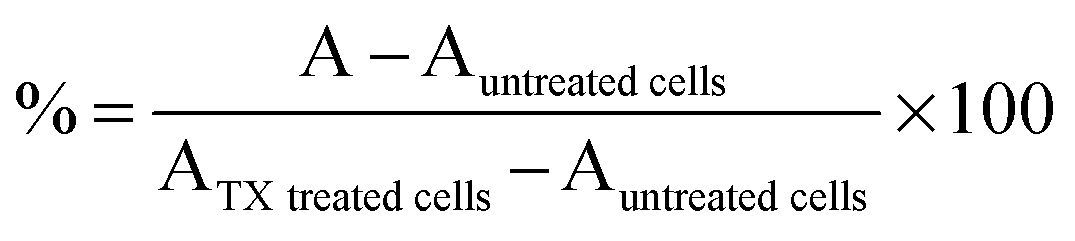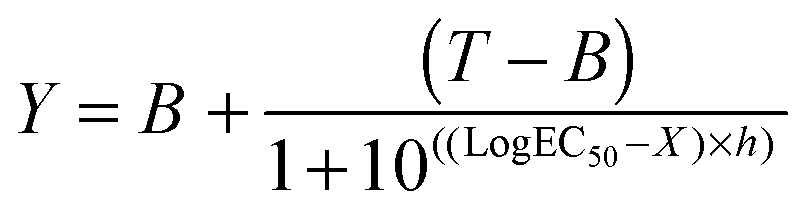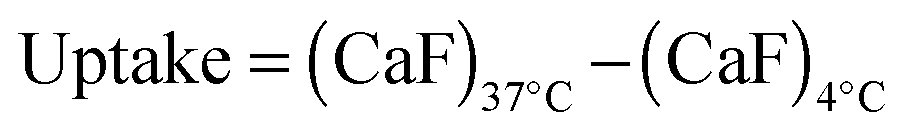Cationic poly(amidoamine) promotes cytosolic delivery of bovine RNase A in melanoma cells, while maintaining its cellular toxicity†
Received
15th December 2014
, Accepted 6th July 2015
First published on 13th July 2015
Abstract
Ribonucleases are known to cleave ribonucleic acids, inducing cell death. RNase A, a member of the ribonuclease family, generally displayed poor in vitro activity. This has been attributed to factors such as low intracellular delivery. Poly(amidoamine)s have been used to promote the translocation of non-permeant proteins to the cytosol. Our objective was to demonstrate that poly(amidoamine)s could potentially promote the delivery of RNase A to selected cell line. Interactions of three cationic poly(amidoamine)s (P1, P2 and ISA1) with wild-type bovine RNase A were investigated using gel retardation assays, DLS and microcalorimetry. Although the polymers and the protein are essentially cationic at physiological pH, complexation between the PAAs and RNase A was observed. The high sensitivity differential scanning calorimetry (HSDSC) thermograms demonstrated that the thermal stability of the protein was reduced when complexed with ISA1 (Tmax decreased by 6.5 °C) but was not affected by P1 and P2. All the polymers displayed low cytotoxicity towards non-cancerous cells (IC50 > 3.5 mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mL−1). While RNase A alone was not toxic to mouse melanoma cells (B16F1), P1 was able to promote cytosolic delivery of biologically active RNase A, increasing cell death (IC50 = 0.09 mgmL−1).
mL−1). While RNase A alone was not toxic to mouse melanoma cells (B16F1), P1 was able to promote cytosolic delivery of biologically active RNase A, increasing cell death (IC50 = 0.09 mgmL−1).
Introduction
Studies in the early 1950s reported the anticancer activity of high doses of bovine ribonuclease A (RNase A).1 However, over the years, the cytotoxicity of RNase A has been controversial and efforts have been made to develop more potent ribonucleases such as Onconase. Still, Onconase is of non-mammalian origin and is known to display some nephro- and neurotoxicity.2 The low efficiency of RNase A has been attributed to poor intracellular delivery, inhibition by the ribonuclease inhibitor (RI) and short half-life.2 Bovine seminal RNase (BS-RNase) is a naturally occurring dimeric ribonuclease.3 It is composed of two subunits (M), which are either linked by simple disulphide bonds (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) M form) or via additional non-covalent interactions, resulting from the swapping of the N-terminal α-helices (M×M form). These dimeric structures favour evasion from the RI and confers enhanced cytotoxicity to the M×M form, more stable under the reducing environment of the cytosol.3
M form) or via additional non-covalent interactions, resulting from the swapping of the N-terminal α-helices (M×M form). These dimeric structures favour evasion from the RI and confers enhanced cytotoxicity to the M×M form, more stable under the reducing environment of the cytosol.3
Although dimerisation may not always grant the highest cytotoxicity,4 strategies to obtain RI resistant RNase A, have led to the synthesis of dimeric RNase A endowed with different degree of cytotoxicity3,5–8. Microinjection of ribonuclease A into the cytosol of Xenopus oocytes and NIH/3T3 cells confirmed its cytotoxicity and demonstrated that the protein was potent at low doses.9,10 More recent studies have revealed that enhanced cytosolic delivery can compensate the inhibitory action of the RI.11,12 To that effect, different methods such as cationisation13–17 or conjugation to non-biodegradable synthetic polymers18–20 have been developed. Encouraging results were obtained but the catalytic activity of the ribonuclease bioconjugates was often decreased.15,16
Poly(amidoamine)s (PAAs) are biodegradable polymers and are generally less cytotoxic compared to most polycations used as nanocarriers.21 They are synthesised by Michael addition polymerisation and possess secondary or tertiary amines in the polymer backbone. Poly(amidoamine)s are pH-responsive polyelectrolytes and upon acidification of their environment their conformation will change from a coiled structure to a more extended structure with increased radius of gyration. This pH responsiveness has also been observed in a biological environment and increased haemoglobin release has been noticed at acidic pH when red blood cells were incubated with some poly(amidoamine)s.21 The capacity of the PAAs to be protonated at acidic pH allows them to potentially destabilise the endosomal or lysosomal membranes and act as endosomolytic (or lysosomolytic) polymers.22 Hence, some poly(amidoamine)s have been used to promote the cytosolic delivery of drugs21,23 and biomacromolecules,24–26 including non-permeant toxins27–29i.e. protein toxins which are not able to translocate to the cytosol of the cells.
We recently synthesised some cationic poly(amidoamine)s able to interact with and stabilise BSA.30 We hypothesised that these PAAs could be used to promote the translocation of wild-type RNase A into the cytosol of the cells. Using gel retardation assays and high sensitivity differential scanning calorimetry (HSDSC), we evaluated the interaction of the polymers with the protein. The complexes obtained upon incubation were characterised by DLS. The cellular uptake of the protein in presence or absence of the PAAs was evaluated at 37 °C and 4 °C using flow cytometry. RNase A induces cell death by cleaving RNA located in the cytosol. The retention of its biological activity and the cytosolic delivery of the protein was determined indirectly by measuring the cells viability following incubation with the PAA/RNase A complexes, using a non-toxic concentration of RNase A.
Results and discussion
Cytotoxicity of the synthesised poly(amidoamine)s
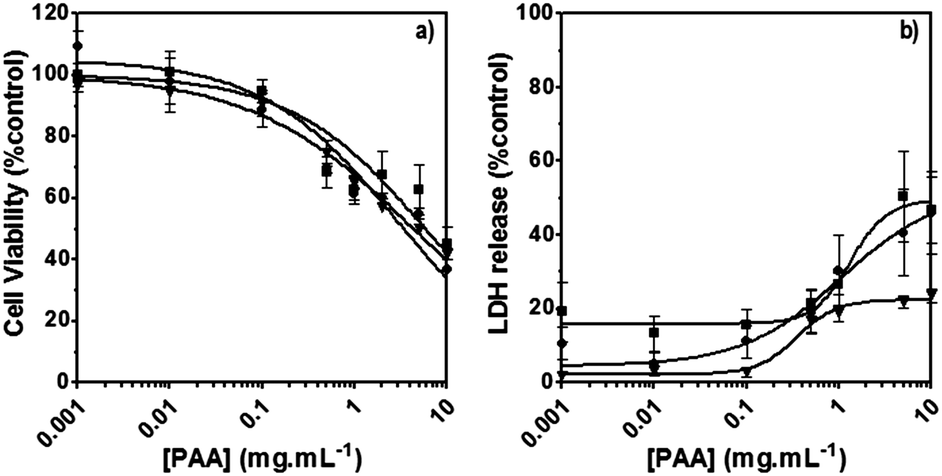
The poly(amidoamine)s used in this study were synthesised by Michael-type polyaddition (Table 1). P1 and P2 were derived from the structure of ISA1 to modulate the polymer chain flexibility and hydrophobicity. We previously demonstrated that the polymers were cationic at physiological pH.30 Polycations tend to display relatively high cytotoxicity, which often make them unsuitable carriers for macromolecule delivery, especially if a biological function needs to be restored.31,32 Ribonuclease A possesses some potential anticancer activity, however cancerous cells are not appropriate model cell lines to evaluate the biocompatibility of nanocarriers, as they are known to display different sensitivity compare to healthy cells. Therefore, in order to evaluate the potential biocompatibility of the PAAs, the cytotoxicity of the polymers towards non-cancerous cells was evaluated. Their effect on the metabolic activity of HEK293 cells was assessed by measuring the mitochondrial activity after 24 h incubation. The viability of the cells was concentration-dependent (Fig. 1). The PAAs were not toxic (i.e. viability >70%)33 up to a concentration of 1 mgmL−1. All IC50 were above 3.5 mgmL−1 (Table 1) which is lower than for polycations such as poly(ethyleneimine)32 and is similar to what has been reported for other poly(amidoamine)s.21 No significant influence of the polymer chemical structures was noticed. P2 seemed to display a lower IC50 value, which may be attributed to the smaller molecular weight for this polymer, however the difference was not significant. Lactate dehydrogenase (LDH) release assay has also been used as a measure of cell viability and gives additional information to mitochondrial activity-based assays.34 It can be used to evaluate the integrity of the cell membrane and reflect the damage caused by the polymers. Peng et al.35 evaluated the interaction of PAA/DNA polyplexes with bone marrow stromal cells and reported low percentage of LDH release (i.e. <10%) at low N/P ratio. However, polyplexes may display lower cytotoxicity. We measured the LDH released after 24 h in presence of the PAAs alone and found it was also concentration-dependent (Fig. 1) with LDH20 above 0.4 mg mL−1 (Table 1). LDH10, the PAAs concentration at which only 10% of LDH is released, was determined for P2 (0.09 mg mL−1) and ISA1 (0.28 mg mL−1) indicating the destabilising effect of these two polymers was lower in comparison to P1.
Table 1 Physicochemical30 and biological properties of the poly(amidoamine)s
| Polymers |
|
Mw |
Mn |
PDI |
ζ (mV) |
IC50 |
LDH20 |
| (g mol−1) |
(g mol−1) |
(mg mL−1) |
(mg mL−1) |
|
M
w: weight average molecular weight; Mn: number average molecular weight; PDI: polydispersity index; ζ: zeta potential. Mean value of two independent replicates ± SEM; IC50 represents the PAAs concentration at which 50% of the cell population is dead relative to untreated control cells. LDH20 represents the PAAs concentration at which 20% of LDH is released relative to control cells treated with triton X100. |
| P1 |
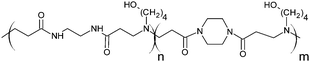
|
10400 |
5100 |
2.0 |
27.6 ± 0.2 |
6.0 |
0.5 |
| P2 |
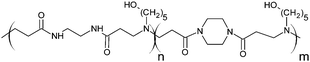
|
5700 |
3000 |
1.9 |
26.6 ± 0.4 |
3.5 |
0.4 |
| ISA1 |
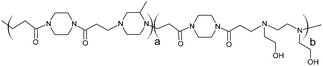
|
16300 |
8200 |
2.0 |
27.6 ± 0.5 |
4.5 |
1 |
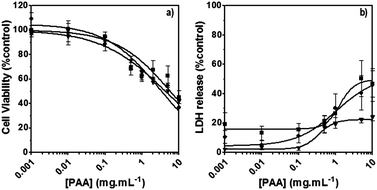 |
| | Fig. 1 Cytotoxicity of the synthesised poly(amidoamine)s (ISA1 (▼), P1 (■) and P2 (●)) was assessed using HEK293 cells (105 cells per well). Panel (a): viability of the cells estimated at 24 h using an MTS assay. Panel (b): membrane integrity of the cells estimated at 24 h using an LDH release assay. Mean values ± SEM, n = 6. | |
Interaction of the poly(amidoamine)s with RNase A
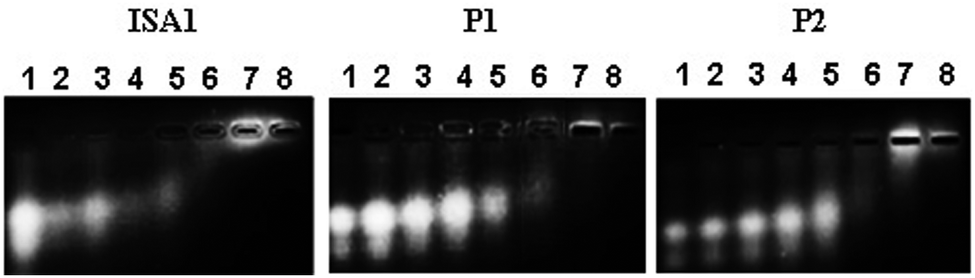
Interactions between polyelectrolytes and proteins are generally due to electrostatic forces.36 Evidence of complex formation, at physiological pH, between the cationic poly(amidoamine)s and BSA (pI ∼ 4.7), an acidic model protein, has been reported.30 RNase A is a 13.7 kDa basic protein (pI ∼ 9.5).37,38 Contrary to BSA, it has an overall cationic charge at physiological pH. The mobility of RNase A in an agarose gel retardation assay was progressively altered with increasing PAA:RNase A weight ratios (Fig. 2) suggesting some complexation despite repulsive forces between the protein and the polyelectrolytes. It is usually considered that complex formation between two macromolecules requires opposite net charges. However, complexation between macromolecules bearing similar net charges (i.e. both anionic or both cationic) has been reported.39–41
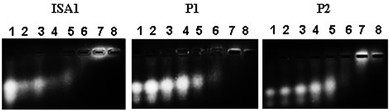 |
| | Fig. 2 Complexation of RNase A–OG with the poly(amidoamine)s at different PAA: RNase A weight ratios (w/w). Lane (1) RNase A alone; (2) 10:1; (3) 50:1; (4) 100:1; (5) 200:1; (6) 500:1; (7) 1000:1; (8) 2000:1. | |
In general, this has been attributed to the anisotropic distribution of the charges on the protein surface which, depending on the nature of the protein, leads to the formation of either positive or negative “patches”.42 Such “negative patches” have been observed for RNase A using computer modelisation39,43 and make binding of the positively charge protein to cationic PAAs possible (Fig. 3). Although Coulombic forces are considered the driving force for protein/polymer interactions, the role of hydrophobicity has also been demonstrated.44,45 “Hydrophobic patches” on the surface of RNase A have been identified46,47 and could also be involved in its association with the poly(amidoamine)s via hydrophobic interactions (Fig. 3).
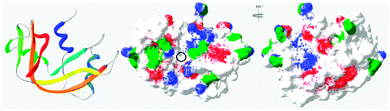 |
| | Fig. 3 Ribbon representation of bovine RNase A (PDB: 1kf5) and modelisation of the electrostatic potential and hydrophobic patches. Positive and negative potential patches are represented in blue and red, respectively. Hydrophobic patches are represented in green. The black circle indicates the binding cavity of the protein (see ESI† for 3D animated structure). | |
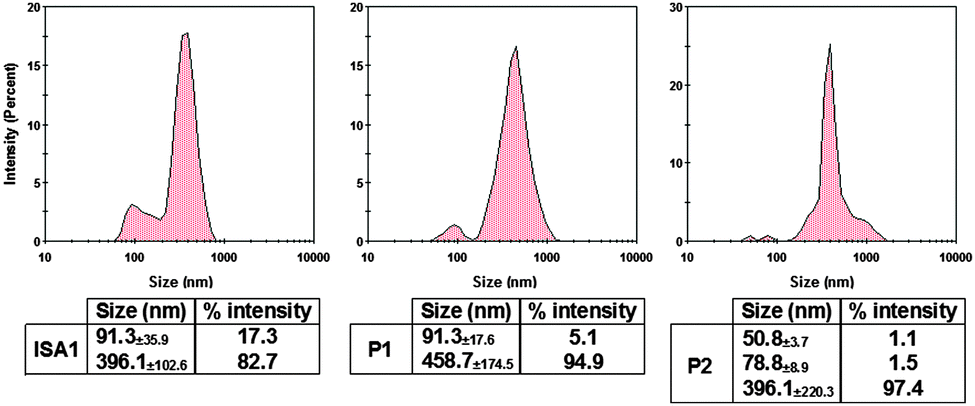
The size of the complexes were initially measured at PAA:RNase A 500:1 weight ratio for which full retardation was observed. The results, reported as size distribution by intensity, indicated the presence of a heterogeneous population of polydisperse nanoparticles with some potential aggregation (Fig. 4). Although full retardation of the protein was not achieved at lower weight ratios (Fig. 2), a number of additional peaks in the 10 to 100 nm range were observed by DLS for all the polymers. These peaks suggested the formation of small complexes and indicated an increase in the heterogeneity of the samples.
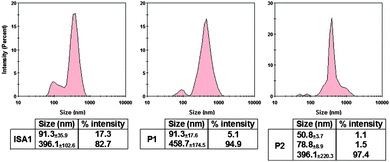 |
| | Fig. 4 DLS characterisation of the complexes. [RNase A] = 25 μg mL−1; PAA:RNase A complexes were prepared at 500:1 (w/w) ratio. Values represent the modal size ± width of the peak, n = 3. | |
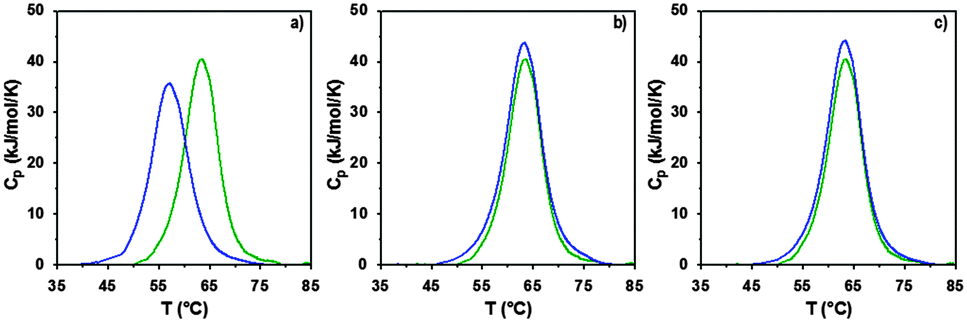
The interaction of RNase A with the poly(amidoamine)s was further investigated by microcalorimetry. High sensitivity DSC is generally used to monitor protein thermal unfolding under different experimental conditions. The denaturation temperature at maximum transition (Tmax) is an indicator of the protein thermostability and depends on factors such as the ionic strength or the pH of the samples.36 The higher Tmax is, the more stable the protein is. The thermal stability of RNase A has been evaluated in different environments and Tmax values ranging from 56.5 to 63.9 °C have been reported.37,48–50 Under our experimental conditions, Tmax was measured at 63.3 °C (Fig. 5 and Table 2) with a corresponding unfolding enthalpy of 382.3 kJ mol−1. DSC can also be used to investigate the thermal denaturation of proteins in complexes. While some studies have demonstrated that polymers can promote or maintain the thermal stability of model proteins,51,52 others have reported a destabilisation effect.36,53,54 Glycosylation, trimethylamine N-oxide (TMAO) and lipids48,50,55 have improved the thermal stability of RNase A to different extent, whereas complexation with polyanions such as heparin or adsorption on silica nanoparticle had essentially a negative effect.37,38
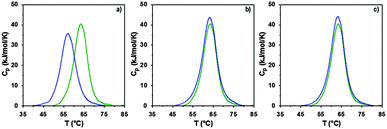 |
| | Fig. 5 DSC profiles following normalisation and baseline correction of RNase A alone (green) and in the presence (blue) of (a) ISA1; (b) P1 or (c) P2. Heating rate 1 °C min−1; [RNase A] = 0.5 mg mL−1; PAA:RNase A complexes were prepared at ratio 10:1 (w/w). | |
Table 2 Thermodynamic properties of RNase A in the absence and presence of the PAAs
| |
T
max (°C) |
ΔHcal (kJ mol−1) |
ΔScal (kJ mol−1 K−1) |
dHV (kJ mol−1) |
n
|
ΔT (°C) |
| Tmax: temperature of denaturation. Tmax was determined as the temperature corresponding to the maximum heat capacity (Cp); ΔHcal: calorimetric enthalpy change; ΔScal: calorimetric entropy change; dHV: Van't Hoff enthalpy change; n: cooperative units; ΔT: width at half maximum. Mean value of two independent replicates ± SEM. |
| RNase A |
63.32±0.02 |
382.3±30.3 |
1.14±0.09 |
396.6±11.8 |
1.04±0.05 |
7.5±0.2 |
| P1:RNase A |
63.11±0.02 |
426.7±7.9 |
1.27±0.02 |
408.1±4.5 |
0.96±0.01 |
7.9±0.0 |
| P2:RNase A |
63.13±0.00 |
419.1±5.7 |
1.25±0.02 |
404.6±1.4 |
0.97±0.02 |
7.6±0.3 |
| ISA1:RNase A |
56.81±0.12 |
353.5±16.2 |
1.07±0.05 |
363.6±6.3 |
1.03±0.03 |
8.2±0.0 |
In a previous study, we demonstrated that P1 and P2 could stabilise BSA, whereas ISA1 had an adverse effect.30 The influence of the poly(amidoamine)s on the thermal stability of RNase A, was investigated under similar conditions (Fig. 5). The temperature at maximum transition for RNase A, decreased by 6.5 °C in the presence of ISA1 (Table 2). This is similar to what we previously observed with BSA, although in the case of RNase A the width of the peak at half maximum transition (ΔT) remained constant, suggesting no intermediate in the unfolding process.37 On the contrary, Tmax was not affected in the presence of P1 or P2 indicating RNase A maintained its thermal stability (Table 2). We previously found that P1 and P2 displayed increased chain flexibility in comparison to ISA1.30 The increased rigidity of the polymer backbone could potentially explain that under similar experimental conditions, ISA1 destabilises the protein. Further analysis of all the thermograms assuming a two-state model of denaturation (Native protein ⇆ Unfolded protein), demonstrated the equivalence of the calorimetric and Van't Hoff enthalpies, which further confirmed the absence of intermediate. This also indicated some cooperativity in the unfolding process of RNase A (i.e. n = dHv/ΔHcal ∼ 1) and has been previously observed for the native protein or in presence of TMAO and lipids.48,50
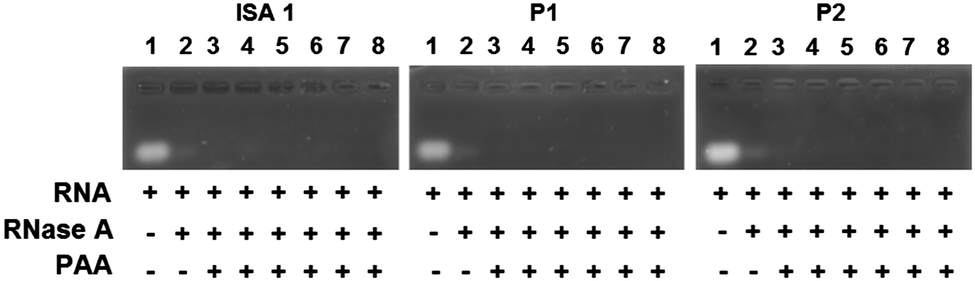
Complex formation between cationic poly(amidoamine)s and an acidic protein revealed that in some cases, the activity of the protein could be inhibited.24 We therefore assessed whether the enzymatic activity of RNase A would be altered upon interaction and complexation with our poly(amidoamine)s (Fig. 6). Yeast RNA was incubated for 30 min. at 37 °C in the presence or absence of RNase A. The enzyme was able to degrade the ribonucleic acids in the absence or in the presence of the poly(amidoamine)s, even at higher ratio corresponding to full complexation of the protein (Fig. 2) indicating the catalytic site of RNase A was accessible to the substrate.
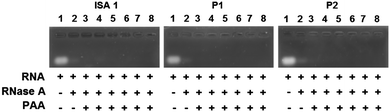 |
| | Fig. 6 Enzymatic activity of RNase A in the absence or presence of poly(amidoamine)s. Lane (1) RNA alone; (2) RNA + RNase A; Lanes (3–8) RNA + PAA:RNase A at different weight ratios: (3) 1:1; (4) 5:1; (5) 10:1; (6) 50:1; (7) 100:1; (8) 500:1. | |
Cellular uptake and cytotoxicity of RNase A
Ribonucleases capable to reach cellular ribonucleic acids are known to induce cell death.2 Although the intracellular pathway of RNase A is still subject to discussion, some aspects have been identified.56 However, following interaction with the cell membrane and endocytosis, only a small amount of RNase A reaches the cytosol where it eventually binds the ribonuclease inhibitor (RI).57 After binding, the catalytic activity of RNase A is totally inhibited due to obstruction of its active site, hence eliminating any cytotoxic effect. B16F1 cells were initially subjected to increasing concentration (0–100 μg mL−1) of wild-type RNase A. As expected, no effect on the viability of the cells was observed (Fig. 7c – red line). Aside from direct cytosolic injection,9,10 most approaches to improve the efficiency of RNase A have involved the development of RI evasive mutant proteins.57 However, a few techniques involving the chemical derivatisation of RNase A, including cationisation, have been used with varied success.15–20 More recently, an alternative method has been developed to co-incubate RNase A with PAMAM dendrimers58 but under the experimental conditions reported, only RNase A variants were successfully delivered. Poly(amidoamine)s are endosomolytic polymers and have been used to promote the intracellular delivery of macromolecular drugs such as plasmid DNA, siRNA and proteins.21,22 RNase A exerts its cytotoxicity by cleavage of cytosolic RNA, which requires escape from the endosomes to the cytosol.
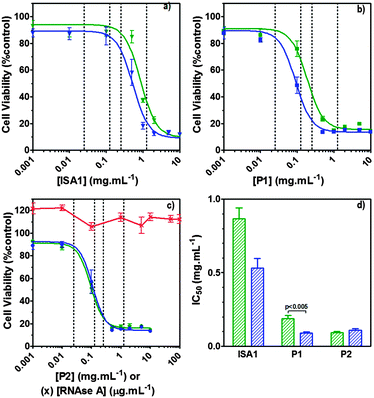 |
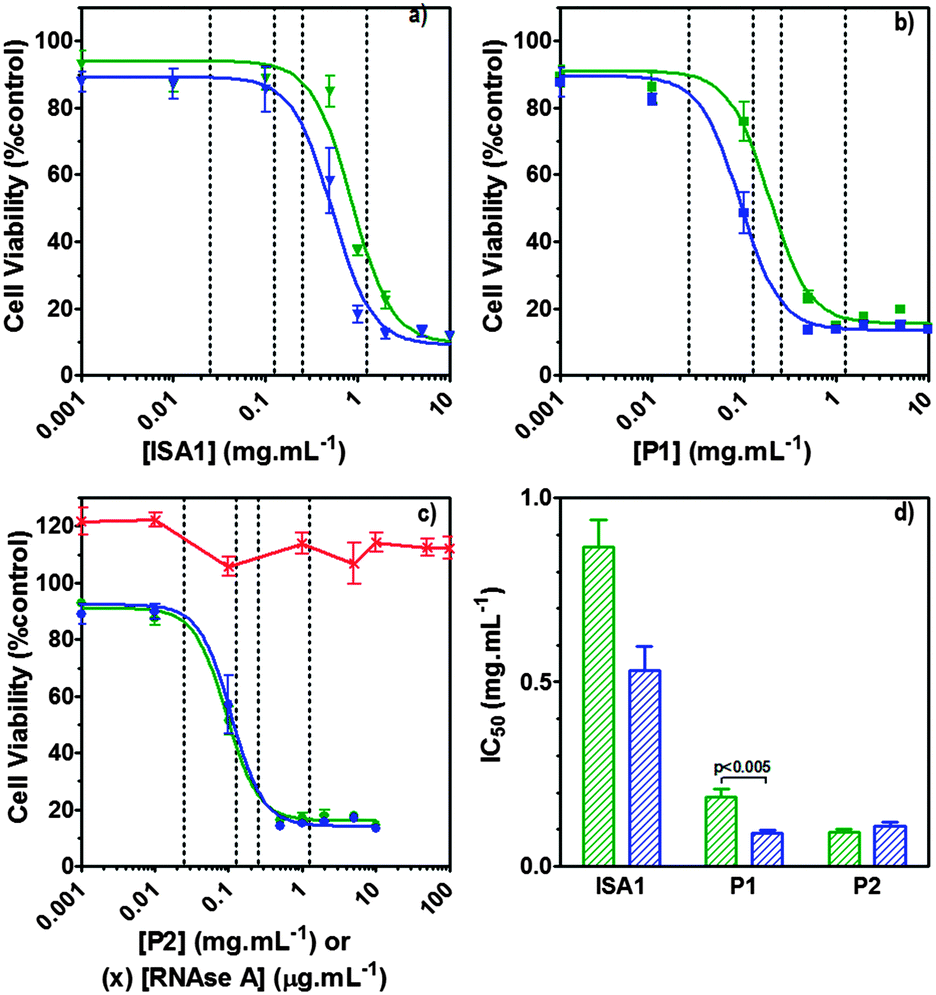
| | Fig. 7 Intracellular delivery and cytotoxicity of RNase A. Panels (a), (b) and (c) show the viability of B16F1 cells (104 cells per well) at 48 h following 5 h incubation with poly(amidoamine)s ISA1 (a), P1 (b) and P2 (c) in the absence (green) or presence (blue) of a non-toxic concentration of RNase A (25 μg mL−1). Panel (d) shows the corresponding IC50 in the absence (green) or presence (blue) of RNase A; IC50 values were determined by nonlinear regression using a sigmoidal dose–response curve (eqn (2)) (goodness of fit, R2 > 0.9 for all fits). Panel (c) also shows the viability of B16F1 with increasing concentration of RNase A (red). Mean values ± SEM, n = 6. | |
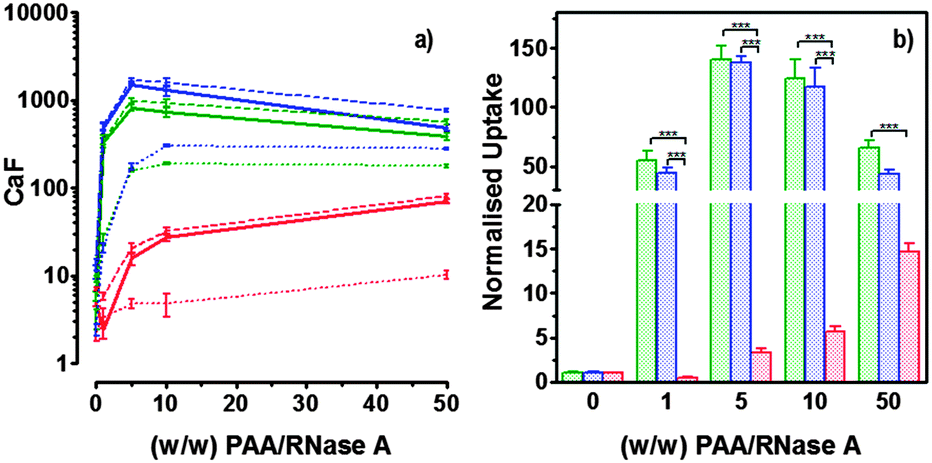
Hence, to assess the ability of the synthesised linear poly(amidoamine)s to deliver RNase A inside the cytosol, B16F1 cells were incubated for 5 h with the polymers alone or the complexes. The complexes were prepared using a non-toxic concentration of RNase A and different PAA:RNase A weight ratios. Following removal of the complexes, the cell viability was assessed at 43 h post-transfection using an MTS assay. Concentrations of 5 and 10 μgmL−1 of RNase A were initially used but no effect was observed (data not shown). The concentration was increased to 25 μg mL−1 (Fig. 7). Comparison of the IC50 values obtained for the polymers alone and the complexes demonstrated that for P2 no increase in cytotoxicity was observed (Fig. 7c and d). ISA1, which has been previously used to deliver non-permeant proteins,27–29 seemed to be more efficient as a decrease of the IC50 was noticed for the complexes (Fig. 7a and d). However, the difference was considered not significant (p > 0.005). P1 was the only poly(amidoamine) able to induce efficient intracellular release of RNase A, as indicated by a significant two-fold increase of the complexes cytotoxicity (IC50 = 0.09 mg mL−1; p < 0.005) (Fig. 7b and d). To further investigate the cellular delivery of wild-type RNase A by the PAAs, we used a protein labelled with oregon green (OG) and incubated the B16F1 cells with complexes prepared at different PAA/RNase A–OG weight ratios (RNase A–OG: 25 μg mL−1), selected to match different sections of the dose–response curves (Fig. 7a–c – vertical dotted black lines). The cell associated fluorescence (CaF) at 37 °C was quantified after 5 h using flow cytometry (Fig. 8). At this temperature, the observed fluorescence corresponded to the fraction of protein taken up by the cell via endocytosis but also to the protein simply adsorbed on the external surface of the cell membrane. This latter non-specific binding (NSB) can be estimated by reducing the metabolic activity of the cells at low temperature, inhibiting endocytosis.59,60 NSB was quantified at 4 °C and was subsequently subtracted to determine the total uptake of the protein at 37 °C.22 All the polymers promoted increased delivery of the protein at all ratios. Uptake of the protein increased with the proportion of PAA levelling off at higher ratios, perhaps indicating some saturation of the intracellular pathway.
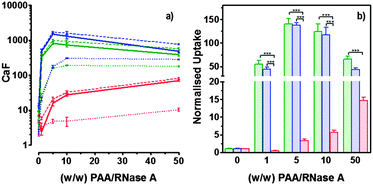 |
| | Fig. 8 Cellular uptake of fluorescently labelled RNase A (25 μg mL−1) at several PAA/RNase A (w/w) ratios: ISA1 (red), P1 (green) and P2 (blue). Panel (a) shows the B16F1 (105 cells per well) cell associated fluorescence at 37 °C (dashed lines) and 4 °C (dotted lines) after 5h incubation. Total uptake at 37 °C (plain lines) was calculated by subtracting values at 4 °C. Panel (b) shows the total uptake at 37 °C normalised to the uptake of RNase A alone. Mean values ± SEM, n = 6, *** p <0.001. | |
The use of wild-type RNase A as a potential therapeutic agent has been essentially impaired due to its inefficient intracellular delivery and inhibition by the RI.2 However, some studies have recently demonstrated that RI evasion might not be the only prerequisite to induce some cytotoxicity, improving translocation of the protein into the cytosol after endocytosis could be sufficient to saturate the RI.11,12 Although, P2 was able to promote cellular uptake of the protein to similar level as P1 (Fig. 8) we did not observe any increased toxicity for the delivery of RNase A with this polymer (Fig. 7c). The mechanism by which poly(amidoamine)s can promote macromolecule translocation into the cytosol of the cells has not been fully elucidated. However, it is generally considered that the buffer capacity of the polymer is an important factor.21 The “proton sponge effect” is the current standard theory to explain the endosomolytic properties of most polycations.61 The hypothesis relies on the buffer capacity of the polymers, higher buffer capacity corresponding to higher transfection efficiency. P2 displayed a lower buffer capacity in comparison to both P1 and ISA1,30 this seems to have impaired its endosomolytic capacity and potentially limited the cytoplasmic translocation of RNase A. On the contrary, the efficiency of ISA1 seemed to be limited not by its buffer capacity, similar to P1, but by its ability to promote cellular uptake significantly lower than both P1 and P2 (Fig. 8b). The amount of protein reaching the cytosol may not have been appropriate to saturate the RI to a significant extend. The results observed in the presence of P1 indicated sufficient amount of RNase A was delivered into the cytosol of the B16F1 cells to increase cell death (Fig. 7b). However, at the ratios for which the effect is observed, a population of complexes co-exist with free protein. It is therefore likely that the intracellular delivery taking place is promoted by both complexation with P1 and the effect of the polymer and/or complexes “in trans”. Overall, our results indicate that a minimum concentration of RNase A was required to observe an effect and correlate with previous observations indicating that provided the appropriate amount of RNase A reaches the cytosol, the RI could be saturated.11,12
Experimental
Materials
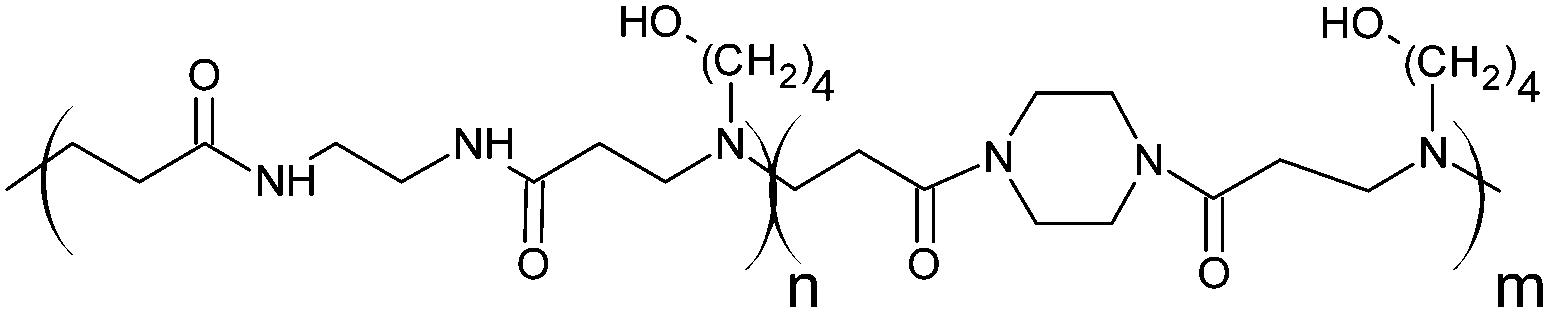
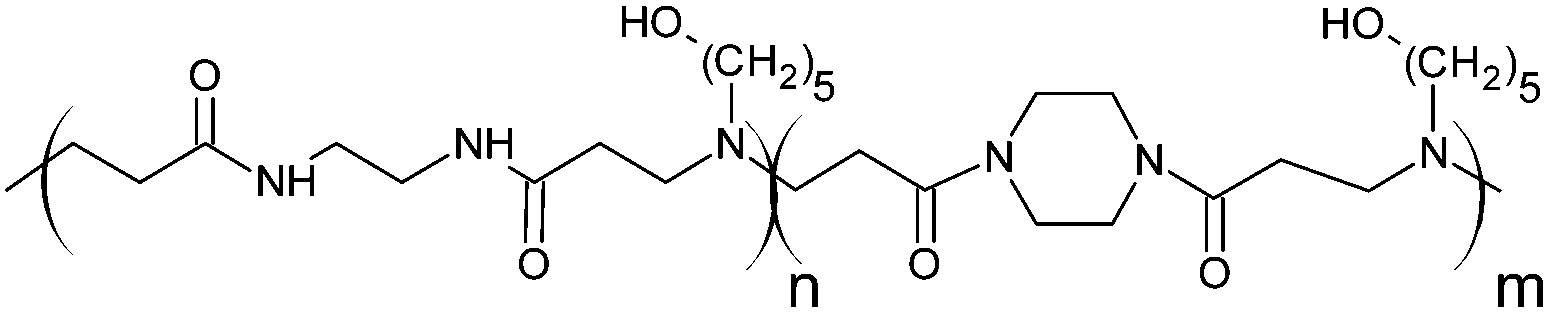
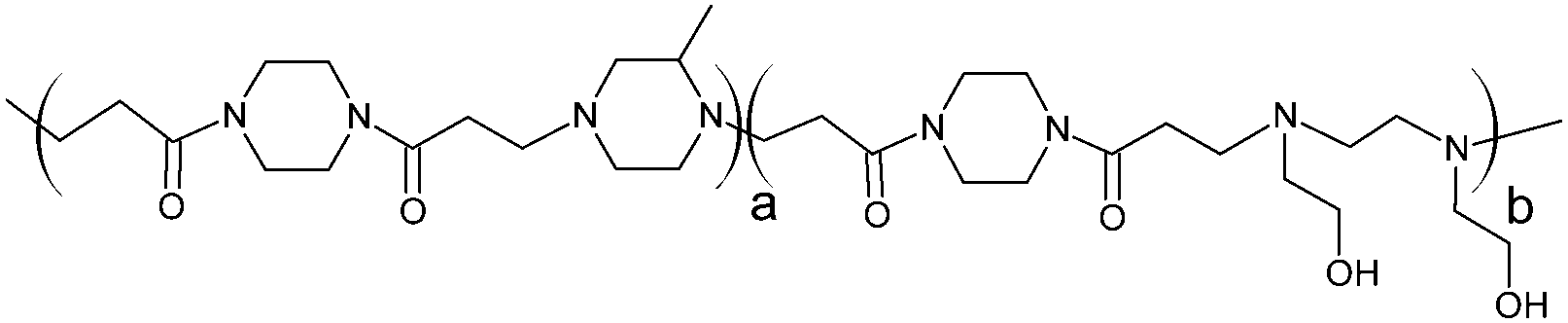
RNase A from bovine pancreas, NaCl and triton X-100 were from Sigma (Gillingham, UK). TRIS buffer, nicotinamide adenine dinucleotide (NAD), HEPES, D-glucose, DMSO, HCl, phenazine methosulfate (PMS) and TAE were from Fisher (Loughborough, UK). Sodium-L-lactate was from Alfa Aesar (Heysham, UK). Iodonitrotetrazolium salt (INT) was from TCI (Zwijndrecht, Belgium). Agarose was from Roche (Burgess Hill, UK). DMEM, RPMI-1640, DPBS, FBS, penicillin/streptomycin/glutamine (PSG), minimum essential medium non-essential amino acids (MEM NEAA) and succinimidyl ester oregon green 488 (OG) were from Life Technologies (Paisley, UK). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was from Promega (Mannheim, Germany). Mouse melanoma (B16F1) and human embryonic Kidney 293 (HEK293) cells were from the European Collection of Cell Cultures. The synthesis and physicochemical characterisation of the poly(amidoamine)s (P1: poly[(N,N′-ethylenebisacrylamide)-co-(N,N′-bis(acryloyl)piperazine)-co-(4-amino-1-butanol)]; P2: poly[(N,N′-ethylenebisacrylamide)-co-(N,N′-bis(acryloyl)piperazine)-co-(5-amino-1-pentanol)] and ISA1: poly[(N,N′-bis(acryloyl)piperazine)-co-(2-methylpiperazine)-co-(N,N′-bis(2-hydroxyethyl)ethylenediamine)]) have been reported elsewhere.30
Methods
Agarose gel retardation assay.
Samples of PAA:RNase A–OG were prepared by mixing 18.7 μL of PAAs (2.4–480 mg mL−1 in 10 mM HEPES, pH = 7.4, 5 wt% D-glucose) with 3.8 μL of RNase A–OG (1.18 mg mL−1). The mixtures were left at room temperature (r.t.) for 30 min. and 21.5 μL of HEPES buffer was added. Samples were applied onto a 0.7% (w/v) agarose gel. Electrophoresis was run at 60 V for 45 min. in a 1× TAE buffer. RNase A–OG was visualised using a G:BOX transilluminator (Syngene). Free protein was used as a control. [Oregon green (OG) labelling of RNase A (RNase A–OG) was carried out according to Life Technologies protocol (degree of labelling found: 0.33 mol OG mol−1 RNase A). No free dye was detected following thorough purification by PD10 column and ultracentrifugation (MWCO 3 kDa)].
High sensitivity differential scanning calorimetry.
Thermal stability of RNase A was determined using a Nano DSC microcalorimeter (TA Instruments) equipped with capillary cells (300 μL). PAA:RNase A samples at ratio 10:1 (w/w) were prepared by mixing RNase A (400 μL, 2.5 mg mL−1, 10 mM HEPES pH = 7.4, 0.15 M NaCl) with appropriate PAA solutions (1.6 mL, 6.25 mg mL−1, 10 mM HEPES pH 7.4, 0.15 M NaCl). Final concentration of RNase A was 0.5 mg mL−1. The samples were left at room temperature for 30 min. and were degassed for 10 min., at 10 °C and under vacuum (23 mmHg). During the analysis, a pressure of 4 atm was applied over the reference and sample cells. Thermograms were recorded from 10 °C to 100 °C with a scanning rate of 1 °C min−1. Data were analysed using the Launch Nanoanalyze software from TA Instrument. No thermal transition were detected for the PAAs alone.30
Dynamic light scattering.
Measurements were carried out at 25 °C using a nanoZS zetasizer (Malvern). Complexes were prepared as previously described at PAA:RNase A (w/w) ratios from 10:1 to 500:1 with an RNase final concentration of 25 μgmL−1. In order to increase the resolution of the peak observed, the analysis of the results was carried out using narrow mode.
Enzymatic activity.
Complexes (11 μL) were prepared as previously described at PAA:RNase A (w/w) ratios from 1:1 to 500:1 using 40 ng of RNase A. The mixtures were left at r.t. for 30 min. 4 μg of RNA (5 μL, 0.8 mg mL−1) were added and the samples were further incubated for 30 min. at 37 °C. Samples were then applied onto a 1% (w/v) agarose gel. Electrophoresis was run at 40 V for 30 min. in a 1× TAE buffer. The gels were stained for 30 min. in 200 mL of 1× TAE buffer containing ethidium bromide (0.5 μg mL−1) and visualised using the transilluminator.
Cells and culture conditions.
HEK293 cells were cultured in DMEM supplemented with 10% FBS, 1× PSG and 1 × MEM NEAA. B16F1 cells were cultured in RPMI supplemented with 10% FBS and 1× PSG. All cells were maintained in a humidified 5% CO2-containing atmosphere at 37 °C and passaged when reaching 80% confluence.
Cytotoxicity of the PAAs.
HEK293 cells (105 cells per well) were seeded in 96-well plates. After 24 h, the medium was replaced by filtered (0.2 μm) polymer solutions (100 μL; 0–20 mg mL−1) and fresh medium (100 μL). MTS assay: After 21 h incubation, PAAs solutions were removed and fresh medium without FBS was added (200 μL). MTS:PMS reagent (20 μL; 2:0.92 mg mL−1 in DPBS) was added and cells were left at 37 °C for 3 h. Absorbance was read at 492 nm. Viability was expressed as percentage of untreated control cells. IC50 values were determined graphically. LDH release assay: After 24 h incubation, the cells were centrifuged at 1000 rpm for 5 min. and 100 μL of the supernatant was transferred to clean 96-well plates. 90 μL of fresh reagent I and 10 μL of fresh reagent II were sequentially added to each well [reagent I: sodium-L-lactate (800 μL; 0.54 mM), NAD (800 μL; 1.30 mM) diluted into 5.6 mL TRIS buffer (0.2 M) pH = 8.2; reagent II: INT in DMSO (1 mL; 66 mM) and PMS in TRIS buffer (1 mL; 28 mM)]. The plates were incubated for 30 min. in the dark and at room temperature. The reaction was stopped by adding 15 μL of HCl (1 M). Absorbance was read at 492 nm. LDH release was expressed as percentage release (1) relative to control cells treated with 2% triton X (TX) and LDH20/10 values were determined graphically.| |  | (1) |
Cytotoxicity of RNase A.
B16F1 cells (104 cells per well) were seeded in 96-well plates. After 24 h, the medium was replaced by fresh medium (100 μL). 100 μL of filtered (0.2 μm) RNase A solutions (0.2 ng mL−1–200 μg mL−1) or polymer solutions (0–40 mg mL−1) without or incubated with RNase A (30 min. at r.t.; 25 μg mL−1 final concentration) were added to the cells. After 5 h of incubation at 37 °C, the solutions were removed and fresh medium (200 μL) was added. Following a further 40 h, an MTS assay was carried out as described above. IC50 values were determined by nonlinear regression using a sigmoidal dose–response curve and eqn (2) with a slope h = −2 as a constraint (goodness of fit, R2 > 0.9 for all fits). Data represent mean value of two independent experiments with triplicates and were further analysed using an unpaired t-test to calculate two-tailed p values with a confidence of interval set as 99%. IC50 were considered significantly different if p < 0.005 (GraphPad Prism).| | 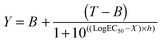 | (2) |
where B and T are the bottom and the top of the curve, respectively and h the Hill slope.
Cellular uptake.
B16F1 cells (105 cells per well) were seeded in 24-well plates. After 24 h, the medium was replaced (200 μL) and 200 μL of PAA:RNase A–OG complexes (1:1 to 50:1 (w/w) ratios; 25 μg mL−1 RNase A–OG final concentration) were added. The cells were incubated at 37 °C for 5 h, rinsed with ice-cold PBS and recovered by trypsinisation. The samples were then centrifuged for 3 min at 800 g. The supernatant was removed and the cells suspended in ice-cold PBS (300 μL) for analysis by flow cytometry. For each sample 10000 events were acquired, untreated cells were used as reference and RNase A–OG and polymer solutions were used as control. The same experiments were also conducted at 4 °C in which case the cells were left at 4 °C for 1 h prior addition of the complexes. The cellular uptake was calculated using eqn (3).| | 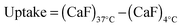 | (3) |
where CaF represents the cell associated fluorescence at 37 °C or 4 °C calculated as previously described.22
Protein surface modelling.
Modelisations were carried out using the Swiss-Pdb viewer (v4.1.0). Coordinates for RNase A were those deposited in the Protein Data Bank (PDB) as data set “1KF5”.62 The electrostatic potential surrounding RNase A was calculated by the Poisson–Boltzmann equation using a dielectric constants of 2 for the protein interior, 80 for the solvent region and an ionic strength of 0.15 mM.
Conclusions
We demonstrated the ability of linear cationic poly(amidoamine)s ISA1, P1 and P2 (ζ = + 26.6 to + 27.6 mV at pH 7.4) to interact with RNase A on the “wrong side” of the isoelectric point due to the presence of “negative patches” on the surface of the protein at physiological pH. Upon interaction, a heterogeneous population of nanocomplexes (10 to 100 nm) were observed. Microcalorimetry further indicated that at PAA:RNase A - 10:1 (w/w) ratio, P1 and P2 did not destabilise RNase A whereas ISA1 did. Upon 24 h incubation all the PAAs displayed low cytotoxicity towards non-cancerous cells (IC50 = 3.5 to 6 mg mL−1; LDH20 = 0.5 to 1 mg mL−1). Free wild-type RNase A was not toxic to B16F1 cells up to a concentration of 100 μg mL−1. However, when complexed with P1, increased cytotoxicity and cellular uptake was observed indicating that P1 was able to promote intracellular delivery of biologically active RNase A (IC50 = 0.09 mg mL−1) and potentially helped to bypass or overpower the inhibitory action of the RI found in the cytosol of most cells.
Acknowledgements
We would like to thank the Medway School of Pharmacy for financial support and Pr. B. Chowdhry (University of Greenwich) for providing access to the HSDSC.
References
- L. Ledoux, Nature, 1955, 176, 36–37 CrossRef CAS PubMed.
-
A. W. Nicholson, Ribonucleases, Springer, Berlin Heidelberg, 2011 Search PubMed.
- C. Ercole, R. A. Colamarino, E. Pizzo, F. Fogolari, R. Spadaccini and D. Picone, Biopolymers, 2009, 91, 1009–1017 CrossRef CAS PubMed.
- J. E. Lee and R. T. Raines, Biochemistry, 2005, 44, 15760–15767 CrossRef CAS PubMed.
- A. Di Donato, V. Cafaro and G. D'Alessio, J. Biol. Chem., 1994, 269, 17394–17396 CAS.
- V. Cafaro, A. Bracale, A. Di Maro, S. Sorrentino, G. D'Alessio and A. Di Donato, FEBS Lett., 1998, 437, 149–152 CrossRef CAS.
- P. Pouckova, M. Morbio, F. Vottariello, D. V. Laurents, J. Matousek, J. Soucek, G. Gotte, M. Donadelli, C. Costanzo and M. Libonati, Bioconjugate Chem., 2007, 18, 1946–1955 CrossRef CAS PubMed.
- F. Vottariello, C. Costanzo, G. Gotte and M. Libonati, Bioconjugate Chem., 2010, 21, 635–645 CrossRef CAS PubMed.
- S. K. Saxena, S. M. Rybak, G. Winkler, H. M. Meade, P. Mcgray, R. J. Youle and E. J. Ackerman, J. Biol. Chem., 1991, 266, 21208–21214 CAS.
- M. R. Smith, D. L. Newton, S. M. Mikulski and S. M. Rybak, Exp. Cell Res., 1999, 247, 220–232 CrossRef CAS PubMed.
- F. Leich, N. Stohr, A. Rietz, R. Ulbrich-Hofmann and U. Arnold, J. Biol. Chem., 2007, 282, 27640–27646 CrossRef CAS PubMed.
- C. De Lorenzo, C. Di Malta, G. Cali, F. Troise, L. Nitsch and G. D'Alessio, FEBS Lett., 2007, 581, 296–300 CrossRef CAS PubMed.
- D. Wang and S. Moore, Biochemistry, 1977, 16, 2937–2942 CrossRef CAS.
- J. Futami, E. Nukui, T. Maeda, M. Kosaka, H. Tada, M. Seno and H. Yamada, J. Biochem., 2002, 132, 223–228 CrossRef CAS.
- J. Futami, T. Maeda, M. Kitazoe, E. Nukui, H. Tada, M. Seno, M. Kosaka and H. Yamada, Biochemistry, 2001, 40, 7518–7524 CrossRef CAS PubMed.
- J. Futami, M. Kitazoe, T. Maeda, E. Nukui, M. Sakaguchi, J. Kosaka, M. Miyazaki, M. Kosaka, H. Tada, M. Seno, Y. Sasaki, N. H. Huh, M. Namba and H. Yamada, J. Biosci. Bioeng., 2005, 99, 95–103 CrossRef CAS.
- J. Zhang, J. Du, M. Yan, A. Dhaliwal, J. Wen, F. Liu, T. Segura and Y. Lu, Nano Res., 2011, 4, 425–433 CrossRef CAS.
- J. Matousek, P. Pouckova, J. Soucek and J. Skvor, J. Controlled Release, 2002, 82, 29–37 CrossRef CAS.
- P. Pouckova, M. Zadinova, D. Hlouskova, J. Strohalm, D. Plocova, M. Spunda, T. Olejar, M. Zitko, J. Matousek, K. Ulbrich and J. Soucek, J. Controlled
Release, 2004, 95, 83–92 CrossRef CAS PubMed.
- S. X. Liu, L. J. Sun, J. Wang, G. H. Ma, Z. G. Su and T. Hu, Process Biochem., 2012, 47, 1364–1370 CrossRef CAS PubMed.
- P. Ferruti, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2319–2353 CrossRef CAS PubMed.
- S. C. W. Richardson, N. G. Pattrick, N. Lavignac, P. Ferruti and R. Duncan, J. Controlled Release, 2010, 142, 78–88 CrossRef CAS PubMed.
- N. Lavignac, J. L. Nicholls, P. Ferruti and R. Duncan, Macromol. Biosci., 2009, 9, 480–487 CrossRef CAS PubMed.
- G. Coué and J. F. J. Engbersen, J. Controlled Release, 2011, 152, 90–98 CrossRef PubMed.
- S. Cohen, G. Coué, D. Beno, R. Korenstein and J. F. J. Engbersen, Biomaterials, 2012, 33, 614–623 CrossRef CAS PubMed.
- G. Coué, C. Freese, R. E. Unger, C. J. Kirkpatrick and J. F. J. Engbersen, Acta Biomater., 2013, 9, 6062–6074 CrossRef PubMed.
- N. G. Pattrick, S. C. W. Richardson, M. Casolaro, P. Ferruti and R. Duncan, J. Controlled Release, 2001, 77, 225–232 CrossRef CAS.
- N. Lavignac, M. Lazenby, P. Foka, B. Malgesini, I. Verpilio, P. Ferruti and R. Duncan, Macromol. Biosci., 2004, 4, 922–929 CrossRef CAS PubMed.
- N. Lavignac, M. Lazenby, J. Franchini, P. Ferruti and R. Duncan, Int. J. Pharm., 2005, 300, 102–112 CrossRef CAS PubMed.
- J. L. N. Dubois and N. Lavignac, Polym. Chem., 2014, 5, 1586–1592 RSC.
- A. C. Hunter, Adv. Drug Delivery Rev., 2006, 58, 1523–1531 CrossRef CAS PubMed.
- L. Parhamifar, A. K. Larsen, A. C. Hunter, T. L. Andresen and S. M. Moghimi, Soft Matter, 2010, 6, 4001–4009 RSC.
- International Organization for Standardization, ISO 10993-5:2009(E), Biological evaluation of medical devices Part 5: Tests for in vitro cytotoxicity, Switzerland. 2009.
- L. Parhamifar, H. Andersen and S. M. Moghimi, Methods Mol. Biol., 2013, 948, 13–22 CAS.
- L. Peng, Y. Gao, Y.-N. Xue, S.-W. Huang and R.-X. Zhuo, Biomaterials, 2010, 31, 4467–4476 CrossRef CAS PubMed.
- M. Karayianni, S. Pispas, G. D. Chryssikos, V. Gionis, S. Giatrellis and G. Nounesis, Biomacromolecules, 2011, 12, 1697–1706 CrossRef CAS PubMed.
- H. Larsericsdotter, S. Oscarsson and J. Buijs, J. Colloid Interface Sci., 2001, 237, 98–103 CrossRef CAS PubMed.
- E. Sedlak, D. Fedunova, V. Vesela, D. Sedlakova and M. Antalik, Biomacromolecules, 2009, 10, 2533–2538 CrossRef CAS PubMed.
- J. L. Xia, P. L. Dubin, Y. Kim, B. B. Muhoberac and V. J. Klimkowski, J. Phys. Chem., 1993, 97, 4528–4534 CrossRef CAS.
- K. W. Mattison, P. L. Dubin and I. J. Brittain, J. Phys. Chem. B, 1998, 102, 3830–3836 CrossRef CAS.
- T. Hattori, R. Hallberg and P. L. Dubin, Langmuir, 2000, 16, 9738–9743 CrossRef CAS.
- E. Seyrek, P. L. Dubin, C. Tribet and E. A. Gamble, Biomacromolecules, 2003, 4, 273–282 CrossRef CAS PubMed.
- K. J. Yeo, E. Hwang, K. M. Min, J. G. Jee, C. K. Lee, K. Y. Hwang, Y. H. Jeon, S. I. Chang and H. K. Cheong, Chem. Commun., 2014, 50, 12966–12969 RSC.
- C. Tribet, I. Porcar, P. A. Bonnefont and R. Audebert, J. Phys. Chem. B, 1998, 102, 1327–1333 CrossRef CAS.
- M. E. Lienqueo, A. Mahn, J. C. Salgado and J. A. Asenjo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 849, 53–68 CrossRef CAS PubMed.
- A. Mahn, M. E. Lienqueo and J. A. Asenjo, J. Chromatogr. A, 2004, 1043, 47–55 CrossRef CAS PubMed.
- A. J. Wilson, J. Hong, S. Fletcher and A. D. Hamilton, Org. Biomol. Chem., 2007, 5, 276–285 CAS.
- Y. L. Lo and Y. E. Rahman, Int. J. Pharm., 1998, 161, 137–148 CrossRef CAS.
- P. Sasisanker, A. Oleinikova, H. Weingartner, R. Ravindra and R. Winter, Phys. Chem. Chem. Phys., 2004, 6, 1899–1905 RSC.
- F. Foglia, P. Carullo and P. Del Vecchio, J. Therm. Anal. Calorim., 2008, 91, 67–72 CrossRef CAS.
- N. L. Almeida, C. L. P. Oliveira, I. L. Torriani and W. Loh, Colloids Surf., B, 2004, 38, 67–76 CrossRef CAS PubMed.
- D. Romanini, M. Braia, R. G. Angarten, W. Loh and G. Picó, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 857, 25–31 CrossRef CAS PubMed.
- O. N. Ivinova, V. A. Izumrudov, V. I. Muronetz, I. Y. Galaev and B. Mattiasson, Macromol. Biosci., 2003, 3, 210–215 CrossRef CAS PubMed.
- M. van de Weert, M. B. Andersen and S. Frokjaer, Pharm. Res., 2004, 21, 2354–2359 CrossRef CAS.
- W. Pfeil, Thermochim. Acta, 2002, 382, 169–174 CrossRef CAS.
- T. Y. Chao and R. T. Raines, Biochemistry, 2011, 50, 8374–8382 CrossRef CAS PubMed.
- P. A. Leland, L. W. Schultz, B. M. Kim and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10407–10412 CrossRef CAS.
- G. A. Ellis, M. L. Hornung and R. T. Raines, Bioorg. Med. Chem. Lett., 2011, 21, 2756–2758 CrossRef CAS PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS PubMed.
- X. W. Ding, Y. Liu, J. H. Li, Z. Luo, Y. Hu, B. L. Zhang, J. J. Liu, J. Zhou and K. Y. Cai, ACS Appl. Mater. Interfaces, 2014, 6, 7395–7407 CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- R. Berisio, F. Sica, V. S. Lamzin, K. S. Wilson, A. Zagari and L. Mazzarella, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 441–450 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: MP4 file for the 3D animation representing the surface of bovine RNase A (PDB: 1kf5) including the electrostatic potential and hydrophobic patches. See DOI: 10.1039/c4tb02065k |
| ‡ Current address: Laboratoire ERRMECe, Université de Cergy-Pontoise, 2 av. Adolphe Chauvin BP222, 95302 Cergy-Pontoise Cedex, France. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*