LSM analysis supported the LDH analysis and further indicated that AgNP/CeO2 containers could also induce necrosis in the A549 cells, showing effects similar to the AgNO3/CeO2 sample, although in a slower fashion (Fig. 10D). It is important to note, however, that the AgNP/CeO2 containers elucidated a slower cytotoxicity than the AgNO3/CeO2 containers due to a gradual, instead of immediate, silver release from the nanocontainers.
More detailed descriptions of AgNP effects on cells can be found elsewhere.32 As an example, Mukherjee et al.36 have demonstrated that biosynthesized AgNPs are more toxic towards cancerous cells, such as A549 cells, due to the cancerous cells' tendency to acidify their environment. The development of silver-containing materials can therefore find use in biomedical applications as an antibacterial or anticancer agent.
4. Conclusions
In conclusion, it was demonstrated that it is possible to use CeO2 nanocontainers to efficiently encapsulate either AgNO3 or AgNPs. These materials demonstrated an enhanced antimicrobial activity against E. coli, whereas in human cells (A549 epithelial cells) all three samples, when tested in their raw form, were observed to induce a form of cell death (i.e. apoptosis (CeO2 nanocontainers and AgNP/CeO2 nanocontainers) or necrosis (AgNO3/CeO2 and AgNP/CeO2 nanocontainers)). The AgNP/CeO2 nanocontainers are particularly interesting for the prevention of implant-related infections thanks to their long-term silver release.These effects, however, which can be attributed to the rough surface of the CeO2 nanocontainers and the kinetics of silver release, could be controlled by encapsulating the materials into a biomaterial (i.e. a polymer). From these findings, it can be suggested that with further emphasis towards increasing their biocompatibility, these newly designed AgNP/CeO2 nanocontainers may offer an advantageous strategy towards potentially preventing bacterial infections following implant surgery.
Acknowledgements
We are grateful to the Swiss National Science Foundation, the University of Fribourg, the Adolphe Merkle Foundation, FriMat and the NCCR “Bioinspired Materials” for generously supporting this project. We also thank Dr N. H. Kwon and Dr P. S. Brunetto for the SEM images and Y. Umehara for her help with the eukaryotic cell culture.Notes and references
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780 RSC.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422–1429 CrossRef CAS PubMed.
- R. M. Donlan and J. W. Costerton, Clin. Microbiol. Rev., 2002, 15, 167–193 CrossRef CAS.
- A. G. Gristina, Science, 1987, 237, 1588–1595 CAS.
- (a) K. A. Poelstra, N. A. Barekzi, A. M. Rediske, A. G. Felts, J. B. Slunt and D. W. Grainger, J. Biomed. Mater. Res., 2002, 60, 206–215 CrossRef CAS PubMed; (b) M. Emmerson, New Horizons: Science & Practice of Acute Medicine, 1998, 6, S3–S10 CAS.
- P. Gilbert, P. J. Collier and M. R. W. Brown, Antimicrob. Agents Chemother., 1990, 34, 1865–1868 CrossRef CAS.
- S. Hackenberg, A. Scherzed, M. Kessler, S. Hummel, A. Technau, K. Froelich, C. Ginzkey, C. Koehler, R. Hagen and N. Kleinsasser, Toxicol. Lett., 2011, 201, 27–33 CrossRef CAS PubMed.
- (a) T. V. Slenters, I. Hauser-Gerspach, A. U. Daniels and K. M. Fromm, J. Mater. Chem., 2008, 18, 5359–5362 RSC . http://pubs.rsc.org/en/content/articlepdf/2008/jm/b813026d; (b) O. Gordon, T. Vig Slenters, P. S. Brunetto, A. E. Villaruz, D. E. Sturdevant, M. Otto, R. Landmann and K. M. Fromm, Antimicrob. Agents Chemother., 2010, 54, 4208–4218 CrossRef CAS PubMed.
- P. S. Brunetto and K. M. Fromm, Chimia, 2008, 62, 249–252 CrossRef CAS.
- (a) A. Montali, Injury, 2006, 37(suppl. 2), S81–S86 CrossRef PubMed; (b) M. J. Santillan, N. E. Quaranta and A. R. Boccaccini, Surf. Coat. Technol., 2010, 205, 2562–2571 CrossRef CAS PubMed; (c) J. Wu, S. Hou, D. Ren and P. T. Mather, Biomacromolecules, 2009, 10, 2686–2693 CrossRef CAS PubMed.
- V. Alt, T. Bechert, P. Steinrucke, M. Wagener, P. Seidel, E. Dingeldein, E. Domann and R. Schnettler, Biomaterials, 2004, 25, 4383–4391 CrossRef CAS PubMed.
- A.-H. Pei, Z.-W. Shen and G.-S. Yang, Mater. Lett., 2007, 61, 2757–2760 CrossRef CAS PubMed.
- I. Kartsonakis, I. Daniilidis and G. Kordas, J. Sol–Gel Sci. Technol., 2008, 48, 24–31 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- (a) A. Madadlou, D. Iacopino, D. Sheehan, Z. Emam-Djomeh and M. E. Mousavi, Biochim. Biophys. Acta, 2010, 1800, 459–465 CrossRef CAS PubMed; (b) Y. Urabe, T. Shiomi, T. Itoh, A. Kawai, T. Tsunoda, F. Mizukami and K. Sakaguchi, ChemBioChem, 2007, 8, 668–674 CrossRef CAS PubMed.
- S. S. Hardas, D. A. Butterfield, R. Sultana, M. T. Tseng, M. Dan, R. L. Florence, J. M. Unrine, U. M. Graham, P. Wu, E. A. Grulke and R. A. Yokel, Toxicol. Sci., 2010, 116, 562–576 CrossRef CAS PubMed.
- F. R. Cassee, E. C. van Balen, C. Singh, D. Green, H. Muijser, J. Weinstein and K. Dreher, Crit. Rev. Toxicol., 2011, 41, 213–229 CrossRef PubMed.
- (a) A. S. Karakoti, N. A. Monteiro-Riviere, R. Aggarwal, J. P. Davis, R. J. Narayan, W. T. Self, J. McGinnis and S. Seal, JOM, 2008, 60, 33–37 CrossRef CAS PubMed; (b) T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed; (c) D. Schubert, R. Dargusch, J. Raitano and S.-W. Chan, Biochem. Biophys. Res. Commun., 2006, 342, 86–91 CrossRef CAS PubMed.
- I. A. Kartsonakis, P. Liatsi, I. Daniilidis and G. Kordas, J. Am. Ceram. Soc., 2008, 91, 372–378 CrossRef CAS PubMed.
- D. D. Evanoff and G. Chumanov, J. Phys. Chem. B, 2004, 108, 13948–13956 CrossRef CAS.
- A. S. Kumbhar and G. Chumanov, Chem. Mater., 2009, 21, 2835–2839 CrossRef CAS.
- B. Bonev, J. Hooper and J. Parisot, J. Antimicrob. Chemother., 2008, 61, 1295–1301 CrossRef CAS PubMed.
- K. E. Cooper, Nature, 1955, 176, 510–511 CrossRef CAS.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- J. McFarland, JAMA, J. Am. Med. Assoc., 1907, 14, 1176–1178 CrossRef.
- B. M. Rothen-Rutishauser, S. G. Kiama and P. Gehr, Am. J. Respir. Cell Mol. Biol., 2005, 32, 281–289 CrossRef CAS PubMed.
- A. Lehmann, C. H. Brandenberger, F. Blank, P. Gehr and B. Rothen-Rutishauser, A 3D model of the human epithelial airway barrier, in Alternatives to Animal Testing, ed. M. L. Yarmush and R. S. Langer, Artech House, 2010, vol. 239–260 Search PubMed.
- (a) F. Chen, Y. Cao and D. Jia, Appl. Surf. Sci., 2011, 257, 9226–9231 CrossRef CAS PubMed; (b) Y. S. Chaudhary, S. Panigrahi, S. Nayak, B. Satpati, S. Bhattacharjee and N. Kulkarni, J. Mater. Chem., 2010, 20, 2381 RSC; (c) K. Kalimuthu, R. S. Babu, D. Venkataraman, M. Bilal and S. Gurunathan, Colloids Surf., B, 2008, 65, 150–153 CrossRef CAS PubMed.
- (a) C. Damm, H. Münstedt and A. Rösch, J. Mater. Sci.: Mater. Med., 2007, 42, 6067–6073 CrossRef CAS PubMed; (b) M. Z. Kassaee, A. Akhavan, N. Sheikh and A. Sodagar, J. Appl. Polym. Sci., 2008, 110, 1699–1703 CrossRef CAS; (c) D. Zampino, T. Ferreri, C. Puglisi, M. Mancuso, R. Zaccone, R. Scaffaro and D. Bennardo, J. Mater. Sci.: Mater. Med., 2011, 46, 6734–6743 CrossRef CAS.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Navěčná and R. Zbořil, J. Phys. Chem., 2006, 110, 16248–16253 CrossRef PubMed.
- C. Yao, V. Perla, J. L. McKenzie, E. B. Slamovich and T. J. Webster, J. Biomed. Nanotechnol., 2005, 1, 68–73 CrossRef CAS PubMed.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- L. L. Wong and J. F. McGinnis, Adv. Exp. Med. Biol., 2014, 801, 821–828 CrossRef.
- S. Mittal and A. K. Pandey, BioMed Res. Int., 2014, 2014, 891934 CrossRef PubMed.
- X. Cheng, W. Zhang, Y. Ji, J. Meng, H. Guo, J. Liu, X. Wu and H. Xu, RSC Adv., 2013, 3, 2296–2305 RSC.
- S. Mukherjee, D. Chowdhury, R. Kotcherlakota, S. Patra, B. Vinothkumar, M. P. Bhadra, B. Sreedhar and C. R. Patra, Theranostics, 2014, 4, 316–335 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb02079k |
| This journal is © The Royal Society of Chemistry 2015 |