
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-polymerization functionalization of poly(3,4-propylenedioxythiophene) (PProDOT) via thiol–ene “click” chemistry†
Bin
Wei
a,
Liangqi
Ouyang
a,
Jinglin
Liu
a and
David C.
Martin
*ab
aMaterials Science and Engineering, the University of Delaware, Newark, DE 19716, USA. E-mail: milty@udel.edu
bBiomedical Engineering, the University of Delaware, Newark, DE 19716, USA
First published on 25th February 2015
Abstract
The surface functionalization of conjugated polymers such as the poly(alkoxythiophenes) poly(3,4-ethylenedioxythiophene) (PEDOT) and poly(3,4-propylenedioxythiophene) (PProDOT) provides a potential means for systematically tailoring their physical properties. We previously reported the synthesis of an alkene-functionalized 3,4-propylenedioxy-thiophene (ProDOT) derivative that could be readily modified through thiol–ene “click” chemistry. Here, we investigated the post-polymerization modification of PProDOT surfaces by using a dialkene functionalized variant (ProDOT-diene). The chemical structure of the ProDOT-diene monomer was confirmed by Nuclear Magnetic Resonance (NMR) and Fourier Transform Infrared spectroscopy (FTIR). The ProDOT-diene monomer was either chemically or electrochemically polymerized into the PProDOT-diene polymer, and then subsequently modified with alkyl, PEG, or ferrocene moieties via radical-based thiol–ene chemistry. We found that the normally insoluble PProDOT-diene could be converted into a soluble derivative by grafting alkyl groups onto the polymer chains after chemical polymerization. When electrochemically deposited on indium-tin oxide (ITO) glass electrodes, the conductivity, electroactivity and contact angles of the modified PProDOT-diene films could be tuned over a broad range. Scanning Electron Microscopy (SEM) revealed that post-polymerization modification did not significantly alter the surface morphology of the PProDOT-diene films. Overall, this method allows for efficient, facile tuning of the surface chemistry of poly(alkylthiophene) films, making it possible to tailor properties such as conductivity and wettability for different applications.
Introduction
Poly(3,4-alkylenedioxythiophenes) such as poly(3,4-ethylenedioxythiophene) (PEDOT) and poly(3,4-propylenedioxythiophene) (PProDOT) and their derivatives have been widely used in organic solar cells,1,2 electrochromic devices,3,4 biosensors5 and neural electrodes.6,7 The interest in using polydioxythiophenes for these applications is related to their low oxidation potential, relatively high chemical and thermal stability, and high conductivity. For example, electrochemically deposited PEDOT films on metal electrodes have shown to dramatically decrease the impedance of the microfabricated neural probes.8 Polythiophenes are also of interest for energy storage and conversion devices. Liu and coworkers designed and characterized an electrochemical supercapacitor based on PProDOT-Me2.9The performance of conjugated polymers depends largely on the ability to optimize their properties for a given application. To improve conductivity, for example, the films are often annealed (either thermally or in solvent vapor)10 or small molecule additives are incorporated.11 However, the processability, surface chemistry, and biocompatibility of conjugated polymer films can only be tuned within a limited range without additional synthesis or functionalization. Fast and convenient chemical functionalization of conjugated polymers is still a challenge and of considerable continued interest for a wide variety of applications.
Conjugated polymers are often polymerized electrochemically instead of chemically due to their typically low solubility and the common requirement of a thin-film geometry. Electrochemical polymerization has many advantages over traditional chemical polymerization such as its speed, reproducibility, and ability to create films with precisely controlled film thickness and morphology.12 Functionalized conducting polymers can be made by incorporating specific small molecules or even polymers of interest into the film as dopants during electropolymerization.13 However, since the dopants are only physically entrapped in the films, the desired functionality may be lost after dopant release or exchange during extended use. There still remains a need for reliable, stable film chemical functionalization strategies. Covalently incorporating functional groups onto the monomers themselves is an attractive strategy for the film functionalization. By doing so, it is ensured that the chemical functionality will remain in the film and not be lost upon electrochemical cycling. Various previous efforts have been directed toward the synthesis and electrochemical polymerization of polymers based on modified 3,4-alkylenedioxythiophenes. Balog et al. reported the synthesis of a functionalized EDOT derivative bearing a highly nucleophilic thiolate group as the side chain.14 Guittard synthesized and characterized a fluorinated EDOT derivative and made superhydrophobic films by electrochemically polymerizing this new monomer.15 Malmström synthesized a new EDOT derivative with an ATRP-initiating side group and grafted poly(acrylic acid) (PAA) brushes on PEDOT films via ATRP.16 Povlich successfully introduced a carboxylic acid side group to EDOT and created peptide-functionalized PEDOT films.17
Despite the simplicity and success of using functionalized thiophene monomers to make copolymers, it is not applicable when the introduced side groups are too bulky or have competing redox reactions under the same conditions with the monomer polymerization.18,19 Side groups with low oxidation potentials may interfere with, or even inhibit, the thiophene polymerization reaction.
Post-polymerization functionalization of appropriately designed precursor conjugated polymers is a possible means to overcome this problem. Post-polymerization functionalization has been widely used in conventional polymer surface modifications, and has also been applied to conjugated polymers.20,21 However, in macromolecular reactions, the low reaction rate and the formation of byproducts can often be drawbacks. Therefore, the use of a clean, efficient reaction that could be carried out under mild conditions with high yield and easy separation would be even more advantageous. In this respect, the radical based thiol–ene “click” reaction is a particularly attractive candidate for polymer functionalization. This reaction has received considerable attention in recent years due to its many advantages over other available coupling reactions.22–25 Thiol–ene “click” reactions usually tolerate a variety of solvents in the presence of oxygen. They give high, often quantitative yields, and produce limited byproducts, leading to straightforward purification. They have been shown to be particularly useful in polymer surface functionalization and macromolecular synthesis including block copolymers and dendrimers, as well as in more traditional applications ranging from crosslinked networks to functionalized biomaterials. Thiol–ene “click” chemistry also provides an additional advantage over other “click” methods that various cysteine-containing peptides or proteins (i.e., with terminal thiols) could be easily introduced to the polymer without extra chemical modification or toxic catalysts. This would be especially beneficial for bio-functionalizing conjugated polymers on biomedical devices such as neural electrodes.17,26,27
In this article, we report the post-polymerization functionalization of poly(3,4-propylenedioxythiophene) (PProDOT) via thiol–ene “click” chemistry. Various terminal thiols were surface immobilized onto PProDOT-diene films under mild reaction conditions with high conversion efficiencies. The resulting functionalized PProDOT polymers were examined by a variety of analytical techniques.
Experimental
3,4-Dimethoxythiophene, diethyl diallylmalonate, lithium aluminum hydride, tetrahydrofuran (THF), toluene, p-toluene sulfonic acid (p-TSA), acetonitrile (ACN), tetrabutyl ammonium perchlorate (TBAP), 2-ethylhexanethiol, o-(2-mercaptoethyl)-o′-methyl-hexa(ethylene glycol) 6-(ferrocenyl) hexanethiol were purchased from Sigma-Aldrich. All reagents and solvents were used without further purification, unless otherwise noted. Samples were irradiated using a UVP Black Ray UV Bench Lamp XX-15L, emitting 365 nm light at 15 W.For oxidative chemical polymerization, FeCl3 (112 mg, 0.69 mmol) was suspended in 2 mL of CHCl3 with stirring. A solution of ProDOT-diene (55 mg, 0.23 mmol) dissolved in 1 mL of CHCl3 was added dropwise, upon which the solution immediately turned dark purple. After stirring for 18 h at room temperature, excess methanol was added to precipitate the polymer and wash away any remaining FeCl3. The resulting solid was filtered and washed with methanol, then dried under vacuum to give 30 mg (55% yield) of a purple powder.
The electrochemical polymerization of ProDOT-diene and characterization of the resulting PProDOT-diene polymer films were performed with an Autolab PGstat12 Potentiostat/Galvanostat (EcoChemie) controlled by a personal computer using the Nova 1.8 electrochemical software in a three-electrode cell. ITO-coated glass (Delta Technologies) served as the working electrode, a platinum plate of 1 cm2 surface area was the counter electrode, with Ag/AgNO3 as the reference electrode. The electrochemical polymerization of ProDOT-diene was studied in a dilute (5 mM) acetonitrile solution containing 0.1 M tetrabutyl ammonium perchlorate (TBAP) as supporting electrolyte. The electrodeposited and surface modified polymer films were investigated in neat electrolyte solution free of monomer.
ITO electrodes coated with PProDOT-diene (P1) thin films with thickness ranging from 0.5 μm to 1.5 μm were immersed into vials containing appropriate thiols (2 mmol) in acetonitrile and 0.1 wt% DMPA as catalyst. After 1 hours of UV (365 nm) exposure at room temperature, the functionalized films were rinsed with copious amounts of acetonitrile and dried in vacuum.
NMR spectra (1H and 13C) were acquired on a Bruker DRX-400 spectrometer. Chemical shifts were reported in parts per million, referenced to chloroform solvent as internal standard (CHCl3: 7.24 ppm for 1H and 77.2 for 13C). FTIR spectra were collected on a Perkin Elmer Spectrum 100 spectrometer fitted with a Universal ATR accessory. Scanning Electron Microscopy (SEM) images and Energy-dispersive X-ray Spectroscopy (EDS) analysis were acquired using a Zeiss Auriga 60 Focused Ion Beam-Scanning Electron Microscope (FIB-SEM) operating at 3 kV (SEM) and 10 kV (EDS), respectively. Static water contact angles measurements were performed by applying a 5 μL drop of deionized water to a film set on a levelled base. Photographs were taken of the drops and their contact angles measured using ImageJ software with a Drop Shape Analysis plug-in and the DropSnake method.
Results and discussion
Synthesis and chemical polymerization of ProDOT-diene
The bi-functional precursor monomer ProDOT-diene was synthesized via the p-TSA catalyzed transetherification route from 3,4-dimethioxythiophene and 2,2-diallyl-1,3-propanediol (Scheme 1) as previously described.28 The dialkene functionalized monomer offers a number advantages over the mono-functional monomer with either alkene, alkyne or azide as the side group. Compared to the mono-functional monomer we previously reported, the bi-functional derivative would largely increase the graft density of the surface modification and thus the properties of the conjugated polymers could be more widely tuned. In addition, functional polymers with pendant electroactive side groups could be produced via either thiol–ene “click” or azide–alkyne “click” step growth polymerization with a bi-functional precursor monomer. The dialkene functionalized monomer also opens the possibility of directly polymerization via ADMET polymerization.29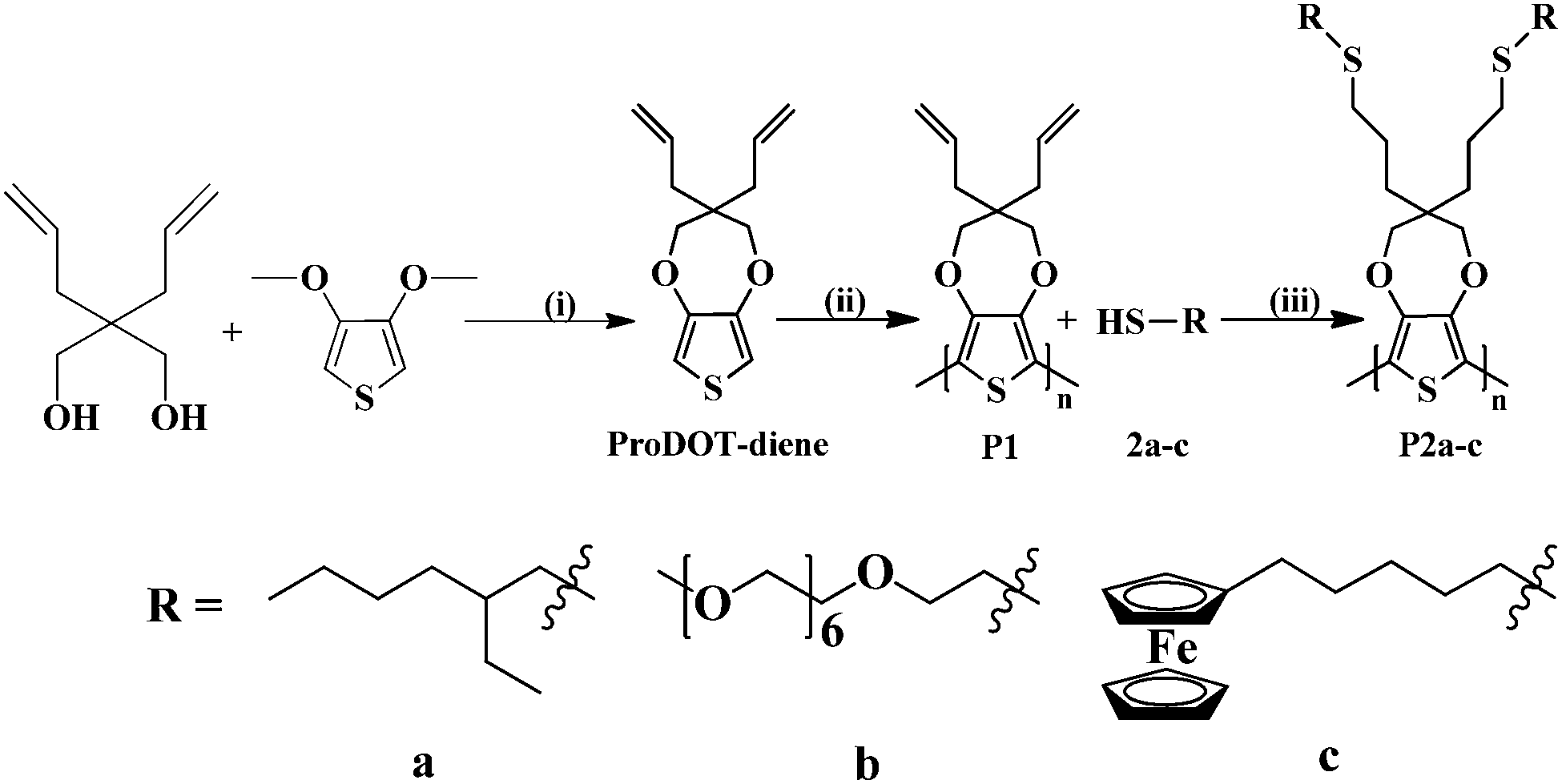 | ||
| Scheme 1 Synthesis of ProDOT-diene (1), electrochemical polymerization of (1) to form poly(ProDOT-diene) (P1) and “click” reaction of P1 with various thiols 2a–c to form P2a–c. (i) pTSA in toluene at 100 °C for 2 days (ii) ACN–TBAP; (iii) 0.1% DMAP, UV 365 nm, room temperature. | ||
The ProDOT-diene was purified as a light yellow viscous liquid and the chemical structure was confirmed by 1H NMR, 13C NMR and FTIR. In Fig. 1A, all the chemical shifts corresponding to each proton on the monomer are clearly assigned. The two peaks located at 5.2 ppm and 5.8 ppm in 1H NMR correspond to the protons on the ene-moiety. Fig. 2A displays the FTIR spectrum of ProDOT-diene. The peaks around 1374 and 1451 cm−1 are the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C stretches of the thiophene ring. The stretching absorption of the alkylenedioxy group and the C–S stretch of the thiophene ring are found at 1189 and 847 cm−1, respectively.
C and C–C stretches of the thiophene ring. The stretching absorption of the alkylenedioxy group and the C–S stretch of the thiophene ring are found at 1189 and 847 cm−1, respectively.
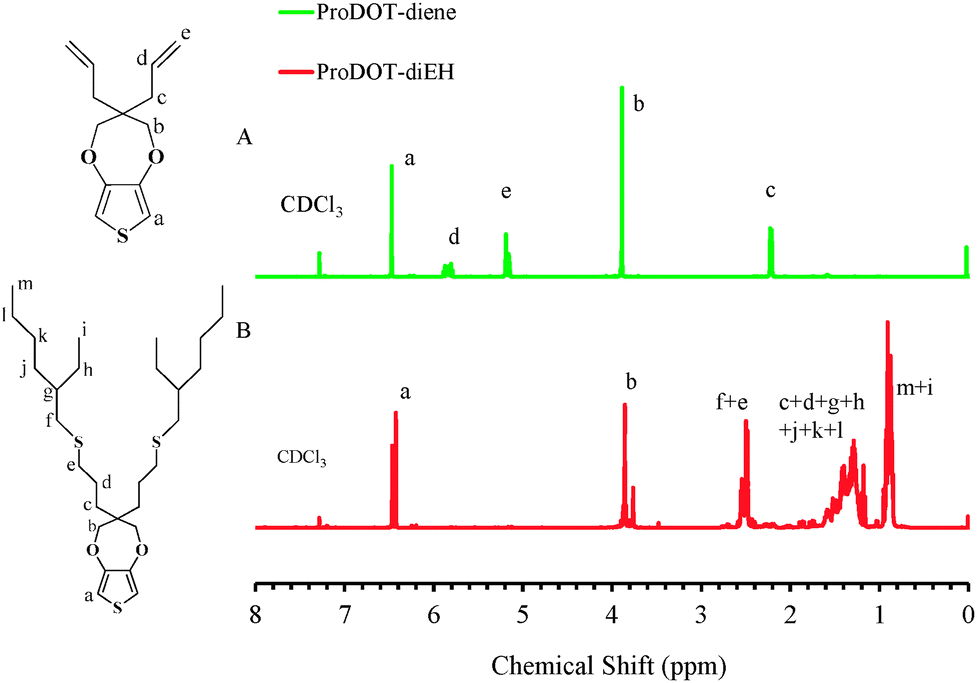 | ||
| Fig. 1 Chemical structure and 1H NMR spectra in CDCl3 of (A) ProDOT-diene and (B) ProDOT-diEH. | ||
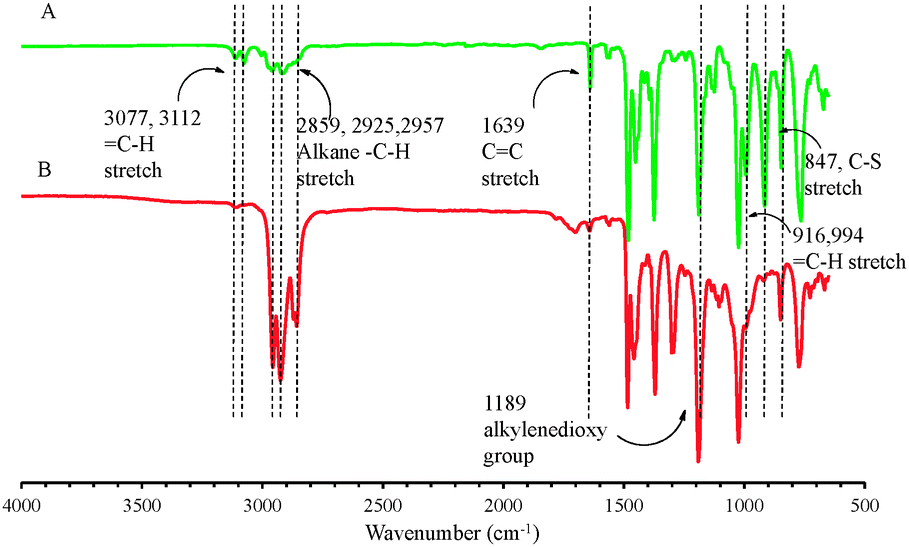 | ||
| Fig. 2 FTIR spectra of ProDOT-diene and ProDOT-diEH. Characteristic absorption bands are labelled with their corresponding wavenumbers. | ||
To verify that the thiol–ene “click” chemistry was feasible for this molecule, ProDOT-diene was subjected to various terminal thiols in the presence of a radical initiator. As confirmed by both 1H NMR (Fig. 1B) and FTIR (Fig. 2B), the disappearance of the two peaks located at 5.2 ppm and 5.8 ppm in 1H NMR and changes of the characteristic peaks associated with the alkene group on the ProDOT-diene (C–H bend at 916 and 994 cm−1 and CC stretch around 1639 cm−1) indicated that the alkene side groups were consumed during the thiol–ene addition. This clearly shows that the ProDOT-diene derivative structure allows various functionalized monomers to be attached through this highly efficient chemistry. The synthesis, polymerization and post-polymerization functionalization process of ProDOT-diene are shown in Scheme 1.
We previously reported the polymerization of various mono-functionalized ProDOTs including ProDOT-ethylhexane and ProDOT-SO3H.28 We also successfully polymerized difunctionalized ProDOT with different small alkyl side groups, for example butanethiol. However, it is not possible to polymerize monomers with electroactive side groups that have lower oxidation potentials than the monomer backbone. Bulky, sterically hindered side groups can also interfere with thiophene polymerization.19,30 In these cases, the direct modification of the conducting polymer products, instead of the monomer precursors, allows for fine-tuning of the ultimate materials surface chemistry without compromising the polymerization and fabrication processes.
PProDOT-diene was chemically polymerized in chloroform with ferric chloride as oxidant, precipitated and then rinsed with copious amounts of methanol to give a dark purple powder. The unmodified polymer had limited solubility in all kinds of organic solvents, however, after modification with alkyl thiols using “click” chemistry the final product showed solubility in tetrahydrofuran, chloroform and toluene. This successfully proved that the thiol–ene “click” chemistry could also be performed on solid polymers as well as the liquid monomers.
Electrochemical polymerization and characterization
The ProDOT-diene monomer was electrochemically polymerized with a potentiodynamic method, scanning between −0.60 V and +1.30 V (vs. Ag/AgNO3). Cyclic voltammograms from a typical experiment are shown in Fig. 3. The irreversible oxidation of ProDOT-diene at +1.20 V is seen in the first anodic scan (red curve). In the subsequent scans, new electroactive species appeared at lower potentials with two small peaks at +0.05 V and +0.35 V corresponding to the formation of polarons and bipolarons31,32 during the anodic scan and two reduction peaks at +0.10 V and −0.28 V during the cathodic scan. The two anodic peaks gradually merged into one broad anodic peak after further electrochemical scans, probably due to the post-polymerization of the polymer. These peaks demonstrated the formation and growth of the PProDOT-diene conjugated polymer film. The thickness of the polymer film steadily increased and could be precisely controlled by the number of scans (Fig. S2, ESI†). The resulting polymer thin films were well adhered to the ITO substrate.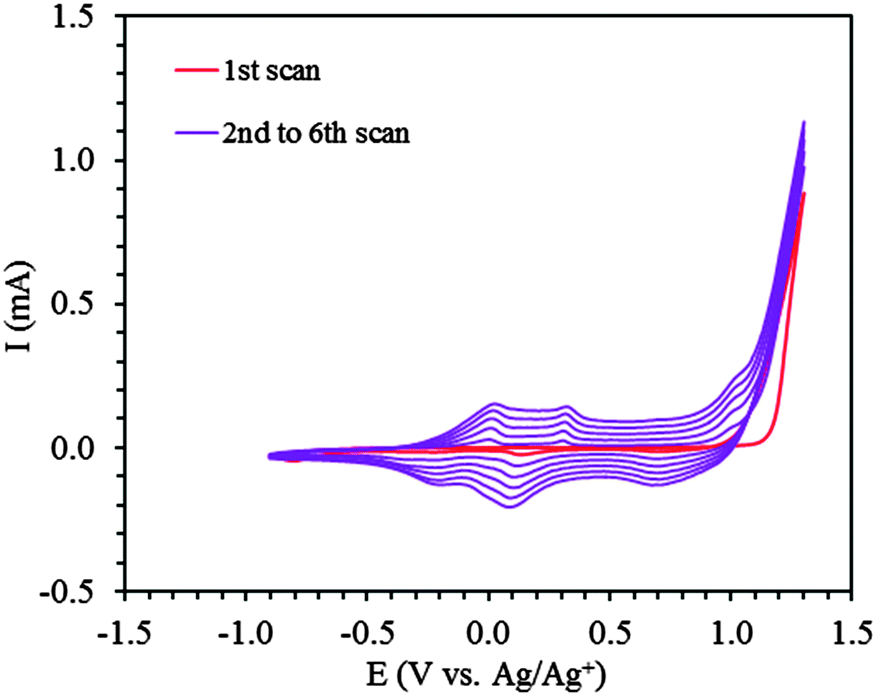 | ||
| Fig. 3 Electrochemical polymerization of ProDOT-diene (5 mM) in ACN–TBAP (0.1 M). 1st scan (red), subsequent scans, 2nd to 6th (purple). | ||
In order to study the redox behavior of the polymer, the films (0.6 μm thick) were rinsed with acetonitrile to eliminate any residual monomer and then cyclic voltammetric studies were performed with the electrodeposited polymer in 0.1 M solution of TBAP in monomer-free acetonitrile by cycling the potential between −0.60 V and +0.80 V at scan rates of 50, 100, 150, 200, 250, 300 and 350 mV s−1 (Fig. 4a). The polymer oxidation and reduction current increased linearly as a function of voltage scan rate33,34 indicating that the coating was electroactive (Fig. 4b). For subsequent surface modification of conducting polymers, electrochemically stable films are a prerequisite. The electrochemical stability of the polymer film P2 was examined by continuous redox cycling. The CV curves of the 1st scan and 100th scan are plotted in Fig. 5. By comparing the charge storage capacity (CSC) of the two curves, it was found that 93% of the CSC was retained after 100 electrochemical scans, indicating that the polymer film is indeed electrochemically stable and would be an excellent platform for post-polymerization functionalization.
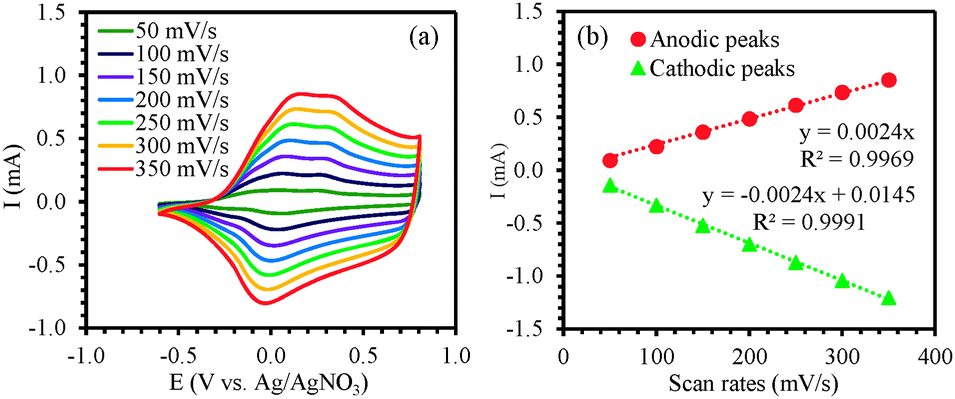 | ||
| Fig. 4 (a) Scan rate dependence of P(ProDOT-diene) film on an ITO electrode in 0.1 M TBAP–ACN at different scan rates. (b) Relationship of anodic and cathodic current peaks as a function of scan rate of P(ProDOT-diene) in 0.1 M TBAP–ACN. | ||
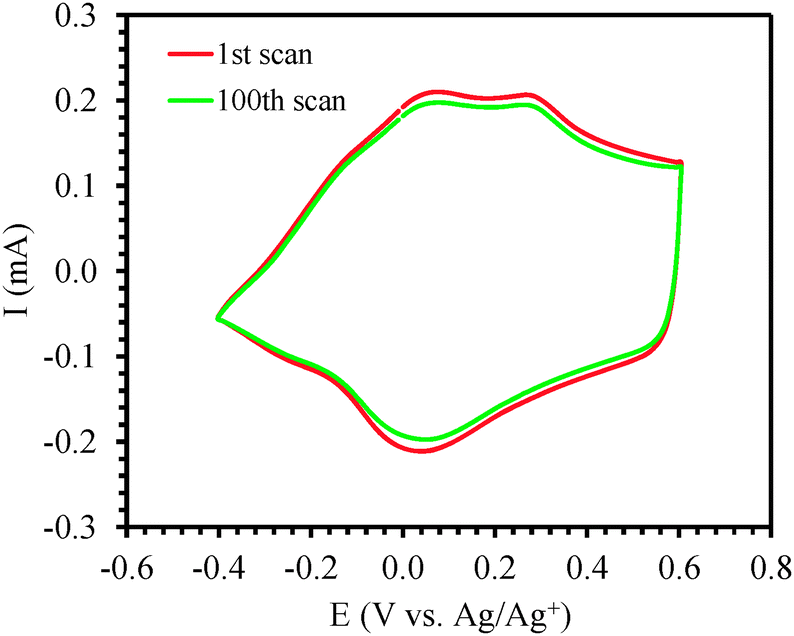 | ||
| Fig. 5 Cyclic voltammograms of P(ProDOT-diene) film: 1st and 100th cycle is plotted. | ||
Post-polymerization functionalization and electrochemical characterization
Based on the high yield of the “click” reaction at the monomer stage and the highly stable thin polymer films that were generated by electrodeposition, it was possible for us to modify the surface of the poly(ProDOT-diene) with various terminal thiols. The protocol for post polymerization functionalization of PProDOT-diene (P1) by thiol–ene “click” chemistry was as follows: ITO electrodes coated with PProDOT-diene thin films were immersed in vials containing various thiols (a–c in Scheme 1) in acetonitrile as solvent and 2,2-dimethyl-2-phenyl-acetophenone (DMPA) as photo-initiator. After being exposed to UV 365 nm for 3 hours at room temperature with gentle swirling, the films were rinsed with copious amounts of acetonitrile to remove excess thiols35 and then dried in vacuum.
Fig. 6 compares the FTIR spectra of the polymer films before and after surface immobilization from 1900 cm−1 to 600 cm−1.36–38 By comparing the FTIR spectrum of P1 with that of P2a, characteristic changes in the spectral features can be found. The stretching band of the –CC backbone at 1638 cm−1 and C–H stretch at 916 cm−1 were found in P1, but almost completely disappeared in the FTIR spectrum of P1a. This observation indicates that the attachment of the EH-thiol onto the P1 side chain is associated with the consumption of the –CHCH2 moieties of P1, which can be ascribed to the thiol–ene addition in the present case.
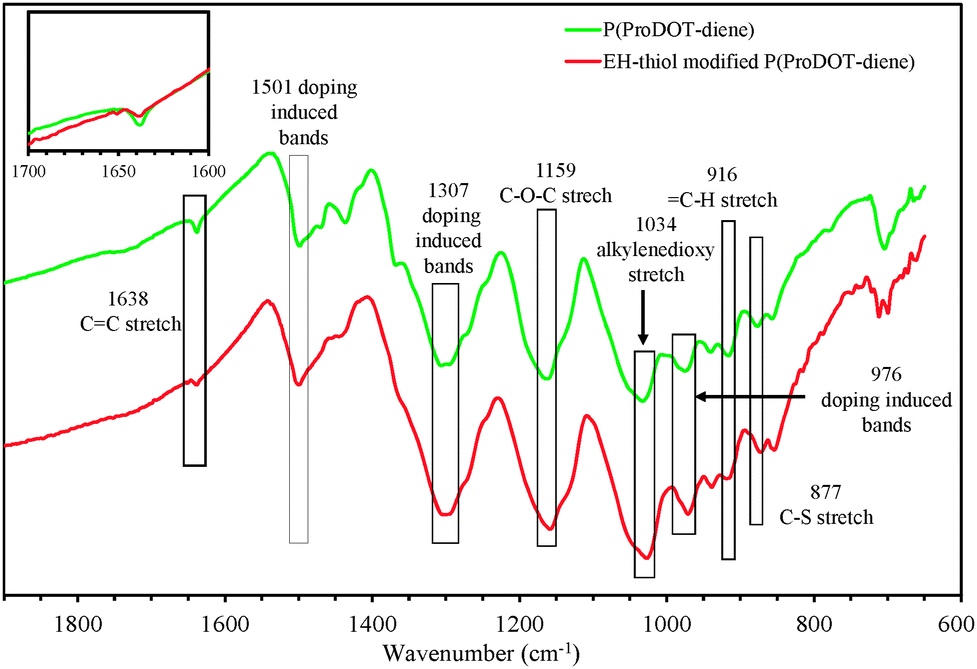 | ||
| Fig. 6 FTIR spectra of P(ProDOT-diene) and EH-thiol modified P(ProDOT-diene). Part of the characteristic absorption bands are labelled with wavenumbers. | ||
In order to demonstrate the broad scope of our synthetic strategy, three moieties with different conductivities and functionalities were chosen. The ethyl-hexyl moiety has alkyl side groups (a) that are charge blocking, so we expected that this group would create an electrically insulating layer, leading to a dramatic decrease in the charge transport properties. While not useful for devices, it would demonstrate that the chemistry was proceeding as anticipated. For the PEG thiol (b) we expected to form a thin, uniform, hydrophilic surface layer. Such layers are known to be useful for functionalizing biomedical devices.39 Ferrocene (c) is an electroactive substituent that can easily change its oxidation state. Attaching a moiety such as this to the polymer films, and showing that the modified material responds dramatically to the electrical activity of the environment, would show the potential of similar chemistries for making chemical sensors based on these materials.40 The electrochemical behavior of the resulting polymer films was investigated by CV. The comparison of the CV curves of the pristine and modified films is shown in Fig. 7. It is clear that the redox behavior of the PProDOT films was considerably altered by grafting the functional moieties onto their surfaces. The disappearance of the broad redox peaks located at +0.1 V that were observed in the pristine polymer film P1 and the significantly reduced charge storage capacity of the modified film P2a (Fig. 7a) are evidently due to the relatively insulating nature of the 2-ethylhexane thiol (a). The electroactivity of the PProDOT was maintained when modified with a relatively short alkane chain,21 whereas longer alkyl side groups block charge transport more effectively.41 In our experiment, the bi-functionality of the side chain presumably allowed a high surface density of grafted alkyl chains. Fig. 7b showed that both the redox peak potentials and the oxidation onset potentials in the o-(2-mercaptoethyl)-o′-methyl-hexa(ethylene glycol) (b) modified polymer film P2b shifted in the positive direction. The oxidation peak potential shifted from +0.1 V to +0.5 V and the oxidation onset potential shift from −0.35 V to +0.15 V. The higher charge storage capacity of polymer film P2b as compared to polymer film P2a is evidently due to the enhanced ion transport enabled by thiol (b)42 considering the almost tripled chain length of thiol (b) than that of thiol (a). The CV of polymer P2c which was modified by electroactive 6-(ferrocenyl) hexanethiol (c) clearly showed the reversible redox behavior of the ferrocene overlaid with the broad peaks of the PProDOT conducting polymer backbone. The potential of the reduction peak in polymer film P2c was the same as for thiol (c) while the potential of the oxidation peak of the modified polymer film P2c (Epox = +0.22 V) was slightly higher than that of thiol (c) (Epox = +0.15 V). This may be due to interference with the conducting polymer itself. Similar to P2b, the oxidation onset potential also shifted to the positive direction moving from −0.35 V to 0 V.
 | ||
| Fig. 7 Cyclic voltammograms of P(ProDOT-diene) films after post-polymerization functionalization with various terminal thiols (a–c) via thiol–ene “click” chemistry in acetonitrile containing 0.1 M TBAP as electrolyte. | ||
The successful surface immobilization on the PProDOT-diene thin films was also confirmed by static contact angle measurements. The pristine PProDOT-diene films were quite hydrophobic, with a water contact angle of 130°. This high contact angle value is likely due to both the intrinsic hydrophobicity of PProDOT polymer films, as well as the rough surface structure of the polymer films as shown in Fig. 8.43,44 Due to the similarly non-polar EH-thiol and ferrocene thiol groups, not much difference was found between the corresponding modified and unmodified PProDOT films. However, when highly hydrophilic PEG was grafted onto the surface, the contact angle of the polymer films decreased to 50°, confirming the ability to dramatically tune the surface wettability of these conjugated polymer coatings through surface chemistry.
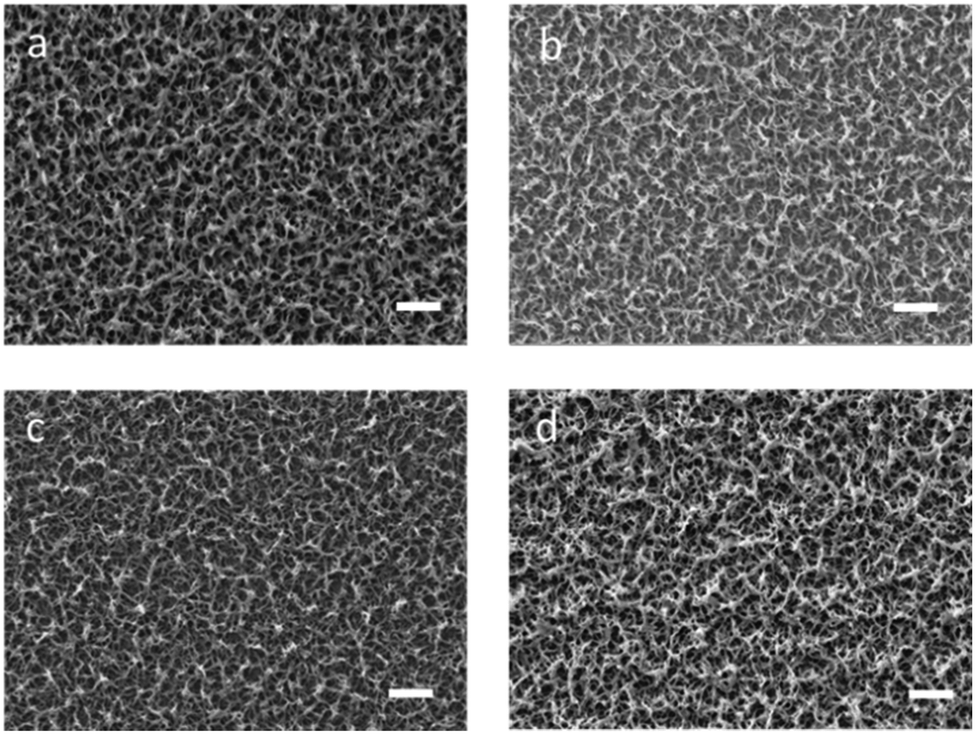 | ||
| Fig. 8 SEM images of (a) P(ProDOT-diene); (b) EH-thiol modified PProDOT-diene; (c) PEO–thiol modified P(ProDOT-diene); (d) ferrocene–thiol modified PProDOT-diene (scale bar represents 2 μm). | ||
The morphologies of the polymers before and after modification were examined using SEM as shown in Fig. 8. Similar to the PProDOT films as shown in Fig. S5 (ESI†), the PProDOT-diene films also showed a rough, network-like structure composed of small fibers and interfibrillar pores. In these networks, the diameter of the pores was between 100 and 300 nm and the diameter of these fibers was around 100 nm. Morphological studies of PProDOT-diene films prepared with different numbers of deposition scans revealed an initially dense surface layer followed by a more articulated, rough surface structure. The effective surface area is significantly increased by this open, rough morphology, and this facilitates charge transport.45,46 Another potential advantage is that rough surfaces may enhance cell adhesion strength and thus promote growth and proliferation.47,48 The rough surface morphology of the PProDOT-diene films investigated were all well retained even after the modification with various thiols, as seen in Fig. 8. This makes it feasible for us to consider immobilizing other moieties of interest such as catalysts or biologically active molecules that could find potential applications in novel biosensors or biomedical devices, while retaining the advantages of the rough, articulated surface morphology.
Conclusions
We have synthesized and characterized a dialkene-functionalized variant of the ProDOT monomer (ProDOT-diene). The corresponding polymer PProDOT-diene was successfully synthesized via both oxidative chemical polymerization and potentiodynamic electrochemical polymerization. Various terminal thiols were readily attached to the PProDOT-diene films by using thiol–ene “click” reactions under mild conditions. Systematic variations in cyclic voltammetry were observed in the functionalized polymer films, clearly showing how these surface modifications could be used to dramatically alter the charge transport and wetting behavior of the resulting polymer films. This synthetic route also allows the incorporation of pendant electronically active groups such as ferrocene to be covalently attached to the conducting polymer films, which is usually not possible by direct polymerization of the corresponding monomers. This synthetic strategy provides a straightforward means for immobilizing other moieties of interest for conjugated polymer films, such as catalysts or biologically active molecules that could find potential applications in novel biosensors or biomedical devices.Acknowledgements
We would like to acknowledge the National Science Foundation DMR-1103027 and the University of Delaware for partial financial support of this research.Notes and references
- A. J. Mozer, D. K. Panda, S. Gambhir, B. Winther-Jensen and G. G. Wallace, J. Am. Chem. Soc., 2010, 132, 9543–9545 CrossRef CAS PubMed.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- C. A. Cutler, M. Bouguettaya, T.-S. Kang and J. R. Reynolds, Macromolecules, 2005, 38, 3068–3074 CrossRef CAS.
- J. Kim, J. You, B. Kim, T. Park and E. Kim, Adv. Mater., 2011, 23, 4168–4173 CrossRef CAS PubMed.
- M. Gerard, A. Chaubey and B. D. Malhotra, Biosens. Bioelectron., 2002, 17, 345–359 CrossRef CAS.
- S. M. Richardson-burns, J. L. Hendricks, B. Foster, L. K. Povlich, D. Kim and D. C. Martin, Biomaterials, 2007, 28, 1539–1552 CrossRef CAS PubMed.
- M. R. Abidian, J. M. Corey, D. R. Kipke and D. C. Martin, Small, 2010, 6, 421–429 CrossRef CAS PubMed.
- X. Cui and D. Zhou, IEEE Trans. Neural Syst. Rehabil. Eng., 2007, 15, 502–508 CrossRef PubMed.
- D. Y. Liu and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2010, 2, 3586–3593 CAS.
- Z. a. King, C. M. Shaw, S. A. Spanninga and D. C. Martin, Polymer, 2011, 52, 1302–1308 CrossRef CAS PubMed.
- Q. Wei, M. Mukaida, Y. Naitoh and T. Ishida, Adv. Mater., 2013, 25, 2831–2836 CrossRef CAS PubMed.
- L. Groenendaal, G. Zotti, P.-H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855–879 CrossRef CAS.
- R. A. Green, N. H. Lovell and L. A. Poole-Warren, Acta Biomater., 2010, 6, 63–71 CrossRef CAS PubMed.
- M. Balog, H. Rayah, F. Le Derf and M. Sallé, New J. Chem., 2008, 32, 1183–1188 RSC.
- M. Wolfs, T. Darmanin and F. Guittard, Soft Matter, 2012, 8, 9110–9114 RSC.
- J. Malmström, M. K. Nieuwoudt, L. T. Strover and J. TravasSejdic, Macromolecules, 2013, 46, 4955–4965 CrossRef.
- L. K. Povlich, J. C. Cho, M. K. Leach, J. M. Corey, J. Kim and D. C. Martin, Biochim. Biophys. Acta, 2013, 1830, 4288–4293 CrossRef CAS PubMed.
- J. Lyskawa, F. Le Derf, E. Levillain, M. Mazari, P. Viel, C. Bureau and S. Palacin, J. Am. Chem. Soc., 2004, 126, 12194–12195 CrossRef CAS PubMed.
- (a) H. Brisset, A.-E. Navarro, C. Moustrou, I. F. Perepichka and J. Roncali, Electrochem. Commun., 2004, 6, 249–253 CrossRef CAS PubMed; (b) H.-P. Wenzel, G. Kossmehl, J. Schneider and W. Plieth, Macromolecules, 1995, 28, 5575–5580 CrossRef; (c) H.-B. Bu, G. Götz, E. Reinold, A. Vogt, S. Schmid, R. Blanco, J. L. Segura and P. Bäuerle, Chem. Commun., 2008, 1320–1322 RSC.
- M. Besbes, G. Trippé and E. Leviallain, Adv. Mater., 2001, 13, 1249–1252 CrossRef CAS.
- H.-B. Bu, G. Götz, E. Reinold, A. Vogt, S. Schmid, R. Blanco, R. Gómez and P. Bäuerle, Tetrahedron, 2011, 67, 1114–1125 CrossRef CAS PubMed.
- M. Kade, D. Burke and C. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- M. A. Caipa Campos, J. M. M. Paulusse and H. Zuilhof, Chem. Commun., 2010, 46, 5512–5514 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- N. K. Singha, M. I. Gibson, B. P. Koiry, M. Danial and H.-A. Klok, Biomacromolecules, 2011, 12, 2908–2913 CrossRef CAS PubMed.
- K. E. Feldman and D. C. Martin, Biosensors, 2012, 2, 305–317 CrossRef CAS PubMed.
- Y. Qin and M. A. Hillmyer, Macromolecules, 2009, 42, 6429–6432 CrossRef CAS.
- L. Zhai, R. Pilston, K. Zaiger and R. D. McCullough, Macromolecules, 2003, 36, 61–64 CrossRef CAS.
- S. Atak, M. İçli-Özkut, A. M. Önal and A. Cihaner, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4398–4405 CrossRef CAS.
- E. J. Laviron, Electroanal. Chem., 1972, 39, 1–23 CrossRef CAS.
- T. Darmanin and F. Guittard, Chem. Commun., 2009, 2210–2211 RSC.
- S. Sadki and C. Che, Electrochim. Acta, 2003, 48, 733–739 CrossRef CAS.
- Y. Lv, Z. Lin and F. Svec, Analyst, 2012, 137, 4114–4148 RSC.
- D. C. Martin, J. Wu, C. M. Shaw and Z. King, Polym. Rev., 2010, 50, 340–384 CrossRef CAS.
- J. Arias-Pardilla and T. F. Otero, J. Mater. Chem., 2012, 22, 4944–4952 RSC.
- C. Kvarnström, H. Neugebauer, S. Blomquist and N. S. Sariciftci, Synth. Met., 1999, 101, 66 CrossRef.
- L. Rao, H. Zhou, T. Li, C. Li and Y. Y. Duan, Acta Biomater., 2012, 8, 2233–2242 CrossRef CAS PubMed.
- E. Scavetta, R. Mazzoni, F. Mariani and B. Fraboni, J. Mater. Chem. B, 2014, 2, 2861–2867 RSC.
- E. Smela, Langmuir, 1998, 14, 2996–3002 CrossRef CAS.
- C. Chiu, H. Chen and S. Kuo, Macromolecules, 2004, 37, 8424–8430 CrossRef CAS.
- T. Darmanin, M. Nicolas and F. Guittard, Phys. Chem. Chem. Phys., 2008, 10, 4322–4326 RSC.
- M. Wolfs, T. Darmanin and F. Guittard, Eur. Polym. J., 2013, 49, 2267–2274 CrossRef CAS PubMed.
- K.-M. Lee, C.-Y. Hsu, P.-Y. Chen, M. Ikegami and K.-C. Ho, Phys. Chem. Chem. Phys., 2009, 11, 3375–3379 RSC.
- X. Cui, J. F. Hetke, J. a. Wiler, D. J. Anderson and D. C. Martin, Sens. Actuators, A, 2001, 93, 8–18 CrossRef CAS.
- Y. Wan, Y. Wang, Z. Liu, X. Qu, B. Han, J. Bei and S. Wang, Biomaterials, 2005, 26, 4453–4459 CrossRef CAS PubMed.
- X. Cui, V. a. Lee, Y. Raphael, J. a. Wiler, J. F. Hetke, D. J. Anderson and D. C. Martin, J. Biomed. Mater. Res., 2001, 56, 261–272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb02033b |
| This journal is © The Royal Society of Chemistry 2015 |