
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysicochemical and nanomechanical investigation of electrodeposited chitosan:PEO blends
Davide A. D.
Cassani
ab,
Lina
Altomare
c,
Luigi
De Nardo
ac and
Fabio
Variola
*bd
aDepartment of Chemistry, Materials and Chemical Engineering, “G. Natta”, Politecnico di Milano, Milano, Italy
bDepartment of Mechanical Engineering, University of Ottawa, Ottawa, Canada. E-mail: fabio.variola@uottawa.com
cINSTM Local Unit Politecnico di Milano, Milano, Italy
dDepartment of Physics, University of Ottawa, Ottawa, Canada
First published on 18th February 2015
Abstract
Cathodic electrodeposition is a bottom up process that is emerging as a simple yet efficient strategy to engineer thin polymeric films with well-defined physicochemical properties. In particular, this technique offers the distinctive advantage of an easy control over composition, thickness, and morphology of the films by simply adjusting treatment parameters. In this work, cathodic electrodeposition was exploited to engender blends composed by chitosan (CH) and poly-ethylene-oxide (PEO) with different weight ratios. The physicochemical and nanomechanical properties of the resulting films were successively characterized by integrating Raman and Fourier-transform infrared (FT-IR) spectroscopy with Atomic Force Microscopy (AFM). Our findings demonstrate that electro-deposition is an effective technique for the co-deposition of CH:PEO blends. Moreover, spectroscopic and AFM analyses correlated the physicochemical (i.e. structural organization, bond formation and cross-linking) and nanomechanical properties of the blends to the PEO content, ultimately unveiling the molecular interactions and mechanisms involved in the cathodic deposition of CH:PEO films.
Introduction
The ability to precisely assess the physicochemical features and nanomechanical properties of polymers has increasingly become a key aspect in the development and optimization of advanced polymeric materials for a variety of applications, in particular for tissue engineering and drug delivery.1 In these fields, chitosan (hereafter indicated as CH) is undoubtedly one of the most promising and widely used biopolymers, able to conjugate interesting biologically relevant properties such as biocompatibility, non-toxicity and biofunctionality. From a chemical point of view, CH is a linear and high-molecular-weight polysaccharide composed of D-glucosamine (de-acetylated) and N-acetyl-D-glucosamine (acetylated) units linked together by β-(1,4) – glycosidic bonds. At moderately acidic pH (<5), the amine groups acquire a proton, thereby making chitosan soluble in slightly acidic solutions.2,3 Conversely, when the pH increases towards more basic values, the ammonium groups de-protonate, initiating the coagulation of an insoluble structure of chitosan.4 The exploitation of chitosan in tissue engineering applications spans from porous scaffolds capable of promoting tissue regrowth to films/coatings for cartilage, bone, vascular and skin repairs, as well as wound healing and burn treatments.2 Chitosan films have also been successfully exploited as drug delivery systems that provide a controlled and targeted elution of antibiotics and bioactive agents.5,6 In particular, because of its low toxicity, high mucosal tolerance and adhesiveness (due to the electrostatic interaction between the CH amino groups and the sialic acid residues of mucins7), chitosan has rapidly become one of the most promising materials for ocular7–10 and oral delivery systems, enabling, for instance, an excellent permeation of insulin in the buccal mucosa.11 The breath of application of chitosan and chitosan-based hydrogels in drug delivery also extends to stimulus-responsive smart coatings capable of releasing their bioactive content only when instructed by environmental cues, such as pH, temperature and enzymatic activity.12In order to enhance its properties (e.g. greater flexibility, elongation and tensile strength) and ultimately further extend the breath of its applications, CH has been blended with other polymeric materials.13 Among these, poly-ethylene oxide (PEO), a synthetic polymer with a molecular formula of (–CH2CH2O–)n, has emerged as a promising candidate because of its low-toxicity, bioadhesive and mucoadhesive properties as well as a high hydrophilicity.14–18 In particular, pure PEO has been used extensively in drug release and tissue engineering applications.19 Despite these advantages, pure PEO films demonstrated relatively weak mechanical properties, and the high water solubility limits their range of application.14 However, when blended with CH, its overall properties, such as water stability, moisture sorption, elongation at break and tensile strength are enhanced.14–16 CH:PEO blends are thus expected to overcome the limitations of the individual components by synergistically conjugating their properties (e.g. controlled drug elution and physicochemical stability), ultimately resulting in a polymeric material with new capacities and improved performance.
To date, the most widely used methods to manufacture CH:PEO components for various medical applications (e.g. wound healing and tissue engineering) are electrospinning and solvent casting.16–18,20,21 The former permits to draw ultra-fine fibers and weave them into micro- and nano-fibrous structures with an interconnected porosity in the sub-micron and micron scales. Because of these distinctive advantages, electrospinning has been mostly applied to engender biotextile structures and unwoven webs, such as 3-dimensional scaffolds and meshes to support cell growth, among others.22,23 On the other hand, while solvent casting has been successfully employed to generate films with variable thickness for biomedical applications,18 its use may pose challenges in regard to a potential retention of toxic solvent within the film.
While these techniques will keep evolving and improving to address some of the limitations and further enhance their impact, the search is still on for valuable alternatives for the co-deposition of CH:PEO blends. Among these, cathodic electrodeposition promises to combine a relatively fast deposition process with the prospect of controlling the films characteristics (e.g. thickness, pore size).4,24 In addition, this approach relies on a simple experimental setup, can be carried out at room temperature and without the use of toxic reagents/solvents. These characteristics are poised to preserve the natural bioactivity of therapeutic agents to be incorporated in the polymeric network, thereby making electrodeposition a valid new alternative for the production of chitosan-based blends with tunable properties for drug delivery applications. This technique exploits the electrophoretic motion of charged molecules under the influence of an electrical field and their successive coagulation onto a metallic electrode in form of polymeric thin films. While cathodic electrodeposition has been successfully used to engender chitosan thin films and coatings,3,4,25 its capacity to deposit bi-component mixtures has never been investigated. In particular, in the case of CH:PEO blends, the electrically neutral PEO molecules and, as a matter of fact, any electrically neutral molecule, are not expected to respond to the applied field, a circumstance that would ultimately preclude their participation in the coagulation process. Therefore, the question whether polymeric blends composed by a combination of charged and electrically neutral molecules could be created by cathodic electrodeposition was still unanswered to date.
In this work, we validate the potential of cathodic electrodeposition to co-deposit CH:PEO blends and the prospect of controlling the properties of the resulting films by varying the weight ratio of the two components. Physicochemical and nanomechanical characterization was successively carried out by exploiting Raman and Fourier-transform infrared (FT-IR) spectroscopy, as well as Atomic Force Microscopy (AFM), to understand the influence of the PEO content on the properties of the films. In addition, we closed in on the molecular interactions occurring during the co-deposition process, ultimately shedding new light on how these influence the resulting physicochemical and structural properties of CH:PEO films. Our findings highlight the versatility of electrodeposition as a technique to engender CH:PEO blends, paving the way for its broader application in tissue engineering and drug-delivery applications which require the deposition of multi-component polymeric blends.
Experimental
Cathodic electrodeposition
Four mixtures of medium molecular weight chitosan and 106 kDa PEO (both purchased from Sigma Aldrich) with different CH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEO weight ratios (i.e. 100:0, 80:20, 50:50 and 20:80) were diluted in a 1:50 (w/v) citric acid solution with a concentration of 5 g l−1 to create solutions with four different nominal compositions. The resulting CH:PEO solutions were individually used as the electrolyte in a cell composed by a double-faced 1.5 × 1.5 cm2 stainless steel cathode and two graphite rod anodes. A 100 W potentiometer (Keithley Instruments, United States) was used to impose a potential of 20 V between the electrodes for 15 minutes, in order to transport by electrophoresis the positively charged and soluble chitosan molecules to the metallic cathode. Here, they coagulated as a result of the more basic pH at the electrode–electrolyte interface.3 Continuous injection of N2 reduced the O2 partial pressure and in turn improved the films homogeneity. In addition, since it was previously shown that a relatively lower acidic pH (i.e. 3–4) of the chitosan solution promotes the formation interconnected pores,25 the films were deposited at pH = 2 to minimize this phenomenon. At the end of the deposition, the coated cathode was immersed in a 1 M NaOH solution overnight to facilitate the detachment of the films, which were successively dried at 37 °C for 8 h and weighted with a 1 μg resolution scientific scale (FISONS Sartorius, United Kingdom). The average weight of the electrodeposited CH:PEO films varied as a function of the PEO content, decreasing from 6.5 ± 0.2 g for pure chitosan to 5.3 ± 0.3 g, 4.7 ± 0.1 g and 2.6 ± 0.1 g for the 80:20, 50:50 and 20:80 samples, respectively (Table 1).
:PEO ratio
:PEO weight ratios (i.e. 100:0, 80:20, 50:50 and 20:80) were diluted in a 1:50 (w/v) citric acid solution with a concentration of 5 g l−1 to create solutions with four different nominal compositions. The resulting CH:PEO solutions were individually used as the electrolyte in a cell composed by a double-faced 1.5 × 1.5 cm2 stainless steel cathode and two graphite rod anodes. A 100 W potentiometer (Keithley Instruments, United States) was used to impose a potential of 20 V between the electrodes for 15 minutes, in order to transport by electrophoresis the positively charged and soluble chitosan molecules to the metallic cathode. Here, they coagulated as a result of the more basic pH at the electrode–electrolyte interface.3 Continuous injection of N2 reduced the O2 partial pressure and in turn improved the films homogeneity. In addition, since it was previously shown that a relatively lower acidic pH (i.e. 3–4) of the chitosan solution promotes the formation interconnected pores,25 the films were deposited at pH = 2 to minimize this phenomenon. At the end of the deposition, the coated cathode was immersed in a 1 M NaOH solution overnight to facilitate the detachment of the films, which were successively dried at 37 °C for 8 h and weighted with a 1 μg resolution scientific scale (FISONS Sartorius, United Kingdom). The average weight of the electrodeposited CH:PEO films varied as a function of the PEO content, decreasing from 6.5 ± 0.2 g for pure chitosan to 5.3 ± 0.3 g, 4.7 ± 0.1 g and 2.6 ± 0.1 g for the 80:20, 50:50 and 20:80 samples, respectively (Table 1).
:PEO ratio
| Parameter considered | 100:0 |
80:20 |
50:50 |
20:80 |
|---|---|---|---|---|
| Weight (g) | 6.5 ± 0.2 | 5.3 ± 0.3 | 4.7 ± 0.1 | 2.6 ± 0.1 |
| RMS-roughness (nm) (5 μm × 5 μm) | 31.5 ± 9.5 | 14.3 ± 3.4 | 22 ± 4.8 | 20.5 ± 5.6 |
| RMS-roughness (nm) (500 nm × 500 nm) | 8.9 ± 1.8 | 10.9 ± 2 | 11.4 ± 2.3 | 7.3 ± 1.3 |
Scanning electron microscopy
A JSM-7500F Field Emission Scanning Electron Microscope (FE-SEM, JEOL, Japan) was used in secondary electron mode to image the electrodeposited films. Prior to observation, samples were sputter-coated with a thin gold–platinum layer (Gatan Precision Etching Coating System, United States) to reduce surface charging.Raman spectroscopy
An Alpha 300 confocal Raman microscope (WITec, Germany) was used to carry out Raman spectroscopic analysis on five samples for each weight ratio. Ten single spectra were collected in randomly selected regions of the films, for a total of 50 spectra per condition (n = 50). The presence of a high intensity broad band in the 100–3000 rel cm−1 range (data not shown) was attributed to the autofluorescence of the material.26,27 This was minimized by exposing the samples to photobleaching with a 633 nm laser (Helium–Neon Research Electro-Optics laser, Boulder, United States) for 5 minutes prior to each measurement. Optical microscopy inspection with a 20× air objective ensured the absence of any significant damage caused by the laser. The same laser was used for spectra collection. Each spectrum was acquired with a 120-second integration time (single acquisition) to enhance the signal-to-noise ratio. Raman data were processed by the OriginPro software (OriginLab corporation, United States). The Savitzky–Golay filter was used after linear baseline subtraction. Raman spectra were successively normalized to their global maximum peak (i.e. C–H deformation, τC–H/ωC–H, in the 1342–1371 rel cm−1 region) as internal standard and fitted by Guassian functions.28–30 The assignment of the peaks was performed according to previous literature.28–36 The same software was also used to carry out one-way ANOVA test. A p value <0.05 was used to identify statistically significant differences.Fourier transform infrared (FT-IR) spectroscopy
The physicochemical properties of the films were successively analyzed by exploiting an air-purged Thermo Nicolet Nexus 870 FT-IR spectrometer equipped with a grazing-angle SAGA accessory (Thermo Scientific, United States). Four samples (n = 4) per composition were investigated by using polarized light at an angle of 80° with respect to the surface normal and an 8 mm diameter opening. Spectroscopic information was collected in the 800–1900 cm−1 range with a 4 cm−1 resolution (256 scans per spectrum), using a gold substrate for reference. The choice of the spectral range was based on the data interpretation and peak assignment found in previous literature.37–49 Similar to the Raman analysis, infrared data were processed by using the OriginPro software. In particular, spectra were smoothed with a Savitzky–Golay filter after linear baseline subtraction, normalized with respect to a common band (i.e. C–O stretching, 977–1003 cm−1), and fitted with Gaussian functions to resolve secondary vibrational components. In this context, variations in the film thicknesses and the resulting differences in the optical paths affected the FT-IR band absolute intensities. In addition, the significant overlapping between chitosan and PEO infrared signatures, as well as the absence of PEO-exclusive bands, prevented us from directly quantifying the effective chitosan-to-PEO ratio in the thin films.49 These limitations were however overcome by normalizing the intensities to the common C–O stretching band. This allowed us to compare the normalized intensities of different bands, ultimately inferring the presence of PEO in the blends (see Discussion). Noteworthy, the absolute peak shifts are not expected to depend on the thickness of the sample.Atomic force microscopy
The WITec Alpha 300 microscope also features an Atomic Force Microscope which was used in Digital Pulsed Force Mode (DPFM). This technique permitted to simultaneously visualize the nanoscale surface morphology of the thin films and probe their nanomechanical properties. For each composition, three (5 × 5) μm2 DPFM images were obtained on three different samples (n = 9) with a cantilever characterized by a tetrahedral tip (tip radius less than 10 nm), a resonance frequency of 75 kHz and a nominal spring constant of 2.8 N m−1 (Arrow™ FM Nanoworld, Switzerland). DPFM maps were collected on micrometric regions of sample surfaces without pores. Set-point, amplitude and frequency were set at 3 V (equivalent to a measured maximum indentation force of 35 nN), 1.5 V and 1000 Hz, respectively. Each DPFM image contained 2.5 × 105 force–distance curves, which were converted from deflection [V]-phase [°] (AFM output) into force [N]-indentation depth [nm] according to a previously published procedure.50,51 From these curves, we directly calculated the maximum indentation depth, stiffness and compliance.51 In order to extract the Young's modulus, data were fitted with the Hertz model and the Sneddon approximation.52–57Results and discussion
While cathodic electrodeposition has already proven to be an effective strategy to engender thin films of pure CH, its potential and effectiveness for the co-deposition of CH:PEO blends was yet to be validated. Once demonstrated, results from this study will support the future applications of this technique in the production of chitosan-based blends, ultimately becoming a valid alternative to existing approaches such as electrospinning and solvent casting.In this work, we carried out physicochemical and nanomechanical characterization to monitor how the properties of the electrodeposited films vary as a function of the nominal composition. This allowed us to assess the efficiency of the process (i.e. capacity to generate CH:PEO films with controlled composition by electrodeposition) and the characteristics of the films.
A preliminary indication of a successful co-deposition process could be given by the film's weight reduction as a function of the nominal PEO content (Table 1). It was in fact demonstrated that the deposition rate decreases linearly with the CH concentration: the mass of the electrodeposited films should thus linearly follow the reduction of CH content in the electrolyte assuming, by a proof of contradiction, that no PEO is deposited.4 However, our data show that the weight of the blends is always greater than the expected weight of the films if only CH was deposited. It could be conceived that during cathodic co-deposition, chitosan acts as a carrier of PEO molecules. Electrically neutral PEO are in fact expected to form hydrogen bonds with charged chitosan molecules,20,21,58,59 which migrate towards the electrode's surface under the influence of the applied voltage. Therefore, according to this hypothesis, variations in the weight of the deposited film suggest that during the process a second component is also deposited. In addition, such postulate can also explain the reduction of the total weight measured at lower CH:PEO ratios. As the CH content in solution decreases, the amount of carrier molecules in the electrolyte decreases as well, thereby decreasing the efficiency of the process in terms of number of CH and PEO molecules coagulated onto the electrode. Noteworthy, one may also argue that the less heavy films deposited in a given time (15 minutes) result from a lower diffusion rate of the heavier CH:PEO complex in comparison to unbounded CH molecules in solution. Taken together, these considerations indicate that the weight of the films alone cannot provide a direct evidence of a successful co-deposition process, and complementary techniques have been exploited to achieve unambiguous proof of the efficiency of this technique.
Scanning electron imaging revealed that, regardless of the composition, all electrodeposited films generally exhibited a topography characterized by micrometric pores (about 30 μm in diameter) randomly distributed across the entire surface (Fig. 1A and B). These most likely originated from the evolution of hydrogen gas (i.e. reduction of hydrogen ions) during the deposition process, even after the use of N2 and a low pH.25 In this context, more uniform films could be obtained by introducing H2O2 or p-benzoquinone to the deposition solution to allow cathodic reactions at lower potentials, ultimately further reducing bubble formation.60
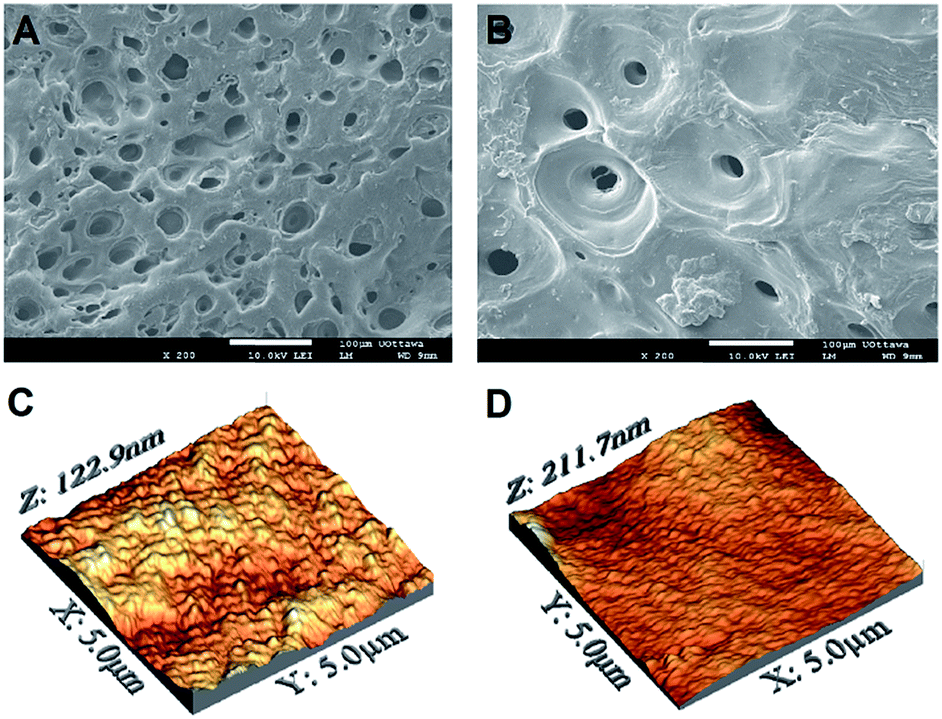 | ||
| Fig. 1 (A–B) SEM and (C–D) AFM-DPFM images of pure chitosan and 20:80 CH:PEO films. | ||
SEM imaging was complemented by AFM investigation, which allowed us to qualitatively and quantitatively close in on the nanotopographical features of the films. Fig. 1C and D display representative AFM micrographs of pure chitosan and 20:80 samples, selected for visual comparison. At low magnification (5 × 5 μm2), while the surface nanotopography of the former appeared relatively rough, that of 20:80 films resulted smoother. These observations were confirmed by the quantification of the root-mean-square (RMS) roughness, which ranged from 14.3 ± 3.4 nm for the 80:20 to 20.5 ± 5.6 nm for 20:80 samples. At a higher magnification (500 × 500 nm2), RMS values were more uniform, spanning from 10.9 ± 2.0 to 7.3 ± 1.3 nm for the 80:20 and 20:80 films, respectively. The complete sets of the measured RMS values are reported in Table 1. While the RMS roughness measured in 5 × 5 μm2 regions significantly varied as a function of the PEO content, smaller areas 500 × 500 nm2 showed more uniform values (Table 1). Only the 20:80 samples showed a significantly lower roughness at this scale. This can be explained by considering the morphological irregularity of surfaces to be more marked in larger areas. At a higher magnification, in fact, alterations of surface topography are less pronounced, as shown by the similar RMS values measured at this scale for the four conditions tested. Although not unequivocal, these findings suggest a correlation between chemical composition and surface morphology, at least in 5 × 5 μm2 regions of blends mainly composed by PEO.
In order to probe the physicochemical characteristics of the thin films, we exploited Raman (Fig. 2) and FT-IR (Fig. 3) spectroscopy, two powerful techniques previously used to investigate in detail the structural properties of chitosan and PEO.37–49
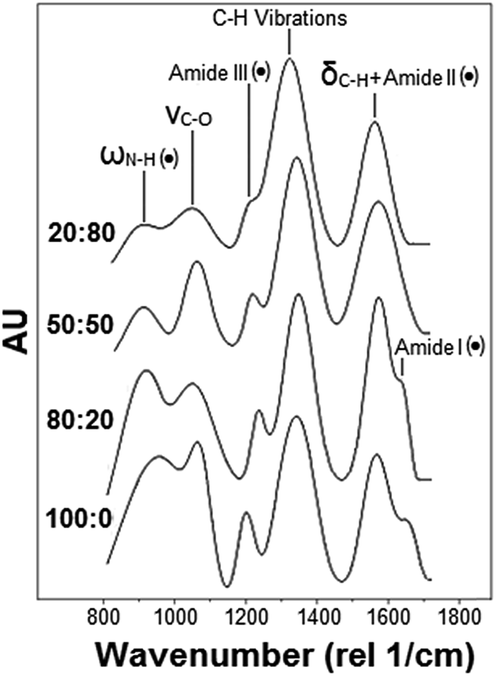 | ||
| Fig. 2 Representative Raman spectra of pure chitosan and CH:PEO films (PEO content increases from bottom to top). Dotted peaks refer to the characteristic peaks of the sole CH. | ||
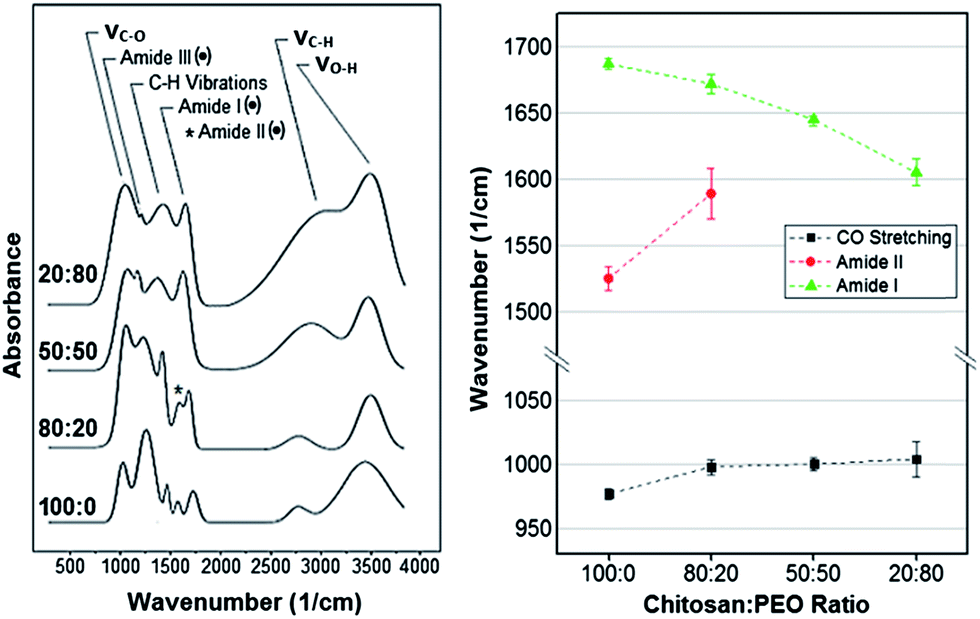 | ||
| Fig. 3 Left: Representative FT-IR spectra of pure chitosan and CH:PEO films (PEO content increases from bottom to top). Dotted peaks refer to the characteristic peaks of the sole CH. Right: Shift of band maxima as a function of the PEO content. | ||
Fig. 2 shows representative normalized Raman spectra of the various films tested. Consistently with previous studies,28–30 the main bands displayed by pure chitosan and PEO are located in the 800–1400 rel cm−1 interval. Among these, the N–H wagging (ωN–H) as well as the amide I, II and III bands uniquely belong to chitosan (black dots in Fig. 2). Conversely, both chitosan and PEO exhibit the C–H bending (δC–H), the C–O stretching (νC–O) and the C–H deformation, which consists of the wagging (ωC–H) and twisting (τC–H) vibration peaks.
Peak deconvolution and fitting allowed us to precisely compare the normalized bands intensity and monitor their variations as a function of the composition. In particular, it resulted that (i) the normalized intensity of the amide III peak, centered in the 1236–1273 rel cm−1 interval,28 as well as (ii) that of the band in the 921–899 cm−1 range, attributed to the N–H wagging vibration (ωN–H),36 significantly decreased as the PEO content increased. The two overlapping vibrational modes, namely (iii) δC–H31,33,34,36 and (iv) the bending vibration of the amide II (δN–H),30,32,35 composing the band in the 1575–1590 rel cm−1 region, were measured but it was not possible to separate the two contributes. Also, despite photobleaching and the use of the 633 nm laser (selected to reduce autofluorescence of the material), we were not able to accurately resolve (v) the amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration) in the 1683–1673 rel cm−1 range28,30,36 in samples with a PEO content higher than 20% because of a relatively high fluorescence background. Finally, (vi) the normalized intensity of the peak centered in the 1050–1061 rel cm−1 interval increased with the PEO content. This peak was previously attributed to the νC–O vibration of the ether bonds31,32,36 or to the stretching/bending vibration of C–C bonds.42
O vibration) in the 1683–1673 rel cm−1 range28,30,36 in samples with a PEO content higher than 20% because of a relatively high fluorescence background. Finally, (vi) the normalized intensity of the peak centered in the 1050–1061 rel cm−1 interval increased with the PEO content. This peak was previously attributed to the νC–O vibration of the ether bonds31,32,36 or to the stretching/bending vibration of C–C bonds.42
Because of the experimental and analytical limitations in resolving all the above Raman bands, only the amide III and ωN–H were considered for comparison. These two bands were first normalized with respect to the C–H deformation peak for all the conditions tested. Results obtained for the three blends were successively expressed in terms of percentage increase/decrease with respect to pure chitosan. The ANOVA test was carried out to identify statistically significant variations. The Raman bands considered in this analysis and their relative normalized intensity variations are shown in Table 2, which shows that the normalized intensity of the ωN–H and amide III peaks decreased as the PEO content increased (Table 2).
| Type of peak | Peak | 80:20 [%] |
50:50 [%] |
20:80 [%] |
|---|---|---|---|---|
| Raman | ω N–H | • | −38 | −45 |
| Raman | Amide III | • | −21 | −29 |
| FT-IR | Amide III | • | −40 | −47 |
| FT-IR | ω/δ/τC–H | +21 | + 43 | + 49 |
| FT-IR | ν C–H | +9 | + 35 | + 41 |
Since these two bands are characteristics of CH only, a reduction of its content in the blend is expected to cause a gradual reduction of these peaks' presence in the Raman spectrum (Fig. 2). In other words, the decrease of the CH content in the electrodeposited film was demonstrated experimentally by monitoring relative variations of the intensity of two bands solely associated to CH.
Similar to Raman analysis, the assignment of the FT-IR absorption bands was based on previous literature.37–49 Representative normalized spectra are shown in Fig. 3. Among the bands observed, only the amide I, II and III bands were characteristic of the sole chitosan (black dots in Fig. 3) while the remaining were in common with PEO.
Spectra processing and peak deconvolution revealed that (i) the band between 1686 cm−1 and 1605 cm−1, attributed to the amide I stretching vibration (νCO)37,43,45,46 underwent a red shift, varying its center from 1687 ± 4 cm−1 for pure chitosan to 1672 ± 7 cm−1, 1645 ± 2 cm−1, 1605 ± 10 cm−1 in 80:20, 50:50 and 20:80 samples, respectively. In addition, (ii) the band centered at 1525 cm−1 and attributed to the amide II (N–H bending vibration, δN–H)37,44–46,49 reduced in normalized intensity and blue shifted as the content of PEO increased. The overlap with the amide I prevented the precise identification of amide II band in the samples with a chitosan:PEO ratio of 50:50 and 20:80. (iii) The third characteristic band of pure chitosan was centered in the 1181–1141 cm−1 region and assigned to the amide III.44,47,48 In this case, the normalized intensity decreased as the PEO content increased. In addition, a shift of the band's center from 977 ± 4 cm−1 for pure chitosan to 1004 ± 13 cm−1 for 20:80 samples was measured (Fig. 3). Also, (iv) the band located in the 1344–1407 cm−1 region increased in normalized intensity with the percentage of PEO in the blend. This band was attributed to the wagging vibration of C–H bonds (ωC–H).37–41 Noteworthy, several authors also assigned it to the bending (δC–H)37,42 and twisting (τC–H)39 vibrations of C–H bonds (C–H deformation in Fig. 3). Moreover, (v) the broad band in the 2784–3057 cm−1 region was attributed to symmetrical and asymmetrical stretching vibrations of C–H bonds (νC–H).37,38,40,42 Similar to the ω/τ/δC–H, the normalized intensity of this band increased with the content of PEO. Finally, (vi) the band centered between 3402 cm−1 and 3514 cm−1 was assigned to the O–H stretching vibrations (νO–H).40,42–46 However, it was not further considered because of its high variability across samples of the same composition. For all the above reasons, only the amide III, ω/τ/δC–H and νC–H were considered for comparison. As for the Raman analysis, results obtained from fitting were expressed in terms of percentage increase/decrease with respect to pure chitosan. The ANOVA test was carried out to identify statistically significant variations. The FT-IR bands considered in this analysis and their relative normalized intensity variations are shown in Table 2. This permitted us to infer an increasing presence of PEO in the deposited films by tracking the FT-IR bands in common between the two components, i.e. νC–H and ω/τ/δC–H, (Table 2) both associated to alkyl groups.14,17,48 In fact, the contribution of PEO molecules to the amount of C–H groups per unit mass is greater than that of chitosan. This in turn translates into the relatively more intense stretching vibration band measured on samples with a higher PEO content. These findings were further supported by the measured reduction of the C–N peak's intensity (amide III band) with the PEO content. In fact, since this band is only associated to CH, a decrease of its content in the blend resulted in a significant decrease of the C–N absorbance intensity.48
The analysis of the FT-IR bands maxima's shifts of the ether (977 cm−1), amide II (1525 cm−1) and amide I peaks (1686 cm−1) allowed us to deduce the structural configuration of CH and PEO molecules in the blends. In the first case, the peak center blue-shifted as a consequence of an increase in the PEO content (Fig. 3). This was related to the establishment of intermolecular hydrogen bonds between the hydroxyl groups of chitosan and the protonated hydroxyl ends and the oxygen in the C–O–C groups in PEO chains.20,21,58,59 The same behavior was observed for the amide II band, which shifted from 1525 ± 9 cm−1 (pure CH) to 1589 ± 19 cm−1 (80:20 composition) (Fig. 3). Like in the case of the ether band, such a shift is an evidence of hydrogen bonds formation between polyether oxygen and amino hydrogen in PEO and CH, respectively (Fig. 4).20,49,58
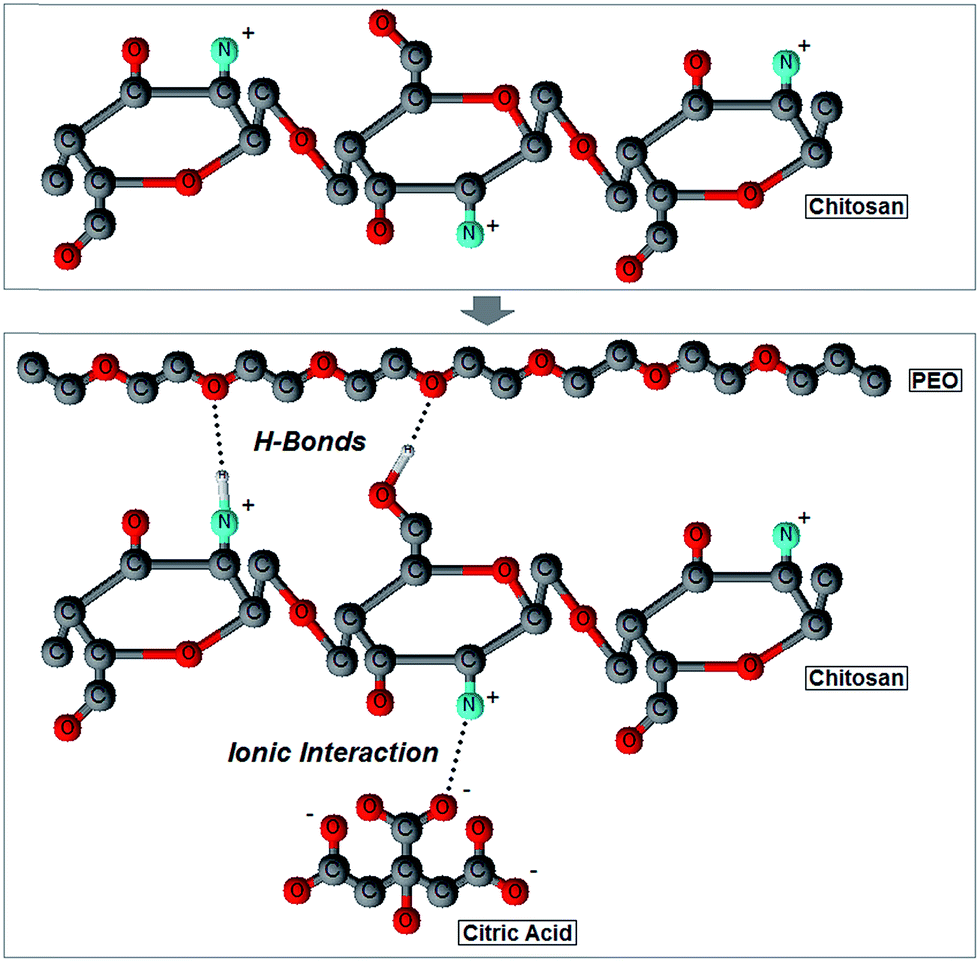 | ||
| Fig. 4 Two-dimensional ball-and-stick representation of the interactions occurring among chitosan, PEO and citric acid molecules during cathodic electrodeposition. | ||
It should be noted that the amide I shift is produced by two different phenomena, namely a low degree of de-acetylation or the formation of ionic interaction with citric acid molecules of the initial electrolytic solution.46,58,61,62 The amide I band results from the vibration of CO bonds in the N-acetyl-D-glucosamine unit of a CH molecule, and it is usually used as an index of the degree of de-acetylation: the greater the amount of CO bonds is, the lower the degree of de-acetylation will be.60,63 As shown in Fig. 3, the amide I band has a relative higher intensity in pure CH films, which thus suggests a low degree of de-acetylation. However, this is in contrast with the high de-acetylation grade chitosan used in this work (see Materials and methods). In addition, the high degree of de-acetylation was also confirmed by Raman spectroscopy: characteristic Raman vibrational bands of the methyl group of acetamide, typical of the acetylated unit of CH (N-acetyl-D-glucosamine), were not in fact detected in the 1379–1397 rel cm−1 range (Fig. 2).28,36,58 We can thus infer that the high presence of CO groups, responsible for the amide I vibration, does not depend on a low degree of de-acetylation but rather on the presence of citric acid molecules which ionically crosslink chitosan by creating a carboxylate connection between the COO− groups of the acid and the NH3+ groups of CH (Fig. 4).46,58,61,62 To further support this observation, the significant large red-shift measured (Fig. 3), an evidence of the reduction of the ionic bonds between the COO− and the NH3+ groups,61 suggests that the number of ionic interactions decreases as the PEO content increases because of the progressive reduction of free NH3+ groups available for ionic interactions. However, it was previously demonstrated that the amide I band can also shift in response to the establishment of hydrogen bonds. In particular, when the band center is located around 1619 cm−1 it can be assigned to the vibration of CO groups hydrogen bonded to hydroxyl groups; conversely, when it is around to 1652 cm−1, the amide I band can be assigned to the vibration of a CO group hydrogen bonded to the amine.37 Taken together, the recorded red shift of the amide I band as a result of an increase in the PEO content most likely results from a combination of the above factors, namely a reduction of ionic interactions with citric acid molecules and the formation of hydrogen bonds between CO, the protonated hydroxyl end groups of PEO and the amine groups of CH.
Spectroscopic results were complemented by the quantification of the nanomechanical properties of the films obtained by Digital Pulsed Force Mode (DPFM) (Table 3). With this technique, stiffness maps are created by assigning a color code to the cantilever's deflection (measured in the linear portion of deflection-phase curves) when indenting the sample's surface. Qualitative comparison of DPFM images revealed a relatively homogeneous stiffness distribution across all samples, without any evident difference associated to their composition (Fig. 5).
| Mechanical properties | 100:0 |
80:20 |
50:50 |
20:80 |
|---|---|---|---|---|
| Stiffness [N m−1] | 1.91 ± 0.32 | 1.92 ± 0.25 | 1.77 ± 0.19 | 1.35 ± 0.12 |
| Young’s Modulus [MPa] | 11.4 ± 2.7 | 11 ± 2.6 | 10.5 ± 1.8 | 8.2 ± 2.2 |
| Compliance [m N−1] | 0.57 ± 0.09 | 0.56 ± 0.08 | 0.59 ± 0.06 | 0.76 ± 0.08 |
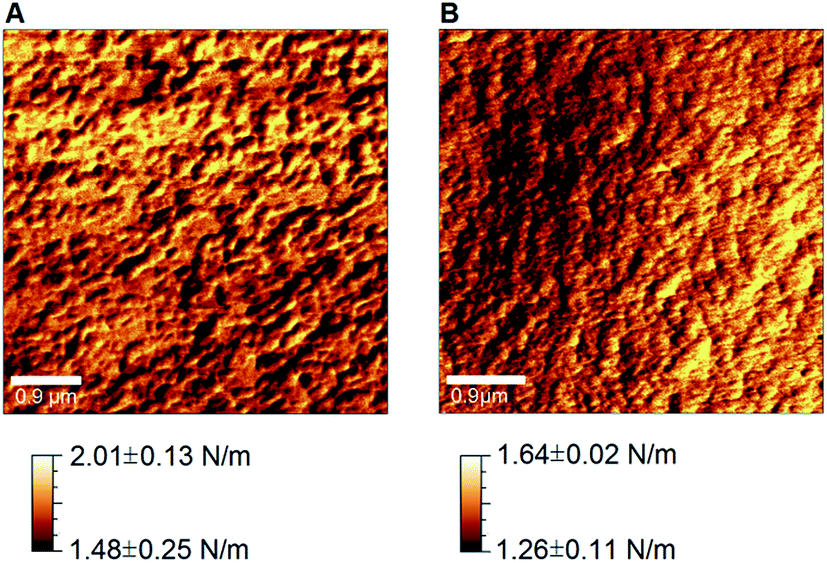 | ||
| Fig. 5 Stiffness maps obtained by AFM-DPFM on pure chitosan (A) and (B) 20:80 CH:PEO films. | ||
Noteworthy, DPFM maps not only qualitatively showed the spatial distribution of stiffness, but they also permitted to quantify it along with additional properties, such as Young's modulus and compliance. Every point of the map can be in fact converted into a force–distance curve that provides deeper information on nanomechanical properties.51
To this end, we collected three DPFM maps for each sample, for a total of 3 samples per condition (n = 9). From each color-coded stiffness map, we selected five points at each of the two ends of the scale (i.e. five highest and lowest stiffness values), which were successively used to calculate the average stiffness (Fig. 6A, black squares) and elastic modulus (Fig. 6B, black squares) for each film composition. It follows that the error bars in Fig. 6 are representative of the intrinsic spatial inhomogeneity of the nanomechanical features of surfaces.
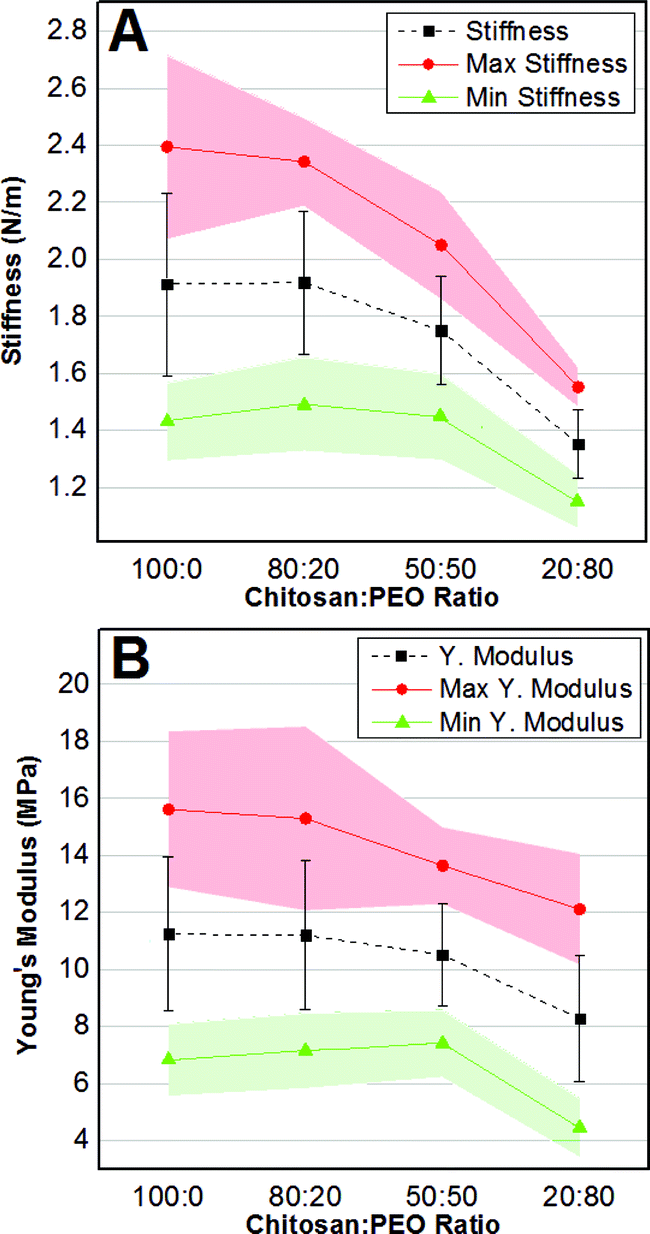 | ||
| Fig. 6 Average ± standard deviation (black squares and bars), average maxima (red dots) and average minima (green triangles) values of stiffness (A) and Young's modulus (B) as a function of the PEO content. The 95% confidence interval is depicted as a red (maxima) and green (minima) shading. | ||
In addition, we monitored how the stiffness' (Fig. 6A) and the elastic modulus' (Fig. 6B) maxima (red dots) and minima (green triangles), expressed in terms of average values with a 95% confidence interval (red and green shading), varied as a function of the composition. The average stiffness gradually decreased from 1.91 ± 0.32 N m−1 for pure chitosan to 1.35 ± 0.12 N m−1 for 20:80 CH:PEO samples. The one-way ANOVA test established that stiffness was only statistically different for the 20:80 samples. Similarly, the average Young's modulus varied from a maximum of 11.4 ± 2.7 MPa for pure CH to a minimum of 8.2 ± 2.2 for the 20:80 composition (Table 3). Also in this case the only significant difference was shown by the 20:80 samples.
The same trend was observed for the average stiffness/Young's modulus maxima and minima. In particular, the average stiffness maxima decreased from 2.4 ± 0.5 N m−1 to 1.5 ± 0.1 N m−1, while the average minima varied from 1.4 ± 0.1 N m−1 to 1.1 ± 0.1 N m−1 as the PEO content increased (Table 3). However, only the nanomechanical parameters of the 20:80 film showed statistical differences. It also followed that the compliance (a more accurate parameter to describe surface nanohardness51) was significantly greater for the 20:80 samples in respect to the other compositions (Table 3).
The comparison of our results with the existing literature was not unequivocal because of the large variability in the published results.64–71 In particular, the mechanical properties of chitosan not only depend on the sample preparation but also on the characterization method and on the scale at which the analysis is carried out. In this context, a correlation between measurements obtained with the same indenter at various scales (i.e. micro and nanoscale) was deemed unreliable, since they depend on various effects associated to the characteristics of the indentation print especially on viscoelastic materials.64 For this reason, the micro- and nano-indentations are generally used for different purposes: the former is considered a measure of the hardness of bulk materials, while the latter is utilized for assessing the nanomechanical surface properties. However, the bulk mechanical properties can nonetheless provide guidance to better understand the nanomechanical behavior at the surface. Translated to our work, the reduction of stiffness/Young's modulus in films with a higher PEO content (Fig. 6) is in good agreement with previous indentation65 and uniaxial tensile test66 measurements even if on a different scale. In addition, an elastic modulus of 9.46 MPa was measured for CH:PEO fibers in ratio 1:1 with a spherical nanoindenter.20 Despite the difference in the sample preparation, this value is consistent with the 10.53 MPa measured for CH:PEO 50:50. Moreover, a stiffness value of 1.71 nN nm−1, a value in the same order of magnitude that calculated for CH:PEO 20:80, was measured for a PEO–PPO–PEO triblock copolymer, a material that, despite a different composition, shows a similar large proportion of PEO.70 On the other hand, inconsistencies between our work and previous literature may originate from different assumptions used in the contact mechanics models68,69 as well as from the strong dependence of stiffness/elastic modulus on the experimental parameters such as indentation depth and frequency.64,71
Sample preparation, and in particular the use of citric acid, is also another variable that influenced the nanomechanical properties by increasing ionic crosslinking between chitosan chains.67 In CH:PEO blends, the crosslinking effect of citric acid, revealed by Raman and FT-IR spectroscopy, was created by ionic interactions between NH3+ of CH chains and COO− of citric acid molecules. Such interactions were not only responsible for the higher Young's modulus compared to literature,68,69 but also for the dependence of the nanomechanical properties on the PEO content. In fact, the increase of PEO content and the consequently reduction of chitosan molecules, produced a reduction of the free NH3+ groups available for ionic interactions with citric acid molecules, thereby resulting in a lower stiffness of the material.
Results obtained by AFM-DPFM imaging (Fig. 5) and measurements (Table 3) also revealed that a phase separation did not occur. In fact, considering the randomized selection of the areas probed by the AFM, if a distinct PEO phase formed, one would expect to see either of the following behavior, as the PEO content increased: (i) the stiffness maxima significantly increase and the minima remain constant (assuming the creation of a PEO second phase stiffer than pure CH) or (ii) the stiffness minima significantly decrease and the maxima remain constant (assuming the creation of a PEO second phase more compliant than pure CH). Since it was previously reported that bulk PEO exhibits a lower stiffness than CH,72,73 the latter outcome was more likely to occur in case of a phase separation. However, since both maxima and minima decreased, we inferred a homogenous distribution of the two molecules, which in turn equally affected the highest and lowest stiffness values towards more compliant films composed by CH and PEO chains bonded via hydrogen bonds and ionic interactions (Fig. 4). The establishment of hydrogen bonds between CH and PEO molecules also plays a fundamental role in determining the blend's crystallinity. While CH shows an amorphous state due to the intramolecular interactions that limit the movement of the chains and thus hamper its crystallization, PEO is characterized by a semicrystalline structure composed by spherulitic crystals embedded in an amorphous matrix.18 It was shown that CH–PEO blends with a CH content higher than 80–90% produced with standard techniques (e.g. solvent casting) do not preserve the crystalline organization of PEO.14,18,65,74 Such effect was ascribed to the interruption of PEO–PEO interactions by the competitive formation of hydrogen bonds between CH and PEO molecules.14 An alternative hypothesis asserts that CH does not affect the ordered structure of PEO but instead limits the nucleation process.18 In this context, the spectroscopic techniques used in this work did not allow to accurately probe the degree of crystallinity of the electrodeposited blends. In particular, although Raman spectroscopy has been successfully exploited to assess the crystallinity of various polymers,75 the background noise generated by the material's autofluorescence may alter the width of the Raman bands, thereby preventing the correct interpretation of the results. Similarly, it could be conceived that crystalline spherulites possess different nanomechanical properties as compared to the surrounding amorphous matrix, and thus could be detected by DPMF-AFM mapping. Although our AFM analysis did not reveal any localized domains with distinctively dissimilar nanomechanical properties (thereby suggesting the absence of such spherulites), we cannot however exclude the existence of crystalline regions with a similar stiffness than that of the matrix. Based on these assumptions and our experimental data, we can only conclude that the amorphous/crystalline character of the electrodeposited blends likely depends on the PEO content,14,18,65,74 but further experimental refinements are required. It will be in fact necessary to resort to additional techniques (e.g. X-ray crystallography, differential scanning calorimetry and polarized optical microscopy) to precisely characterize the crystallinity of the electrodeposited films. It emerges that the processing technique strongly affects the microstructure of the CH–PEO blends, but the comprehension of the underlying mechanisms goes beyond the scope of this work.
Conclusion
In this work, we have exploited cathodic electrodeposition to create biopolymer blends, which we have precisely characterized by spectroscopic and nano-mechanical techniques. Our findings demonstrate the effectiveness of the electrochemical process in generating CH:PEO blends with variable composition and nanomechanical properties, which thus promises to become a valuable tool to engender tailor-made polymeric substrates for in vitro and in vivo applications. In addition, we unveiled the mechanisms at the molecular level that develop during co-deposition, ultimately achieving a better understanding of the interactions between CH and PEO molecules in electrodeposited blends. Our results also suggest that cathodic electrodeposition may lend itself to the creation of multi-component blends, as long as at least one component can be electrically charged and able to establish chemical interactions (e.g. hydrogen bonding) with the other(s).Acknowledgements
D.A.D.C. acknowledges financial support from Politecnico di Milano through the scholarship Tesi Estero. F.V. acknowledges financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery grant, from the Canada Foundation for Innovation (CFI) and the Ontario Ministry of Research and Innovation (MRI) through the Leaders of Opportunity (LOF) fund. L.A. and L.D.N. acknowledge MIUR-FIRB Futuro in ricerca (Surface-associated selective transfection — SAST, RBFR08XH0H) for the economic support.Notes and references
- S. L. Levengood and M. Zhang, J. Mater. Chem. B, 2014, 2, 3161–3184 RSC.
- A. Di Martino, M. Sittinger and M. V. Risbud, Biomaterials, 2005, 26, 5983–5990 CrossRef CAS PubMed.
- I. Zhitomirsky and A. Hashambhoy, J. Mater. Process. Technol., 2007, 191, 68–72 CrossRef CAS PubMed.
- A. Simchi, F. Pishbin and A. R. Boccaccini, Mater. Lett., 2009, 63, 2253–2256 CrossRef CAS PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 CrossRef CAS PubMed.
- A.-Z. Chen, M.-Y. Chen, S.-B. Wang, X.-N. Huang, Y.-G. Liu and Z.-X. Chen, J. Appl. Polym. Sci., 2012, 124, 3728–3736 CrossRef CAS.
- M. J. Alonso and A. Sánchez, J. Pharm. Pharmacol., 2003, 55, 1451–1463 CrossRef CAS PubMed.
- K. Hermans, D. Van den Plas, S. Kerimova, R. Carleer, P. Adriaensens, W. Weyenberg and A. Ludwig, Int. J. Pharm., 2014, 472, 10–19 CrossRef CAS PubMed.
- G. de Oliveira Fulgêncio, F. A. B. Viana, R. O. S. Silva, F. C. F. Lobato, R. R. Ribeiro, J. R. Fanca, R. M. D. Byrro, A. A. G. Faraco and A. da Silva Cunha-Júnior, Vet. Ophthalmol., 2014, 17, 150–155 CrossRef PubMed.
- G. de O. Fulgêncio, F. A. B. Viana, R. R. Ribeiro, M. I. Yoshida, A. G. Faraco and A. da S. Cunha-Júnior, J. Ocul. Pharmacol. Ther., 2012, 28, 350–358 CrossRef PubMed.
- E. Mortazavian, F. A. Dorkoosh and M. Rafiee-Tehrani, Drug Dev. Ind. Pharm., 2014, 40, 691–698 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- Z. Wang, X. Zhang, J. Gu, H. Yang, J. Nie and G. Ma, Carbohydr. Polym., 2014, 103, 38–45 CrossRef CAS PubMed.
- S. Zivanovic, J. Li, P. M. Davidson and K. Kit, Biomacromolecules, 2007, 8, 1505–1510 CrossRef CAS PubMed.
- J. Jin, M. Song and D. J. Hourston, Biomacromolecules, 2004, 5, 162–168 CrossRef CAS PubMed.
- R. S. Jagadish, N. K. Rastogi and B. Raj, J. Polym. Environ., 2010, 18, 266–276 CrossRef CAS PubMed.
- M. Dilamian, M. Montazer and J. Masoumi, Carbohydr. Polym., 2013, 94, 364–371 CrossRef CAS PubMed.
- J. Li, S. Zivanovic, P. M. Davidson and K. Kit, Carbohydr. Polym., 2011, 83, 375–382 CrossRef CAS PubMed.
- A. Apicella, B. Cappello, M. A. Del Nobile, M. I. La Rotonda, G. Mensitieri and L. Nicolais, Biomaterials, 1993, 14, 83–90 CrossRef CAS.
- J. L. Vondran, W. Sun and C. L. Schauer, J. Appl. Polym. Sci., 2008, 109, 968–975 CrossRef CAS.
- M. Pakravan, M.-C. Heuzey and A. Ajji, Polymer, 2011, 52, 4813–4824 CrossRef CAS PubMed.
- M. R. Ladd, T. K. Hill, J. J. Yoo and S. J. Lee, in Nanofibers - Production, Properties and Functional Applications, ed. T. Lin, InTech, 2011 Search PubMed.
- W. E. Teo and S. Ramakrishna, Nanotechnology, 2005, 16, 1878 CrossRef CAS.
- L. Altomare, E. Guglielmo, E. M. Varoni, S. Bertoldi, A. Cochis, L. Rimondini and L. De Nardo, Biomatter, 2014, 4, e29506 CrossRef PubMed.
- L. Altomare, L. Draghi, R. Chiesa and L. De Nardo, Mater. Lett., 2012, 78, 18–21 CrossRef CAS PubMed.
- D. S. Dos Santos, P. J. G. Goulet, N. P. W. Pieczonka, O. N. Oliveira and R. F. Aroca, Langmuir, 2004, 20, 10273–10277 CrossRef CAS PubMed.
- H. L. Vasconcelos, T. P. Camargo, N. S. Gonçalves, A. Neves, M. C. M. Laranjeira and V. T. Fávere, React. Funct. Polym., 2008, 68, 572–579 CrossRef CAS PubMed.
- M. Escamilla-García, G. Calderón-Domínguez, J. J. Chanona-Pérez, R. R. Farrera-Rebollo, J. A. Andraca-Adame, I. Arzate-Vázquez, J. V. Mendez-Mendez and L. A. Moreno-Ruiz, Int. J. Biol. Macromol., 2013, 61, 196–203 CrossRef PubMed.
- G. Keresztury and E. Földes, Polym. Test., 1990, 9, 329–339 CrossRef CAS.
- K.-H. Yang and C.-M. Chang, Anal. Chim. Acta, 2012, 729, 1–6 CrossRef CAS PubMed.
- B. Mattsson, A. Brodin, L. M. Torell, H. Rinne, J. Hamara, F. Sundholm and P. Jacobsson, Solid State Ionics, 1997, 97, 309–314 CrossRef CAS.
- C. E. Orrego, N. Salgado, J. S. Valencia, G. I. Giraldo, O. H. Giraldo and C. A. Cardona, Carbohydr. Polym., 2010, 79, 9–16 CrossRef CAS PubMed.
- A. Aryaei, A. H. Jayatissa and A. C. Jayasuriya, J. Biomed. Mater. Res., Part A, 2014, 102, 2704–2712 CrossRef PubMed.
- A. Synytsya, P. Blafková, A. Synytsya, J. Čopíková, J. Spěváček and M. Uher, Carbohydr. Polym., 2008, 72, 21–31 CrossRef CAS PubMed.
- A. Synytsya, M. Grafová, P. Slepicka, O. Gedeon and A. Synytsya, Biomacromolecules, 2012, 13, 489–498 CrossRef CAS PubMed.
- S. Pèrez, E. Rebollar, M. Oujja, M. Martin and M. Castillejo, Appl. Phys. A: Mater. Sci. Process., 2013, 110, 683–690 CrossRef.
- F. G. Pearson, R. H. Marchessault and C. Y. Liang, J. Polym. Sci., 1960, 43, 101–116 CrossRef.
- S. Morlat and J.-L. Gardette, Polymer, 2001, 42, 6071–6079 CrossRef CAS.
- C. Bergeron, E. Perrier, A. Potier and G. Delmas, Int. J. Spectrosc., 2012, 2012, e432046 Search PubMed.
- L. Xu, Z. Zhang, F. Wang, D. Xie, S. Yang, T. Wang, L. Feng and C. Chu, J. Colloid Interface Sci., 2013, 393, 174–181 CrossRef CAS PubMed.
- H. Zhang and J. Wang, Spectrochim. Acta, Part A, 2009, 71, 1927–1931 CrossRef PubMed.
- W. Cao, K. Tashiro, H. Masunaga, S. Sasaki and M. Takata, J. Phys. Chem. B, 2009, 113, 8495–8504 CrossRef CAS PubMed.
- D. Baskar, R. Balu and T. S. S. Kumar, Int. J. Biol. Macromol., 2011, 49, 385–389 CrossRef CAS PubMed.
- M. H. Uriarte-Montoya, J. L. Arias-Moscoso, M. Plascencia-Jatomea, H. Santacruz-Ortega, O. Rouzaud-Sández, J. L. Cardenas-Lopez, E. Marquez-Rios and J. M. Ezquerra-Brauer, Bioresour. Technol., 2010, 101, 4212–4219 CrossRef CAS PubMed.
- P. Jithendra, A. M. Rajam, T. Kalaivani, A. B. Mandal and C. Rose, ACS Appl. Mater. Interfaces, 2013, 5, 7291–7298 CAS.
- Z. Song, J. Yin, K. Luo, Y. Zheng, Y. Yang, Q. Li, S. Yan and X. Chen, Macromol. Biosci., 2009, 9, 268–278 CrossRef CAS PubMed.
- G. Saraswathy, S. Pal, C. Rose and T. P. Sastry, Bull. Mater. Sci., 2001, 24, 415–420 CrossRef CAS.
- K. Mai-ngam, Colloids Surf., B, 2006, 49, 117–125 CrossRef CAS PubMed.
- N. Rakkapao, V.-s. Visit, Y. Masubuchi and H. Watanabe, Polymer, 2011, 52, 2618–2627 CrossRef CAS PubMed.
- J. Roth and D. Taatjes, Cell Imaging Techniques - Methods and Protocols, Humana Press, 2013 Search PubMed.
- C. A. Rezende, L.-T. Lee and F. Galembeck, Langmuir, 2009, 25, 9938–9946 CrossRef CAS PubMed.
- B. Cappella and G. Dietler, Surf. Sci. Rep., 1999, 34, 1–104 CrossRef CAS.
- M. J. Jaasma, W. M. Jackson and T. M. Keaveny, Ann. Biomed. Eng., 2006, 34, 748–758 CrossRef PubMed.
- D. C. Lin, E. K. Dimitriadis and F. Horkay, J. Biomech. Eng., 2007, 129, 430–440 CrossRef PubMed.
- T. G. Kuznetsova, M. N. Starodubtseva, N. I. Yegorenkov, S. A. Chizhik and R. I. Zhdanov, Micron, 2007, 38, 824–833 CrossRef CAS PubMed.
- M. H. Nurul Hazwani, M. K. Halimah, K. Kaida, W. M. Daud and M. Zaki, AIP Conf. Proc., 2011, 19, 120–127 Search PubMed.
- Jpk Instruments: Application Note, 1–9.
- L. Martinovà and D. Lubasovà, Res. J. Text. Apparel, 2008, 12, 72–79 Search PubMed.
- H. V. Pawar, J. Tetteh and J. S. Boateng, Colloids Surf., B, 2013, 102, 102–110 CrossRef CAS PubMed.
- Q. Zhou, Q. Xie, Y. Fu, Z. Su, X. Jia and S. Yao, J. Phys. Chem. B, 2007, 111, 11276–11284 CrossRef CAS PubMed.
- I. Orienti, T. Cerchiara, B. Luppi, F. Bigucci, G. Zuccari and V. Zecchi, Int. J. Pharm., 2002, 238, 51–59 CrossRef CAS.
- M. F. Cervera, J. Heinämäki, N. de la Paz, O. López, S. L. Maunu, T. Virtanen, T. Hatanpää, O. Antikainen, A. Nogueira, J. Fundora and J. Yliruusi, AAPS PharmSciTech, 2011, 12, 637–649 CrossRef CAS PubMed.
- H. Nazar, D. G. Fatouros, S. M. van der Merwe, N. Bouropoulos, G. Avgouropoulos, J. Tsibouklis and M. Roldo, Eur. J. Pharm. Biopharm., 2011, 77, 225–232 CrossRef CAS PubMed.
- N. Flores-Ramírez, G. Luna-Bárcenas, S. R. Vásquez-García, J. Muñoz-Saldaña, E. A. Elizalde-Peña, R. B. Gupta, I. C. Sanchez, J. González-Hernández, B. Garcia-Gaitan and F. Villasenor-Ortega, J. Biomater. Sci., Polym. Ed., 2008, 19, 259–273 CrossRef PubMed.
- J. Djokic, A. Kojovic, D. Stojanovic, A. Marinkovic, G. Vukovic, R. Aleksic and P. Uskokovic, J. Serb. Chem. Soc., 2012, 77, 1723–1733 CrossRef CAS.
- S. S. Ojha, D. R. Stevens, T. J. Hoffman, K. Stano, R. Klossner, M. C. Scott, W. Krause, L. I. Clarke and R. E. Gorga, Biomacromolecules, 2008, 9, 2523–2529 CrossRef CAS PubMed.
- S. M. Gawish, S. M. Abo El-Ola, A. M. Ramadan and A. A. Abou El-Kheir, J. Appl. Polym. Sci., 2012, 123, 3345–3353 CrossRef CAS.
- Y. Zhu, Z. Dong, U. C. Wejinya, S. Jin and K. Ye, J. Biomech. Eng., 2011, 44, 2356–2361 CrossRef PubMed.
- D. Verma, M. S. Desai, N. Kulkarni and N. Langrana, Mater. Sci. Eng., C, 2011, 31, 1741–1747 CrossRef CAS PubMed.
- M. Tagaya, N. Hanagata and T. Kobayashi, ACS Appl. Mater. Interfaces, 2012, 4, 6169–6175 CAS.
- K. K. M. Sweers, K. O. van der Werf, M. L. Bennink and V. Subramaniam, Nanoscale, 2012, 4, 2072–2077 RSC.
- S. Majd, Y. Yuan, S. Mishra, W. O. Haggard and J. D. Bumgardner, J. Biomed. Mater. Res., Part B, 2009, 90, 283–289 Search PubMed.
- H. Fan, L. Wang, K. Zhao, N. Li, Z. Shi, Z. Ge and Z. Jin, Biomacromolecules, 2010, 11, 2345–2351 CrossRef CAS PubMed.
- M. S. Bostan, E. C. Mutlu, H. Kazak, S. Sinan Keskin, E. T. Oner and M. S. Eroglu, Carbohydr. Polym., 2014, 102, 993–1000 CrossRef CAS PubMed.
- B. H. Stuart, Vib. Spectrosc., 1996, 10, 79–87 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |