
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Histamine-functionalized copolymer micelles as a drug delivery system in 2D and 3D models of breast cancer†
Yuning
Zhang
a,
Pontus
Lundberg
b,
Maren
Diether
a,
Christian
Porsch
c,
Caroline
Janson
b,
Nathaniel A.
Lynd
b,
Cosimo
Ducani
d,
Michael
Malkoch
c,
Eva
Malmström
c,
Craig J.
Hawker
b and
Andreas M.
Nyström
*a
aIMM Institute of Environmental Medicine, Karolinska Institutet, SE-171 77, Stockholm, Sweden. E-mail: andreas.nystrom@ki.se
bMaterials Research Laboratory, University of California, Santa Barbara, CA 93106, USA
cKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Fibre and Polymer Technology, SE-100 44, Stockholm, Sweden
dSwedish Medical Nanoscience Center, Department of Neuroscience, Karolinska Institutet, SE-171 77, Stockholm, Sweden
First published on 12th February 2015
Abstract
Histamine functionalized block copolymers based on poly(allyl glycidyl ether)-b-poly(ethylene oxide) (PAGE-b-PEO) were prepared with different ratios of histamine and octyl or benzyl groups using UV-initiated thiol-ene click chemistry. At neutral pH, the histamine units are uncharged and hydrophobic, while in acidic environments, such as in the endosome, lysosomes, or extracellular sites of tumours, the histamine groups are positively charged and hydrophilic. pH responsible polymer drug delivery systems is a promising route to site specific delivery of drugs and offers the potential to avoid side effects of systemic treatment. Our detailed in vitro experiments of the efficacy of drug delivery and the intracellular localization characteristics of this library of NPs in 2D and 3D cultures of breast cancer revealed that the 50% histamine-modified polymer loaded with DOX exhibited rapid accumulation in the nucleus of free DOX within 2 h. Confocal studies showed enhanced mitochondrial localization and lysosomal escape when compared to controls. From these combined studies, it was shown that by accurately tuning the structure of the initial block copolymers, the resulting self-assembled NPs can be designed to exploit histamine as an endosomal escape trigger and the octyl/benzyl units give rise to a hydrophobic core resulting in highly efficacious drug delivery systems (DDS) with control over intracellular localization. Optimization and rational control of the intracellular localization of both DDS and the parent drug can give nanomedicines a substantial increase in efficacy and should be explored in future studies.
1. Introduction
Polymer based materials and nanoparticles (NPs) are increasingly being investigated in a variety of biomedical applications, such as supporting cell growth,1 as protein carriers,2 gene3–5 and chemotherapeutics delivery systems6,7 especially for cancer treatment. Unlike disaccharides,8 peptides,9 and inorganic NPs10 that require further polymeric or liposomal modifications to serve as a drug delivery system (DDS), the amphiphilic block copolymers can self-assemble into core–shell NPs in aqueous solution without further modification. This combined with their ability to encapsulate a wide range of active compounds offers promise in the design of NPs for effective targeted delivery.11,12 Such NP platforms have been shown to enhance drug solubility,13–15 decrease side effects,16–18 and enhance the blood circulation time,19,20 which facilitates specific tumour uptake via the enhanced permeability and retention (EPR) effect.21–23 However, it is well known that cells protect themselves by entrapping most exogenous substances, including both small molecular drugs and NP-based DDS, in their endo-lysosomal system.24,25 The entrapped substances are then partially or fully degraded which reduces their therapeutic effect.20,26,27 Consequently, a key issue when designing NPs for drug delivery has been enhancing cellular uptake by promoting endo-lysosomal escape of the carriers and their cargo into the cytoplasm.28,29 One widely studied approach for enabling NPs to escape the endo-lysosomal system is to utilize the pH difference between healthy cells and tumour cells.30–32 The pH in the extracellular fluids surrounding healthy tissues and in the blood stream is typically around 7.4, whereas the pH of extracellular fluids of tumour tissues is lower33 as well as intracellular organelles such as early endosomes (6.0–6.5) and lysosomes (4.5–5.5).34 In this acidic environment, positively charged polymers can cause endosomal/lysosomal rupture via the so-called “proton sponge effect”.35,36 However, polymers with a permanent positive charge, like poly(ethylene imine) have been found to be cytotoxic to cells.37,38 Therefore, efforts have been directed toward the design of non-toxic nanocarriers with pH-responsive charge properties. Kataoka et al. have developed various polymer-based nanocarriers for pDNA3,39,40 and siRNA41,42 delivery. For example, the Kataoka group have recently reported a pH-reversible polymer platform based on maleic acid amide (MAA) for endosomal escape of siRNA by switching from negatively to positively charged MAA residues when the compound is exposed to an acidic environment.4 Another interesting alternative is to utilize histidine43 or histamine44 groups as the pH-responsive component due to their enhanced biocompatibility and appropriate pH response in acidic environments. While limited efforts have been made to use histidine-containing polymer NPs as gene45–47 or chemotherapeutic43,48,49 delivery systems, only a few research papers have been published on histamine-functionalized block co-polymers as delivery platforms.44,50We have previously reported on pH-responsive poly(allyl glycidyl ether)-b-poly(ethylene oxide)-b-poly(allyl glycidyl ether) (PAGE-b-PEO-b-PAGE) polymers functionalized with histamine groups.51 These polymers rapidly form NPs in response to changes in pH and proved to be non-toxic to cells. But the hydrophobic components of our previous study needed to be further engineered to achieve the goal of drug delivery, as in the previous study the NPs were incapable of retaining the drug cargo in an efficient way. In the current work, we have elaborated on the performance of histamine-functionalized NPs as DDS which can partly escape from endo-lysosomes and relocate the drug into the mitochondria. Utilization of NPs for mitochondria targeting of chemotherapeutic compounds is a methodology that has emerged as a method for overcoming drug resistance.52–54 A library of different PAGE-b-PEO diblock copolymers were synthesized and functionalized with histamine and/or octyl/benzyl groups to yield a range of functional polymers that can form stable NPs in aqueous solutions. The addition of hydrophobic benzyl or octyl moieties was expected to enhance the drug loading and release behaviour of the NPs due to increased hydrophobicity. The effect of varying histamine content and the hydrophilic/hydrophobic ratio of NPs were thoroughly evaluated in 2D and 3D cell culture models.
2. Experimental
2.1 Materials
Dulbecco's modified Eagle's medium (DMEM), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), phosphate buffered saline (PBS), formaldehyde solution, water, sodium dodecyl sulfate (SDS), methylcellulose powder, ethylene oxide (EO, ≥99.5%), benzyl alcohol (BzOH, 99.8%), 2,2-dimethoxy-2-phenylacetophenone (DMPA, 99%), octanethiol (≥98.5%), and benzyl mercaptane (99%) were purchased from Sigma-Aldrich and stored per the manufacturers' instructions. Phenol red-free DMEM with HEPES was obtained from Gibco. Fetal bovine serum (FBS) and penicillin–streptomycin solution were purchased from Hyclone Laboratories. Specific antibodies – anti-EEA1 and anti-TGN46 – were obtained from Abcam Inc. The fluorescent dyes 4′,6-diamidino-2-phenylindole (DAPI), LysoTracker, MitoTracker, and Donkey Anti-Mouse IgG antibody Alexa 647 were all purchased from Invitrogen and stored at −20 °C as recommended. The CellTiter-Glo Luminescent Cell Viability Assay was obtained from Promega and stock solutions were prepared and stored following the manufacturer's instructions. Methanol (MeOH) was purchased from Fisher Scientific. Allyl glycidyl ether (AGE) ≥99.0% (TCI) was degassed three times and distilled from butyl magnesium chloride before use. Potassium naphthalenide was prepared by dissolving recrystallized naphthalene (10 g) in dry tetrahydrofuran (THF, 250 mL) and then added potassium metal (3 g) under positive argon pressure. The system was closed with a septum and allowed to stir overnight with a glass-coated stir-bar. N-2-(1H-imidazol-4-yl)ethyl-4-mercaptobutanamide (Hist-SH) was synthesized as previously reported.51 MCF-7, MDA-MB-231, and MDA-MB-468 cells were purchased from the American Type Culture Collection (ATCC) and maintained in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin at 37 °C with 5% CO2.2.2 Instrumentation
NMR spectra were recorded on a Varian VNMRS 600 MHz using MeOD (Cambridge Isotope Laboratories) as the solvent. Fluorescence spectroscopy was performed on a Cary Eclipse from Varian using an excitation wavelength of 333.2 nm, and emission spectra were collected between 350 nm and 420 nm. Gel permeation chromatography (GPC) was performed on a TOSOH EcoSEC HLC-8320GPC system equipped with an EcoSEC RI detector and three columns (PSS PFG 5 μm; Microguard, 100 Å, and 300 Å) (MW resolving range: 100–300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) from PSS GmbH, using DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was created using narrow poly(methyl methacrylate) standards ranging from 700 to 2000000 g mol−1, and used to determine the average molar masses (number-average molar mass, Mn and weight-average molar mass, Mw) and the dispersity (ĐM = Mw/Mn). Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process the data. Dynamic light scattering was performed on a Malvern Zetasizer NanoTZ with 10 scans for each sample following a 2 min equilibration. TEM images were obtained with a FEI Morgagni 268(D) transmission electron microscope at 80 kV at magnification 44000. Apoptosis was evaluated on an Accuri C6 flow cytometer using FL-1/FL-3 to collect Annexin V FITC/PI fluorescence separately. MTT absorbance readings were obtained 18 h later with a BioTek Synergy™ MX multi-mode micro plate reader operated by the Gen5™ software at a wavelength of 570 nm. The DOX fluorescence intensity was measured with the same equipment at an excitation wavelength of 485 nm and an emission wavelength of 595 nm. The luminance reading was obtained with an Infinite F200 Tecan plate reader with luminance model. A Zeiss LSM 510 Meta confocal microscope with temperature control was used for both 2D and 3D imaging with lasers for DOX (543 nm laser, BP 560–615), Hoechst 33342 (405 nm laser, BP 420–480), and deep red dyes (633 nm laser, LP 650). A 40× oil-immersion objective was used for the 2D models, and a 10× objective was applied for the 3D model. The Z-stack distance was set in the range of 100 ± 10 μm. Data were processed and 3D images were reconstructed with Imars software. Co-localization analysis was conducted with the ImageJ software with the JACoP plugs in.
000 g mol−1) from PSS GmbH, using DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was created using narrow poly(methyl methacrylate) standards ranging from 700 to 2000000 g mol−1, and used to determine the average molar masses (number-average molar mass, Mn and weight-average molar mass, Mw) and the dispersity (ĐM = Mw/Mn). Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process the data. Dynamic light scattering was performed on a Malvern Zetasizer NanoTZ with 10 scans for each sample following a 2 min equilibration. TEM images were obtained with a FEI Morgagni 268(D) transmission electron microscope at 80 kV at magnification 44000. Apoptosis was evaluated on an Accuri C6 flow cytometer using FL-1/FL-3 to collect Annexin V FITC/PI fluorescence separately. MTT absorbance readings were obtained 18 h later with a BioTek Synergy™ MX multi-mode micro plate reader operated by the Gen5™ software at a wavelength of 570 nm. The DOX fluorescence intensity was measured with the same equipment at an excitation wavelength of 485 nm and an emission wavelength of 595 nm. The luminance reading was obtained with an Infinite F200 Tecan plate reader with luminance model. A Zeiss LSM 510 Meta confocal microscope with temperature control was used for both 2D and 3D imaging with lasers for DOX (543 nm laser, BP 560–615), Hoechst 33342 (405 nm laser, BP 420–480), and deep red dyes (633 nm laser, LP 650). A 40× oil-immersion objective was used for the 2D models, and a 10× objective was applied for the 3D model. The Z-stack distance was set in the range of 100 ± 10 μm. Data were processed and 3D images were reconstructed with Imars software. Co-localization analysis was conducted with the ImageJ software with the JACoP plugs in.
2.3 Methods
300 g mol−1; PAGE20–PEO430 (LH0) = 21200 g mol−1, PAGE41–PEO486 (HH0) = 26100 g mol−1. Molecular weight by SEC (DMF): PAGE22–PEO246 (LL0) = 11000 g mol−1 (PDI = 1.1); PAGE20–PEO430 (LH0) = 19700 g mol−1 (PDI = 1.1); PAGE41–PEO486 (HH0) = 19300 g mol−1 (PDI = 1.1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–), 2.30 (t, J = 12 Hz, –CH2–C(O)–NH–), 2.52–2.60 (m, –S–CH2–CH2–CH2–O–, –S–CH2–CH2–CH2–C(O)– and –S–CH2–CH2–), 2.78 (t, J = 12 Hz, >C–CH2–CH2–NH–C(O)–), 3.43 (t, J = 12 Hz, –CH2–NH–C(O)–), 3.45–3.80 (m, backbone and –O–CH2–CH–), 4.56 (s, Ar–CH2–O–), 6.85 (s, –NH–CH–C<), 7.36 (m, Ar–), 7.59 (s, –NH–CHN–). 1H-NMR (MeOD) δ (ppm) = [P(HGE–BGE)–PEO] = 1.78–1.84 (m, –O–CH2–CH2–CH2–S– and –S–CH2–CH2–CH2–C(O)–), 2.47–2.55 (m, –S–CH2–CH2–CH2–O– and –S–CH2–CH2–CH2–C(O)–), 2.76 (t, J = 12 Hz, >C–CH2–CH2–NH–C(O)–), 3.41 (t, J = 12 Hz, –CH2–NH–C(O)–), 3.45–3.80 (m, backbone, –O–CH2–CH–, and –S–CH2–Ar), 4.52 (s, Ar–CH2–O–), 6.83 (s, –NH–CH–C<), 7.20–7.32 (m, Ar–CH2–S– and Ar–CH2–O), 7.59 (s, –NH–CHN–). Poor solubility made SEC analysis impossible after functionalization.
:1) using a standard curve of DOX absorbance at 490 nm.55,56
:1) was added to each sample to disaggregate the NPs. The resulting solutions were transferred into 96-well fluorescence plates, and the fluorescence intensity was determined with a BioTek Synergy™ MX multi-mode micro plate reader.
000 events in the population. FITC-labelled Annexin V was excited by the 488 nm laser and collected in the FL-1 (533/30 nm) detector, and propidium iodide fluorescence was collected in the FL-3 (>670 nm) detector. Data was analyzed using the BD Accuri C6 Analysis software.
O)–), 2.30 (t, J = 12 Hz, –CH2–C(O)–NH–), 2.52–2.60 (m, –S–CH2–CH2–CH2–O–, –S–CH2–CH2–CH2–C(O)– and –S–CH2–CH2–), 2.78 (t, J = 12 Hz, >C–CH2–CH2–NH–C(O)–), 3.43 (t, J = 12 Hz, –CH2–NH–C(O)–), 3.45–3.80 (m, backbone and –O–CH2–CH–), 4.56 (s, Ar–CH2–O–), 6.85 (s, –NH–CH–C<), 7.36 (m, Ar–), 7.59 (s, –NH–CHN–). 1H-NMR (MeOD) δ (ppm) = [P(HGE–BGE)–PEO] = 1.78–1.84 (m, –O–CH2–CH2–CH2–S– and –S–CH2–CH2–CH2–C(O)–), 2.47–2.55 (m, –S–CH2–CH2–CH2–O– and –S–CH2–CH2–CH2–C(O)–), 2.76 (t, J = 12 Hz, >C–CH2–CH2–NH–C(O)–), 3.41 (t, J = 12 Hz, –CH2–NH–C(O)–), 3.45–3.80 (m, backbone, –O–CH2–CH–, and –S–CH2–Ar), 4.52 (s, Ar–CH2–O–), 6.83 (s, –NH–CH–C<), 7.20–7.32 (m, Ar–CH2–S– and Ar–CH2–O), 7.59 (s, –NH–CHN–). Poor solubility made SEC analysis impossible after functionalization.
:1) using a standard curve of DOX absorbance at 490 nm.55,56
:1) was added to each sample to disaggregate the NPs. The resulting solutions were transferred into 96-well fluorescence plates, and the fluorescence intensity was determined with a BioTek Synergy™ MX multi-mode micro plate reader.
000 events in the population. FITC-labelled Annexin V was excited by the 488 nm laser and collected in the FL-1 (533/30 nm) detector, and propidium iodide fluorescence was collected in the FL-3 (>670 nm) detector. Data was analyzed using the BD Accuri C6 Analysis software.
For the 3D imaging, spheroids were incubated with 2 μg mL−1 DOX or DOX-NPs for 4 h and transferred into an Eppendorf tube and gently spun for 15 s. The supernatant was discarded and PBS was added. The washing step was repeated 3 times. Medium containing Hoechst (5 μg mL−1) and LysoTracker Deep Red (100 nM) or MitoTracker Deep Red FM (100 nM) was then added and incubated for another 15 min. Spheroids were washed with PBS twice then suspended in glass bottom dishes (P35G-0-10-C, MatTek). The prepared samples were visualized with a Zeiss LSM 510 Meta confocal microscope with a temperature control chamber.
3. Results and discussion
3.1 Synthesis of the polymers
We have previously reported on the synthesis of novel histamine-functional poly(allyl glycidyl ether)-b-poly(ethylene oxide)-b-poly(allyl glycidyl ether) (PAGE-b-PEO-b-PAGE) triblock copolymers with interesting pH-responsive hydrogelation behaviours.51 The fact that histamine has a pKa in a physiological relevant pH range (6.0–7.4) in combination with its biocompatibility makes it highly useful in a number of biomedical applications.44,50,60,61 For instance, Wooley et al.44 developed histamine-containing shell cross-linked NPs for siRNA delivery and found that the histamine content contributed to reduced cytotoxicity and assisted in endosomal escape. Liu et al.50 utilized an amphiphilic chondroitin sulfate–histamine conjugate to achieve pH-triggered doxorubicin (DOX) release in vitro. In the present study, structure–property relationships for histamine-functionalized polymers were investigated in the form of NPs for therapeutic delivery with a focus on the effect of histamine protonation on NP performance.In this study, a diverse polymer library of histamine-functionalized amphiphilic copolymers was synthesized by ring opening polymerization followed by post-functionalization through UV-initiated thiol-ene click chemistry.51,62,63 Poly(allyl glycidyl ether)-b-poly(ethylene oxide) (PAGE-b-PEO) diblock copolymers were initially synthesized and the polymerization reactions monitored by 1H-NMR and size exclusion chromatography (SEC) (Scheme 1). Three different well-defined polymer precursors with varying allyl glycidyl (AGE)/ethylene oxide (EO) compositions were targeted (Table 1). The AGE-blocks of the copolymers were subsequently functionalized by photochemical thiol-ene chemistry utilizing thiol-functional building blocks as illustrated in Scheme 1. By targeting different ratios of histamine glycidyl ether (HGE), octyl glycidyl ether (OGE), and benzyl-functional glycidyl ether (BGE) thiols, the role of the hydrophobic core domain on the loading and release of the chemotherapeutic cargo can be explored. These modifications resulted in a diverse library of amphiphilic diblock copolymers with different degrees of histamine functionality of the hydrophobic block, and different lengths of the hydrophilic PEO block as shown in Table 1 and (Table ESI 1†). To condense the naming of each NP's, we created a naming system to represent each material based on the molecular weight of the hydrophobic/hydrophilic components. LL = low PXGE Mw and low PEO Mw group; LH = low PXGE Mw and high PEO Mw group; HH = high PXGE Mw and high PEO Mw group; (X in PXGE = (A) allyl, (O) octyl, (H) histamine, (B) benzyl). The thiol-ene reaction was monitored by 1H-NMR and verified by quantitative functionalization. 1H-NMR spectra of neat PAGE22-b-PEO246 (LL0), P(HGE11-OGE11)-PEO246 (LL3), and P(HGE10-BGE12)-PEO246 (B2) are shown in Fig. 1. The appearance of the characteristic histamine (s and r), octyl (aa and ab), and benzyl (u, v, and x) resonances, as well as the disappearance of the allyl shift (k), can be observed in the spectra. As a result of the complex amphiphilic compositions of the functionalized diblock copolymers complete polymer dissolution only occurred in methanol or in aqueous solution (which induced micelle formation), and this prohibited SEC analysis of the final products. However no chain scission or change in the degree of polymerization is expected when compared to the fully characterized, starting diblock copolymers.
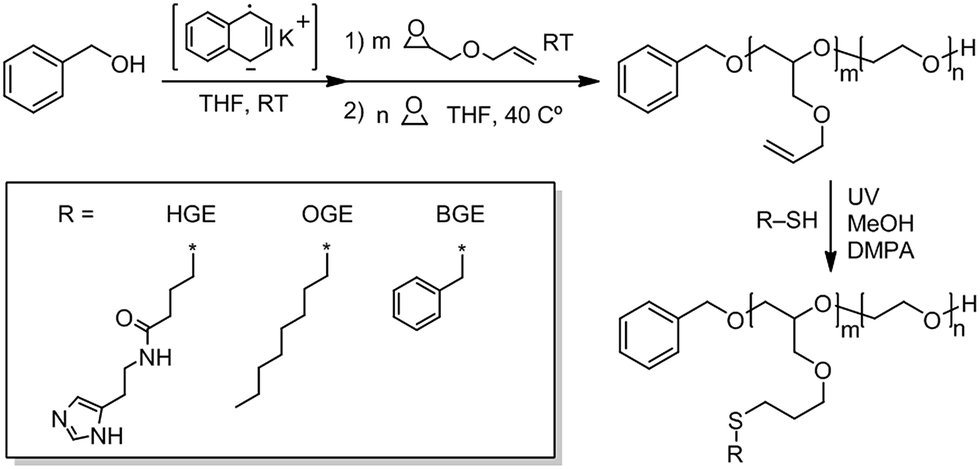 | ||
| Scheme 1 Synthetic route to diblock PAGE-b-PEO copolymer and its functionalized analogues PHGE-b-PEO, P(HGE–OGE)-b-PEO, POGE-b-PEO, P(HGE–BGE)-b-PEO, and PBGE-b-PEO. | ||
| Codesa | Materials | Histamine vs. other thiol (%) | NPs | DOX-NPs | ||
|---|---|---|---|---|---|---|
| Number meanb (d nm) | CMCc (μg mL−1) | Number mean (d nm) | Loading efficiency (%) | |||
| a LL = low PXGE Mw and low PEO Mw group; LH = low PXGE Mw and high PEO Mw group; HH = high PXGE Mw and high PEO Mw group; (X in PXGE = A, O, H). B = BGE unit group. b d.n.m = diameter in nm range. c Values obtained at pH 7.4. | ||||||
| LL1 | POGE22-b-PEO246 | 0 | 16 ± 1 | 4.0 | 80 ± 6 | 23 |
| LL2 | P(HGE7-OGE15)-b-PEO246 | 32 | 16 ± 0 | 3.8 | 260 ± 212 | 28 |
| LL3 | P(HGE11-OGE11)-b-PEO246 | 48 | 28 ± 0 | 4.3 | 42 ± 16 | 15 |
| LL4 | P(HGE18-OGE4)-b-PEO246 | 80 | 39 ± 1 | 10 | 63 ± 2 | 18 |
| LL5 | PHGE22-b-PEO246 | 100 | 17 ± 0 | 9.0 | 60 ± 34 | 17 |
| B1 | PBGE22-b-PEO246 | 0 | 26 ± 5 | 8.1 | 38 ± 1 | 23 |
| B2 | P(HGE10-BGE12)-b-PEO246 | 44 | 18 ± 1 | 4.2 | 25 ± 5 | 44 |
| LH1 | POGE20-b-PEO430 | 0 | 59 ± 1 | 5.7 | 56 ± 3 | 29 |
| LH2 | P(HGE4-OGE16)-b-PEO430 | 22 | 49 ± 1 | 7.9 | 49 ± 2 | 37 |
| LH3 | P(HGE10-OGE10)-b-PEO430 | 51 | 39 ± 2 | 9.4 | 38 ± 4 | 15 |
| LH4 | P(HGE16-OGE4)-b-PEO430 | 82 | 18 ± 5 | 8.4 | 25 ± 4 | 15 |
| LH5 | PHGE20-b-PEO430 | 100 | 16 ± 1 | 140 | 691 ± 1 | 15 |
| HH1 | POGE41-b-PEO486 | 0 | 129 ± 39 | 4.6 | 90 ± 5 | 23 |
| HH2 | P(HGE19-OGE22)-b-PEO486 | 45 | 38 ± 2 | 7.7 | 47 ± 3 | 16 |
| HH3 | PHGE41-b-PEO486 | 100 | 27 ± 1 | 58 | 28 ± 11 | 12 |
 | ||
| Fig. 1 1H-NMR (MeOD) of PAGE22-b-PEO246 (bottom), P(HGE11–OGE11)-b-PEO246 (middle), and P(HGE10–BGE12)-b-PEO246 (top). *Corresponds to resonances from the allyl isomer. | ||
3.2 Characterization of the polymer NPs
Fig. 2 TEM the hydrodynamic sizes of the NPs and DOX-NPs were determined by dynamic light scattering. Apart from HH1, the diameter (by number mean) of most materials ranged in size from 16 nm to 59 nm prior to drug encapsulation and the corresponding drug-loaded DOX-NPs had larger diameters as shown in Table 1. These sizes are larger compared to the size calculated from TEM images (6–20 nm) shown in Fig. 2. This is a result of TEM measuring the dry NP sizes rather than hydrodynamic sizes. It was also noted that samples, such as LH5 and LL2, had aggregation problems upon drug loading as evidenced by their hydrodynamic size increasing to several hundred nm (Table 1) and large aggregated objects seen in the TEM image (Fig. ESI 1†). This indicated that DOX encapsulation induced aggregation of the material, similar to our previous report.55 The loading efficiencies of DOX in the polymer NPs were found to be very similar regardless of the polymer composition and ranged from 15% to 30% (wt/wt) except for HH3 (12%), LH2 (36%), and B2 (44%). These loading efficiencies fall in the middle of other polymer carriers that physically encapsulate DOX, where loading efficiencies from 8% to 51% are typically observed.64–66 | ||
| Fig. 2 TEM images of micelle solutions at pH 7.4. Scale bar = 100 nm. Inset: 2.5 folds magnification of the selected areas. The average size of LL1 = 7 nm, DOX-LL1 = 11 nm; LL3 = 15 nm, DOX-LL3 = 17 nm; LL5 = 10 nm, DOX-LL5 = 19 nm; HH2 = 13 nm, DOX-HH2 = 15 nm. The contrast has been enhanced for clarity. | ||
The critical micelle concentration (CMC) represents the minimal concentration of polymer required to maintain the micellar structure. NPs with lower CMCs are considered better for systemic in vivo drug delivery applications due to reduced risk of structural collapse followed by burst release of the cargo upon dilution in the blood stream. The CMC values of the polymers are given at pH 7.4 in Table 1 and at pH 5.0 in Table ESI 1† and indicate that higher amounts of histamine modification result in higher CMC values. NPs with 0–50% histamine in the hydrophobic cores have CMCs below 10 μg mL−1 at both pH levels investigated, a value sufficiently low for drug delivery applications.67 The fluorescence intensities of free DOX and DOX-LL3 were compared at the same concentration of DOX (50 μg mL−1) by fluorescence measurements (Fig. ESI 2†). This indicated that the fluorescence was quenched to a large extent which suggested that the drug is located in a hydrophobic compartment of the NP.68
3.3 DOX release in vitro
The drug release properties for all DOX-NPs produced in this work were first evaluated in vitro in PBS at pH 6.0 to mimic the pH in the early endosome and pH 7.4 as a general pH value in healthy tissue. This simple model for release at different pH values does not mimic the complex in vivo situation, but it does allow us to compare the release kinetics from these materials to the release kinetics of other compounds under standardized conditions. The histamine groups were expected to be sensitive to differences in pH in the environment, but the release curves at different pH shown in Fig. 3 (Fig. ESI 3A and B†) did not show any significant differences between the same materials after 72 h. However, DOX-NPs with 50% histamine (DOX-LL3 and DOX-HH2) showed lower release rates within 24 and 72 h at pH 6.0 compared to pH 7.4 (Fig. 2 and ESI 3C†). This might be a result of the hydrophobic linker connecting the histamine group to the backbone of the polymer that partly shields water (and, therefore, H+) and limits the histamine responsiveness. However, we found that NPs with 50% histamine tended to have lower release rates (<65% drug release after 72 h at both pH 7.4 and pH 6.0) compared to the other materials in the same group. This indicates that the overall architectural differences might contribute to the drug release kinetics but that the histamine responsiveness does not.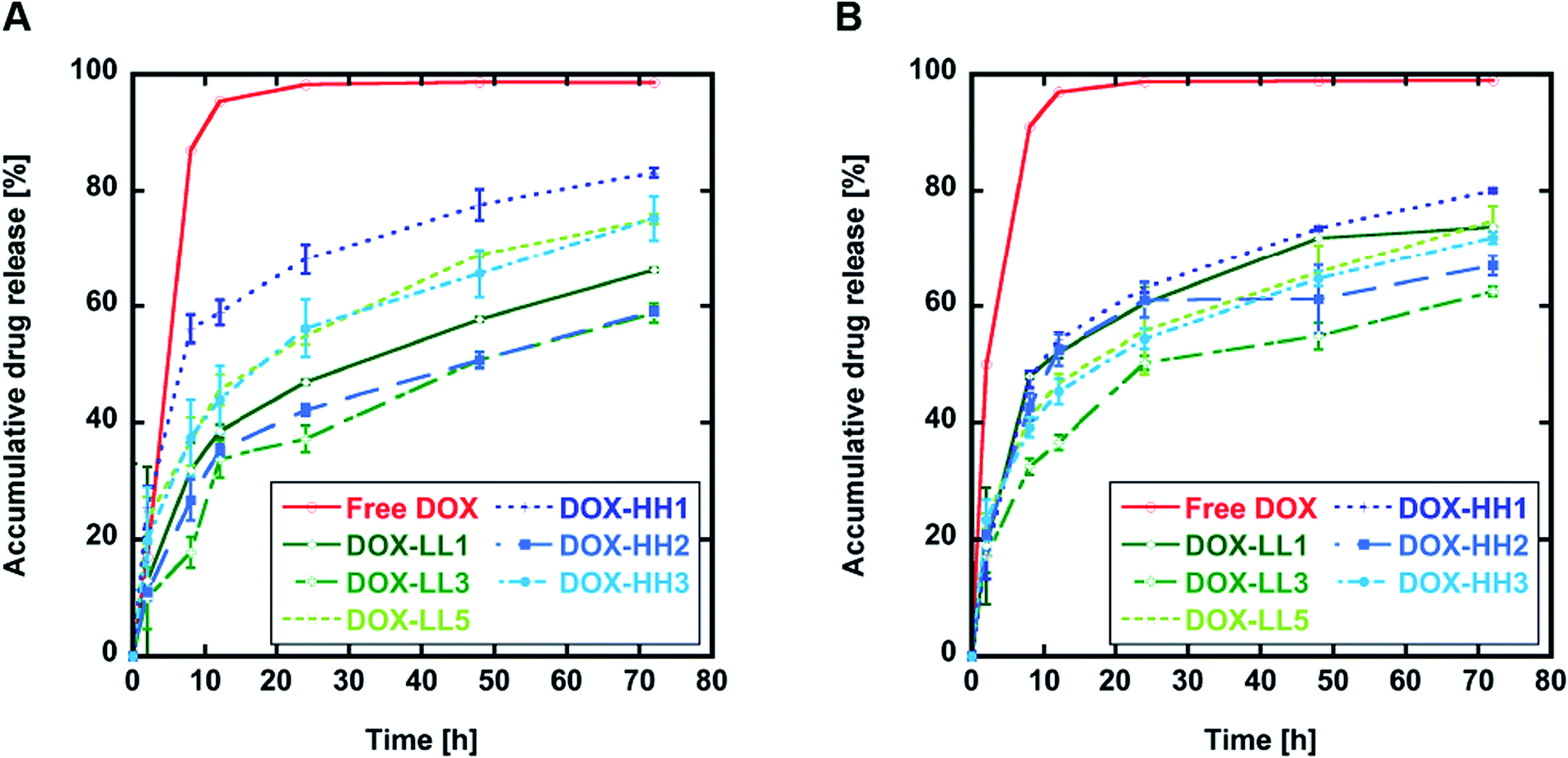 | ||
| Fig. 3 In vitro DOX release at 37 °C. (A) DOX release in sodium phosphate buffer pH 6.0. (B) DOX release in PBS pH 7.4. Results are shown as mean ± SD (n = 3). | ||
3.4 Toxicity evaluation of NPs and DOX-NPs
The initial screening of the NPs was performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays to evaluate the mitochondrial activity and partly reflects cellular viability. Cells were treated with either neat NPs or DOX-loaded NPs for 12 h, 24 h, or 72 h. No obvious loss of mitochondrial function was observed in the three breast cancer cell lines incubated with neat NPs at a high concentration (100 μg mL−1) of polymer after 12 h, 24 h, or 72 h (Fig. ESI 4†). However, significant reductions in mitochondrial activity were observed for the DOX-NP-treated cell lines, and the loss of activity was dependent on both concentration and time (Fig. 4A and ESI 5†). It should be noticed that DOX-LL3 (50% histamine) showed significant (p < 0.001) stronger toxicity compared to free DOX, DOX-LL1 (0% histamine) and DOX-LL5 (100% histamine) in all tested cell lines after 12 or 72 h treatment (Fig. 4A). In order to avoid false positive resulting from a single assay method, another sensitive viability test, the ATP CellTiter-Glo Luminescent cell viability assay, was used to check the viability of MDA-MB-231 cells. This assay measures the amount of ATP from metabolically active cells, which is proportional to the number of living cells.69 Although the absolute values (Fig. ESI 6†) were lower compared to the MTT assay at 72 h (Fig. 4A), the trends of DOX/DOX-NP efficacy towards MDA-MB-231 cells were the same in both assays. | ||
| Fig. 4 Toxicity assays of DOX-NPs. (A) Impairment of mitochondrial function as a result of treatment with DOX-loaded NPs. MTT assays after incubating DOX-NPs for 12 h or 72 h with breast cancer cell lines MDA-MB-231, MDA-MB-468, and MCF-7. Results are shown as mean ± SD (n = 5). (B) Apoptosis & Necrosis assays of DOX-NPs against MDA-MB-468 cells. Cells were incubated with 1 μg mL−1 DOX-NPs for 12 h or 72 h, then collected and stained with PI and Annexin-V FITC. Results are shown as mean ± SD (n = 3). *p < 0.05 and ***p < 0.001 compared to the control/other groups. | ||
Another interesting phenomenon was seen in Fig. ESI 5† where the majority of DOX-NPs induced similar or only slightly stronger effects on mitochondrial function when compared to free DOX after 72 h incubation, while they exhibited significantly stronger effects compared to free DOX at shorter (12 h) incubation times. Significantly, DOX-NPs only induced (p < 0.001) higher degree of cellular apoptosis (PI-Annexin V staining) when compared to free DOX at the longer incubation time (Fig. 4B). This implies that the mechanism of how free DOX and DOX-NPs influences cancer cells might be different and that this difference enhances the efficacy of DOX-NPs in mitochondria function loss over shorter time frames. To verify the early effects of DOX-NPs on these cells and to investigate more subtle differences among the materials, a drug wash-out70,71 experiment was performed on the MDA-MB-468 cell line. This cell line was selected because it is commonly studied as a breast cancer mimic, for which doxorubicin is used as a therapy, as well as the relative sensitiveness to this drug. Three materials with 0%, 50%, and 100% histamine were selected from the lower molecular weight group (with shorter PEO blocks) and three were selected from the high molecular weight group (with longer PEO blocks). As shown in Fig. ESI 7,† after 4 h or 6 h incubation there were some reduced viability effects on the cells. It was, however, obvious after 24 h incubation that all six DOX-NPs had a significant effect (p < 0.001) in reducing mitochondrial function to 40–50% compared to the 20% reduction seen with free DOX with DOX-LL3 and DOX-LH3 having increased efficacy (p < 0.001) compared to the other DOX-NPs. These results imply that mechanisms other than simple inhibition of nuclear DNA replication contribute to early loss of mitochondrial function.
3.5 Intracellular tracking of DOX-NPs in live cells
In our previous study, we found that polymer NPs can be internalized into cells via both clathrin- and macropinocytosis-mediated endocytosis, and the amount of internalized NPs increases during 24 h of studies.59 For materials in this study we did not perform in depth uptake studies but can conclude that the uptake scales with time, and given the size probably via endocytosis mechanisms. In order to further investigate how and where the drug cargo of the NPs is delivered, we performed confocal microscopy investigations of the fluorescence-stained nuclei and organelles. DOX-LL3, DOX-LH3, and DOX-HH2 NPs were used to treat MDA-MB-468 cells to study their specific sub-cellular location. Four subcellular locations were selected based on our previous study,59 including the early endosome (EEA1), the lysosome (stained by LysoTracker), the trans-Golgi network (TGN), and the nucleus (stained by Hoechst 33342). After 2 h treatment with 2 μg mL−1 DOX or DOX-NPs, DOX signals were mainly found overlapping the lysosome, mitochondria, and nucleus rather than in the early endosomes (EEA1) and the trans-golgi network (TGN) (Fig. 5A and ESI 8†), indicating partly evading the early endosome and the lysosome.
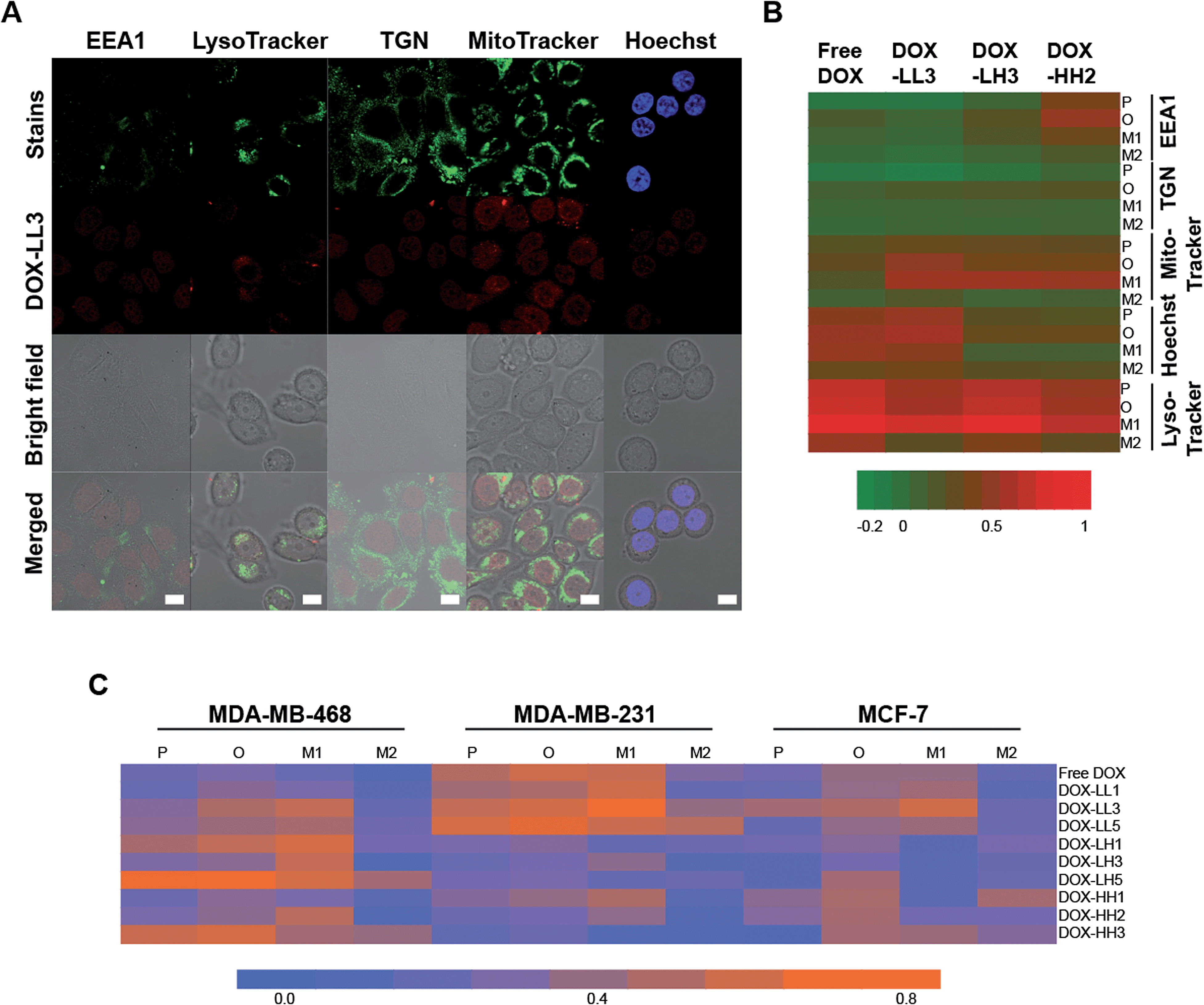 | ||
| Fig. 5 Intracellular distribution of DOX-NPs in 2D culture models. (A) Confocal images of DOX-LL3 in MDA-MB-468 cells. Cells were treated with 2 μg mL−1 DOX-NPs for 2 h. Different cell organelles were labelled with specific dyes with deep red fluorescence represented as false green colour. Early endosomes (EEA1) and Golgi (TGN) were visualized with the primary antibody before labelling with secondary antibody (deep red). Nuclei were stained by Hoechst 33342 dye (blue). Co-localizations between organelles and DOX and between nuclei and DOX in the merged images are seen as yellow and purple, respectively. Scale bar = 10 μm. (B) The heat map of the co-localization parameters of DOX-LL3, DOX-LH3, and DOX-HH2 in different parts of the MDA-MB-468 cells. (C) Heat map of the co-localization between DOX-NPs and the mitochondria in three breast cancer cells. In (B) and (C), P = Pearson's coefficient, O = overlap coefficient, M = Manders' coefficients, M1 = fraction of cell positions overlapping DOX-NPs, and M2 = fraction of DOX-NPs overlapping cell positions. | ||
In order to further investigate and compare the degree of co-localization of DOX to different subcellular compartments, several parameters were calculated including Pearson's coefficient (P), the overlap coefficient (O), and Manders' coefficients M1 and M2.72 M1 reflects the portion of cellular components that co-localize with DOX, and M2 indicates the portion of DOX co-localized with cellular components. All of these coefficients were converted into a heat map (Fig. 5B) in which high values of P, O, and M1 are considered to be indicative of high co-localization while M2 is not. According to the heat map, high values of P, O, and M1 were found in the nuclei, lysosomes, and mitochondria indicating high levels of DOX distribution to these compartments. Lysosomes had the highest degree of co-localization to DOX, which was expected because lysosomes can entrap most exogenous substances, including drugs and smaller nanocarriers.24,25 It is commonly assumed that histidine/histamine functional NPs44,45 can protect active drugs from the acidic environments of the endosomes and lysosomes until endosomal escape, cytosolic release, and uptake into the nucleus. It is very interesting to note that the 50% histamine-containing NPs had strong DOX/nucleus co-localization within a short time period (2 h) which indicates that these NPs can escape from lysosomes.
Additional evidence supporting the hypothesis that DOX-NPs can escape from lysosomes is that a strong co-localization exists between DOX-NPs (50% histamine) and mitochondria in MDA-MB-468 cells. As seen in Fig. 5B, cells treated with free DOX have very strong co-localization with the nuclei and lysosomes but weaker overlap with the mitochondria. However, the three DOX-NPs exhibit stronger co-localization to the mitochondria compared to free DOX because they have higher values of P, O, and M1. Moreover, DOX-LL3 and DOX-HH2 showed decreased co-localization to lysosomes. These observations suggest that histamine modified NPs (50% histamine) can partly avoid entrapment in lysosomes and can deliver the DOX to other regions in the MDA-MB-468 cells within 2 h.
In addition, the enhanced DOX distribution in mitochondria may explain why greater losses of mitochondrial activity are seen when the cells were treated with DOX-NPs compared to free DOX over short time periods (12–24 h, Fig. 4A, ESI 5 and ESI 7†). The mitochondria have their own DNA apart from nuclear DNA, called mitochondria DNA (mtDNA), that encodes 13 polypeptides required for respiratory chain proteins and 12S rRNAs, 16S rRNAs, and 22 tRNAs needed in protein synthesis.73 Because of the irreplaceable role of the mitochondria in cellular metabolism, more and more researchers consider the mitochondria as a potential target for cancer therapy.74,75 As an anthracycline antibiotic, DOX can intercalate into nuclear DNA to inhibit cell proliferation and interfere with topoisomerase II resulting in DNA damage.76 It has also been shown that DOX can also interfere with mtDNA.77 Therefore, we propose that DOX-NPs may be able to localize more DOX to the mitochondria and accelerate the disruption of mtDNA with subsequent loss of mitochondrial function leading to cell death, supported by the significant increase in late apoptosis in cells treated by DOX-NPs compared to free DOX (Fig. 4B). Similar strategies for cisplatin delivery to mitochondria have been explored in an effort to overcome resistance in central nervous system cancers.53 Further toxicological analysis of silver nanoparticles suggests they change the mitochondrial membrane permeability and hence give rise to cell death.78 Similarly, silica NPs have one of their main mechanism of cytotoxicity via induction of oxidative stress and as a consequence result in mitochondrial membrane potential changes and apoptosis.79 Similar results have been found for positively charged triblock copolymer NPs.80 This will contribute to increased cell death when compared to other DOX conjugates and delivery systems.81 Also by localization of the drug carrying NP to the mitochondria instead of the lysosome less DOX will be detoxified by the lower pH in the endo-lysosomal compartment. We also considered investigating the influence of NPs on the levels of reactive oxygen species that is balanced by mitochondria.82 However, due to small numbers of remaining allyl groups in the NPs, which are undetectable by NMR, it is impossible to obtain reliable data on ROS generation by these materials as the allyl groups may act as radical scavengers in these types of assays.
Based on the assumption that His-NPs can increase drug accumulation in mitochondria, we further explored the impact of our NPs on the three breast cancer cell lines in order to find out whether this accumulation is common in breast cancer cells and if there are any variations among the different carriers. According to the merged images in Fig. ESI 8B† and the heat map of the parameters for co-localization to the mitochondria in Fig. 5C, the low PXGE molecular weight (Mw), low PEO Mw group (LL1, LL3, LL5) tends to have enhanced mitochondria localization in the three breast cancer cells compared to free DOX. Among these, DOX-LL3 exhibits the highest co-localization according to the high values of P, O, and M1. The low PXGE, high PEO Mw group (LH1, LH3, LH5) tends to have lower co-localization to the mitochondria in MDA-MB-231 and MCF-7 cells, and this suggests that the molecular weight and structure of His-NPs might influence the distribution of the drug into the mitochondria. Except for DOX-LL3, no obvious differences among different compositions of the NPs in the library were observed.
3.6 3D breast cancer cell culture and viability assays
It have also been suggested that 3D cell models might better represent the in vivo situation than 2D cell cultures.57 This is because the 3D spheroid structure provides increased drug resistance, the formation of oxygen and nutrient gradients, and partially necrotic areas, all of which are features of in vivo tumours. Davis et al.83 utilized such models in NP studies and observed the internalization of chemotherapeutics into the spheroids by confocal microscopy. However, they did not provide any co-localization information in these studies and did not show a reliable measurement of cellular viability in the 3D model. The viability assessment is crucial because such quantitative data can provide convincing evidence for the efficacy of the drug-NPs. Therefore, we developed a 3D cell culture model using MDA-MB-231 cells and assessed cell viability with the ATP CellTiter-Glo® luminescent cell viability assay, which measures the concentration of ATP as a direct reflection of the number of living cells. This method is suitable for measuring cellular viability in 3D spheroids as a function of NP dose because it lyses the cells and the total ATP content can be measured. In contrast, MTT reagents have poor spheroid penetration and false results of mitochondrial function are often observed.In this study, we observed obvious structural damage to the spheroids caused by DOX-NPs after 72 h incubation compared to untreated, free DOX-treated, and neat NP-treated spheroids (Fig. 6A). NPs with 0% histamine led to a total structural collapse of the spheroid, and none of the DOX-NPs with 100% histamine resulted in complete disintegration. It is notable that DOX-HH2 (50% histamine)-treated spheroids also disintegrated and showed the greatest effect in the viability assay. This indicates that the polymer composition has a strong effect on the spheroid morphology and stability that is not related to the viability effects from the delivered drug but rather linked to a surfactant-like effect. In order to evaluate the efficacy of the DOX-NPs and to investigate whether the loss of structural integrity in the spheroids was related to lower viability, we utilized the CellTiter-Glo luminescent cell viability assay. As expected, neat NPs did not introduce any significant viability loss at the highest concentration tested (100 μg mL−1) after 72 h (Fig. 6B). This is similar to the 2D MTT results in Fig. ESI 4.† However, DOX-NPs led to more than 75% viability loss at 10 μg mL−1 DOX-equivalent concentration. In particular, DOX-NPs with 50% and 100% histamine content exhibited significantly stronger toxicity towards spheroids compared to free DOX (p < 0.05 and p < 0.001, respectively) (Fig. 6C). Thus, we can conclude that NPs without histamine have an enhanced effect on breaking down the 3D structures, while NPs with histamine can trigger higher DOX efficacy in MDA-MB-231 3D models.
 | ||
| Fig. 6 The effects of DOX-NPs on the 3D cell culture model. (A) Morphological changes of MDA-MB-231 spheroids treated with 10 μg mL−1 DOX-NPs or DOX or 100 μg mL−1 NPs after 72 h. ATP luminance assays for 3D spheroids after 72 h treatment with pure NPs (B) and DOX-NPs (C). Results are shown as mean ± SD (n = 6). *p < 0.05 and ***p < 0.001 compared to control. | ||
3.7 DOX localization studies in 3D spheroids
In order to further track the localization of the DOX internalized in the 3D spheroids, we performed live cell imaging of 3D spheroids. The projection images were reconstructed with Imars software. Spheroids were treated with DOX or DOX-NPs for 4 h then stained with Hoechst 33342 and LysoTracker or MitoTracker. The fluorescence images in Fig. 7 show that the surfaces of the spheroids are not homogenous and that there are channels in the spheroids. All of the spheroids displayed good co-localization of DOX within the nuclei, but DOX-LL3-treated spheroids demonstrated weaker co-localization between the drug and lysosome – as highlighted by the white arrow in Fig. 7 – indicating possible escape of DOX-LL3 from lysosomes in the surface layer of cells with penetration of the spheroid to a greater degree than free DOX or other DOX-NPs. However, light penetration into the core of the spheroid is not efficient due to its condensed nature, and a complete 3D image of the spheroid cannot be obtained. This limits the full evaluation of the penetration of the spheroid, but this is similar to the case of a real tumour. We also attempted to stain the spheroids with the MitoTracker deep red FM dye.84 Unfortunately, this dye also does not penetrate sufficiently deep into the spheroids (Fig. ESI 9†).
 | ||
| Fig. 7 Live cell imaging of DOX distribution in MDA-MB-231 3D spheroids. Spheroids were treated with 2 μg mL−1 DOX or DOX-NPs for 4 h. Nuclei were stained with Hoechst 33342 represented in blue, and lysosomes were stained with LysoTracker deep red and represented as a false green colour. The white arrow points out the lower level of co-localization between DOX-NPs and Lysotracker. Images were analyzed and reconstructed with Imars software. Scale bar = 70 μm. | ||
Conclusions
In this study, a library of polymer nanoparticles based on structurally different diblock copolymer starting materials, including a range of histamine-functionalized systems, was synthesized via a combination of ring-opening polymerization and thiol-ene click chemistry and used to assemble polymer NPs with charge reversal properties. All materials were found to be non-toxic in three breast cancer cell lines and all were able to encapsulate DOX. No strong pH-responsive properties were found in the in vitro release experiments, but the 50% histamine-modified NPs displayed lower drug release rates after 72 h at both pH 6 and pH 7. Further studies of the drug distribution in cells suggest that DOX-NPs are capable of escaping from endosomes and lysosomes and can deliver DOX into the nucleus and the mitochondria within 2 h. Moreover, DOX-NPs displayed similar efficacy as free DOX after 72 h treatment in the breast cancer cell lines MCF-7, MDA-MB-231, and MDA-MB-468, but were found to be more effective at shorter times due to their mitochondria localization. NPs containing 50% histamine can partly escape from lysosomes and deliver DOX into the nucleus in our established 3D spheroid model. These results suggest that histamine-functionalized NPs are a promising building block/scaffold for drug delivery systems, especially targeting the mitochondria section.Acknowledgements
This work was partially supported by the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) (NAL, CJH) and the National Science Foundation through the MRSEC program DMR-1121053 (NAL, PL, CJH). PL would like to acknowledge the Wenner-Gren and Bengt Lundqvist foundations for financial support. AMN acknowledges financial support from STINT, Karolinska Institutet, Percy Falks Foundation, and The Swedish Research Council (VR) 2009-3259 and 2011-3720.Notes and references
- A. Guiseppi-Elie, C. B. Dong and C. Z. Dinu, J. Mater. Chem., 2012, 22, 19529–19539 RSC.
- J. Wu, N. Kamaly, J. J. Shi, L. L. Zhao, Z. Y. Xiao, G. Hollett, R. John, S. Ray, X. Y. Xu, X. Q. Zhang, P. W. Kantoff and O. C. Farokhzad, Angew. Chem., Int. Ed., 2014, 53, 8975–8979 CrossRef CAS PubMed.
- M. Sanjoh, S. Hiki, Y. Lee, M. Oba, K. Miyata, T. Ishii and K. Kataoka, Macromol. Rapid Commun., 2010, 31, 1181–1186 CrossRef CAS PubMed.
- H. Takemoto, K. Miyata, S. Hattori, T. Ishii, T. Suma, S. Uchida, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2013, 52, 6218–6221 CrossRef CAS PubMed.
- R. G. Qi, S. Liu, J. Chen, H. H. Xiao, L. S. Yan, Y. B. Huang and X. B. Jing, J. Controlled Release, 2012, 159, 251–260 CrossRef CAS PubMed.
- H. H. Xiao, H. Q. Song, Q. Yang, H. D. Cai, R. G. Qi, L. S. Yan, S. Liu, Y. H. Zheng, Y. B. Huang, T. J. Liu and X. B. Jing, Biomaterials, 2012, 33, 6507–6519 CrossRef CAS PubMed.
- H. H. Xiao, H. Q. Song, Y. Zhang, R. G. Qi, R. Wang, Z. G. Xie, Y. B. Huang, Y. X. Li, Y. Wu and X. B. Jing, Biomaterials, 2012, 33, 8657–8669 CrossRef CAS PubMed.
- Z. Q. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- S. Wong, M. S. Shim and Y. J. Kwon, J. Mater. Chem. B, 2014, 2, 595–615 RSC.
- S. Kim, M. J. Stebe, J. L. Blin and A. Pasc, J. Mater. Chem. B, 2014, 2, 7910–7917 RSC.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- A. N. Lukyanov and V. P. Torchilin, Adv. Drug Delivery Rev., 2004, 56, 1273–1289 CrossRef CAS PubMed.
- V. P. Sant, D. Smith and J. C. Leroux, J. Controlled Release, 2004, 97, 301–312 CrossRef CAS PubMed.
- Z. G. Gao, A. N. Lukyanov, A. Singhal and V. P. Torchilin, Nano Lett., 2002, 2, 979–982 CrossRef CAS.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- A. Schroeder, D. A. Heller, M. M. Winslow, J. E. Dahlman, G. W. Pratt, R. Langer, T. Jacks and D. G. Anderson, Nat. Rev. Cancer, 2012, 12, 39–50 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- M. E. Fox, F. C. Szoka and J. M. J. Frechet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- C. L. Tseng, T. W. Wang, G. C. Dong, S. Y. H. Wu, T. H. Young, M. J. Shieh, P. J. Lou and F. H. Lin, Biomaterials, 2007, 28, 3996–4005 CrossRef CAS PubMed.
- H. Y. Nam, S. M. Kwon, H. Chung, S. Y. Lee, S. H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y. K. Oh, I. C. Kwon, K. Kim and S. Y. Jeong, J. Controlled Release, 2009, 135, 259–267 CrossRef CAS PubMed.
- L. A. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS PubMed.
- P. K. Selbo, A. Weyergang, A. Hogset, O. J. Norum, M. B. Berstad, M. Vikdal and K. Berg, J. Controlled Release, 2010, 148, 2–12 CrossRef CAS PubMed.
- J. K. Vasir and V. Labhasetwar, Adv. Drug Delivery Rev., 2007, 59, 718–728 CrossRef CAS PubMed.
- J. Panyam, W. Z. Zhou, S. Prabha, S. K. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217–1226 CrossRef CAS PubMed.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 CAS.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- E. S. Lee, Z. G. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- A. Sorkin and M. von Zastrow, Nat. Rev. Mol. Cell Biol., 2002, 3, 600–614 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- M. Oishi, K. Kataoka and Y. Nagasaki, Bioconjugate Chem., 2006, 17, 677–688 CrossRef CAS PubMed.
- M. P. Xiong, Y. Bae, S. Fukushima, M. L. Forrest, N. Nishiyama, K. Kataoka and G. S. Kwon, ChemMedChem, 2007, 2, 1321–1327 CrossRef CAS PubMed.
- F. Pittella, M. Z. Zhang, Y. Lee, H. J. Kim, T. Tockary, K. Osada, T. Ishii, K. Miyata, N. Nishiyama and K. Kataoka, Biomaterials, 2011, 32, 3106–3114 CrossRef CAS PubMed.
- Y. Kakizawa, S. Furukawa, A. Ishii and K. Kataoka, J. Controlled Release, 2006, 111, 368–370 CrossRef CAS PubMed.
- J. S. Park, T. H. Han, K. Y. Lee, S. S. Han, J. J. Hwang, D. H. Moon, S. Y. Kim and Y. W. Cho, J. Controlled Release, 2006, 115, 37–45 CrossRef CAS PubMed.
- R. Shrestha, M. Elsabahy, S. Florez-Malaver, S. Samarajeewa and K. L. Wooley, Biomaterials, 2012, 33, 8557–8568 CrossRef CAS PubMed.
- K. L. Chang, Y. Higuchi, S. Kawakami, F. Yamashita and M. Hashida, Bioconjugate Chem., 2010, 21, 1087–1095 CrossRef CAS PubMed.
- J. J. Gu, X. Wang, X. Y. Jiang, Y. Z. Chen, L. C. Chen, X. L. Fang and X. Y. Sha, Biomaterials, 2012, 33, 644–658 CrossRef CAS PubMed.
- Y. T. Wen, Z. H. Guo, Z. Du, R. Fang, H. M. Wu, X. Zeng, C. Wang, M. Feng and S. R. Pan, Biomaterials, 2012, 33, 8111–8121 CrossRef CAS PubMed.
- H. Wu, L. Zhu and V. P. Torchilin, Biomaterials, 2013, 34, 1213–1222 CrossRef CAS PubMed.
- R. Liu, D. Li, B. He, X. H. Xu, M. M. Sheng, Y. S. Lai, G. Wang and Z. W. Gu, J. Controlled Release, 2011, 152, 49–56 CrossRef CAS PubMed.
- C. M. Yu, C. M. Gao, S. Y. Lu, C. Chen, J. L. Yang, X. Di and M. Z. Liu, Colloids Surf., B, 2014, 115, 331–339 CrossRef CAS PubMed.
- P. Lundberg, N. A. Lynd, Y. Zhang, X. Zeng, D. V. Krogstad, T. Paffen, M. Malkoch, A. M. Nystrom and C. J. Hawker, Soft Matter, 2013, 9, 82–89 RSC.
- R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS PubMed.
- S. Marrache, R. K. Pathak and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10444–10449 CrossRef CAS PubMed.
- L. M. Wang, Y. Liu, W. Li, X. M. Jiang, Y. L. Ji, X. C. Wu, L. G. Xu, Y. Qiu, K. Zhao, T. T. Wei, Y. F. Li, Y. L. Zhao and C. Y. Chen, Nano Lett., 2011, 11, 772–780 CrossRef CAS PubMed.
- C. Porsch, Y. Zhang, A. Ostlund, P. Damberg, C. Ducani, E. Malmstrom and A. M. Nystrom, Part. Part. Syst. Charact., 2013, 30, 381–390 CrossRef CAS.
- C. Porsch, Y. Zhang, C. Ducani, F. Vilaplana, L. Nordstierna, E. Malmstrom and A. M. Nystrom, Biomacromolecules, 2014, 15, 2235–2245 CrossRef CAS PubMed.
- P. Longati, X. H. Jia, J. Eimer, A. Wagman, M. R. Witt, S. Rehnmark, C. Verbeke, R. Toftgard, M. Lohr and R. L. Heuchel, BMC Cancer, 2013, 13, 95 CrossRef CAS PubMed.
- X. Zeng, R. Morgenstern and A. M. Nystrom, Biomaterials, 2014, 35, 1227–1239 CrossRef CAS PubMed.
- X. Zeng, Y. Zhang and A. M. Nystrom, Biomacromolecules, 2012, 13, 3814–3822 CrossRef CAS PubMed.
- W. C. Shen, Biochim. Biophys. Acta, 1990, 1034, 122–124 CrossRef CAS.
- S. B. Luo, R. Cheng, F. H. Meng, T. G. Park and Z. Y. Zhong, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3366–3373 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- X. Zeng, Y. Zhang, Z. Wu, P. Lundberg, M. Malkoch and A. M. Nyström, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 280–288 CrossRef CAS.
- Y. Hed, Y. Zhang, O. C. J. Andren, X. Zeng, A. M. Nystrom and M. Malkoch, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3992–3996 CrossRef CAS.
- Z. Wu, X. Zeng, Y. Zhang, N. Feliu, P. Lundberg, B. Fadeel, M. Malkoch and A. M. Nystrom, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 217–226 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- E. R. Gillies and J. M. J. Frechet, Bioconjugate Chem., 2005, 16, 361–368 CrossRef CAS PubMed.
- T. L. Riss and R. A. Moravec, Assay Drug Dev. Technol., 2004, 2, 51–62 CrossRef CAS PubMed.
- A. Azzariti, G. Bocci, L. Porcelli, A. Fioravanti, P. Sini, G. M. Simone, A. E. Quatrale, P. Chiarappa, A. Mangia, S. Sebastian, D. Del Bufalo, M. Del Tacca and A. Paradiso, Br. J. Cancer, 2011, 104, 769–780 CrossRef CAS PubMed.
- L. Wang, J. Wang, B. W. Blaser, A. M. Duchemin, D. F. Kusewitt, T. Liu, M. A. Caligiuri and R. Briesewitz, Blood, 2007, 110, 2075–2083 CrossRef CAS PubMed.
- S. Bolte and F. P. Cordelieres, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- P. Huang and J. S. Carew, Mol. Cancer, 2002, 1, 9 CrossRef PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- E. L. Zhang, C. Zhang, Y. P. Su, T. M. Cheng and C. M. Shi, Drug Discovery Today, 2011, 16, 140–146 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS.
- N. Ashley and J. Poulton, Biochem. Biophys. Res. Commun., 2009, 378, 450–455 CrossRef CAS PubMed.
- J. S. Teodoro, A. M. Simoes, F. V. Duarte, A. P. Rolo, R. C. Murdoch, S. M. Hussain and C. M. Palmeira, Toxicol. in Vitro, 2011, 25, 664–670 CrossRef CAS PubMed.
- L. Sun, Y. Li, X. M. Liu, M. H. Jin, L. Zhang, Z. J. Du, C. X. Guo, P. L. Huang and Z. W. Sun, Toxicol. in Vitro, 2011, 25, 1619–1629 CrossRef CAS PubMed.
- S. Bhattacharjee, D. Ershov, K. Fytianos, J. van der Gucht, G. M. Alink, I. M. C. M. Rietjens, A. T. M. Marcelis and H. Zuilhof, Part. Fibre Toxicol., 2012, 9, 11 CrossRef CAS PubMed.
- X. B. Xiong, Z. S. Ma, R. Lai and A. Lavasanifar, Biomaterials, 2010, 31, 757–768 CrossRef CAS PubMed.
- J. F. Turrens, J. Physiol., 2003, 552, 335–344 CrossRef CAS PubMed.
- J. S. Basuki, H. T. T. Duong, A. Macmillan, R. B. Erlich, L. Esser, M. C. Akerfeldt, R. M. Whan, M. Kavallaris, C. Boyer and T. P. Davis, ACS Nano, 2013, 7, 10175–10189 CrossRef CAS PubMed.
- K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information regarding material synthesis and characterization, TEM images, MTT assays, drug wash-out MTT results, ATP assays for MDA-MB-231 cells in 2D culture, confocal images for heat map calculations, and mitochondria-stained 3D images can be found. See DOI: 10.1039/c4tb02051k |
| This journal is © The Royal Society of Chemistry 2015 |