
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling colloidal stability of silica nanoparticles during bioconjugation reactions with proteins and improving their longer-term stability, handling and storage†
C. J.
Moore
a,
H.
Montón
b,
R.
O'Kennedy
cd,
D. E.
Williams
ce,
C.
Nogués
b,
C.
Crean (née Lynam)
f and
V.
Gubala
*a
aMedway School of Pharmacy, Universities of Kent and Greenwich, Chatham, Kent, UK. E-mail: V.Gubala@kent.ac.uk
bDepartament de Biologia Ceŀlular, Fisiologia i Immunologia, Universitat Autònoma de Barcelona, Bellaterra, Spain
cNational Centre for Sensor Research, Biomedical Diagnostics Institute, Dublin City University, Dublin 9, Ireland
dSchool of Biotechnology, Dublin City University, Dublin 9, Ireland
eDepartment of Chemistry, MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
fDepartment of Chemistry, University of Surrey, Guildford, Surrey, UK
First published on 13th January 2015
Abstract
Despite the potential of antibody-coated nanoparticles (Ab-NPs) in many biological applications, there are very few successful, commercially available examples in which the carefully engineered nanomaterial has made it beyond the laboratory bench. Herein we explore the robustness and cost of protein-nanoparticle conjugation. Using multivalent polyamidoamine (PAMAM) dendrimers and dextran as crosslinkers, it was possible to retain colloidal stability during (i) NP-linker binding and (ii) the subsequent conjugation reaction between linker-coated NPs and proteins to generate monodisperse Ab-NPs. This was attributed to the physicochemical properties of the linkers, which were inherited by the NPs and thus benefited colloidal stability. Attaching negatively charged, EDC/sulfo-NHS-activated PAMAM to the NPs contributed to overall negative charge of particles, and in turn led to high electrostatic attraction between the protein and PAMAM-coated NPs during the reaction conditions. In contrast, using an uncharged, EDC/NHS-activated PAMAM dendrimer led to NP aggregation and lower protein binding efficiency. Dextran as a cost-effective, uncharged macromolecule allowed for steric repulsions between neighbouring particles during protein binding, thus inducing NP stability in solution, and also produced monodisperse Ab-NPs. By freeze-drying Ab-NPs from a 1% BSA solution it is possible to reconstitute the solid-form colloid back to a stable state by adding solvent and simply shaking the sample vial by hand. The consequences of the different surface chemistries and freeze-drying stabilizers on the colloidal stability of the NPs were probed by dynamic light scattering. The performance of Ab-NPs was compared in a simple fluorescence linked immunoassay in whole serum. Interestingly, the signal-to-noise ratios were similar for Ab-NPs using PAMAM and dextran, despite dextran binding fewer Abs per NP. We believe this work provides researchers with the tools and strategies for reliably generating Ab-NPs that can be used for a variety of biological applications.
Introduction
The rapid growth in nanotechnology research over the past two decades has led to the development of a ‘nano-Particle Zoo’, housing a myriad of nanomaterials (such as nanoparticles, nanotubes, nanoclusters, nanobubbles, nanostars etc.) with exciting ranges of properties. Though these materials can sometimes be difficult to predictably engineer,1–3 many have shown highly desirable characteristics for industrial applications that promise to influence our daily lives.4–6 Unfortunately, very few have been converted from the laboratory benchtop into commercial success. Importantly, two provocatively-titled contributions by Juliano7 and Venditto8 outstandingly describe the difficult path for the clinical development of nanoparticle-based therapies and warned against ‘overpromising’ the benefits of this technology. This slow progress can be attributed to a number of factors, however some stand out as major stumbling blocks: (i) lack of robustness and reproducibility in nanoparticle surface functionalization, (ii) the use of expensive, specialized reagents (iii) inadequate protocols for the nanomaterial handling that result in excessive manipulation of the samples by the end-user and (iv) limited scope for large scale production. Herein we attempt to address the first three issues by efficiently generating colloidally stable antibody-coated fluorescent silica nanoparticles (Ab-NP) through the use of multivalent linker molecules.The most common method of antibody-nanoparticle conjugation is through covalent bond formation via a linker molecule. The use of a linker creates a space between the protein and the nanoparticle surface, thus avoiding biomolecule denaturation or undesirable biomolecule–nanoparticle interactions. One of the major problems with the published protocols describing the bioconjugation reactions with nanoparticles is that they ignore the effect of the crosslinker introduction, which alters the surface chemistry of a given colloid and often leads to particle aggregation. Conventional colloid science prevents aggregation of ‘plain’ NPs by introducing capping agents to the NP surface or by tailoring surface charges to separate NPs via electrostatic repulsions.9
Bagwe et al.10 for example, preserved the colloidal stability of silica nanoparticles by coating their surface with ‘chemically inert’, negatively charged phosphonates alongside ‘reactive’ –NH2 groups. There has, however, been little focus on how the characteristics of the crosslinker influence nanoparticle stability, and whether this, in turn, can play a role in antibody binding efficiency and Ab-NP polydispersity.
Generating Ab-NPs is usually a two-step process: the attachment of crosslinkers to the ‘plain’ NP surface (Fig. 1b, Step 1), followed by the attachment of antibodies to the NPs via the crosslinker (Fig. 1b, Step 2). It is sensible to assume that sample monodispersity is required throughout these steps and appears to be a natural extension of conventional methods of inhibiting ‘plain’ NP aggregation. This implies that linker's physiochemical properties must be carefully considered before performing the chemical reactions. The lack of attention paid to this issue is surprising, especially when the nanoparticle size and its surface properties are considered to be the two most important parameters for the mediation of biocompatibility of new material used in nanomedicine.11 This view was reinforced in several excellent reviews by Stellacci,12 Tan13 and Thompson,14 in which the authors summarized the main forces governing the interfacial interactions between nanoparticles and a variety of biological systems. Previously, we showed the benefits of multivalent, hyperbranched polyamidoamine (PAMAM) dendrimers as crosslinkers.15 The use of such macromolecules improved antibody binding efficiency and subsequently enhanced Ab-NP binding kinetics in immunoassays.16 Persisting with this multivalent approach, we have reviewed and further optimized the surface conjugation strategies to graft full size model proteins (goat anti-human IgG and bovine serum albumin (BSA)) and also single chain antibody fragments (recombinant anti-C-reactive protein ScFv) onto the surface of dye-doped silica NPs. We have employed three different multivalent crosslinkers: generation 4.5 PAMAM dendrimer and two additional inexpensive macromolecules: 40 kDa dextran and BSA (Scheme 1). Dextran and BSA are more than 10 times less expensive than PAMAM dendrimers and therefore contribute to the reduction of the cost of generating Ab-NPs. The different multivalent linkers present important consequences on the colloidal stability of the NPs and are accountable for the overall efficiency and robustness of the bioconjugation protocols. The prepared antibody-coated nanoparticles were scrutinized in a simple fluorescence-linked immunoassay to qualitatively assess their active surface area (i.e. area of the nanoparticle surface that is covered with antibodies in their active state). We also highlight the misleading depictions of Ab-NPs in many articles that regularly show tethered antibodies oriented such that their epitopes are free and available for binding. Although the scenario of predominantly randomly positioned antibodies on the particle surface is more realistic and appropriate (Fig. 1c), many scientists opt to use the ‘ideal’ and quite unrealistic illustrations. For this reason, we stress the importance of Ab-NP's ‘active surface area’ throughout this article. Importantly, we have also investigated the effect of cryoprotectants17 during freeze-drying to improve the long-term storage and colloidal stability upon re-constitution of Ab-NPs. Apart from the traditional sugar-based protectants, we have also used BSA in an attempt to reduce the different physical and chemical factors that may destabilize the system, notably the freezing and dehydration stresses during the phase separation into ice and cryo-concentrated solution. Two commonly available analytical techniques, dynamic light scattering and fluorescence spectrophotometry, were used to characterize and analyze the prepared nanomaterials. Other techniques such as transmission electron microscopy, scanning electron microscopy, and confocal scanning laser microscopy have been used to support and corroborate the results.
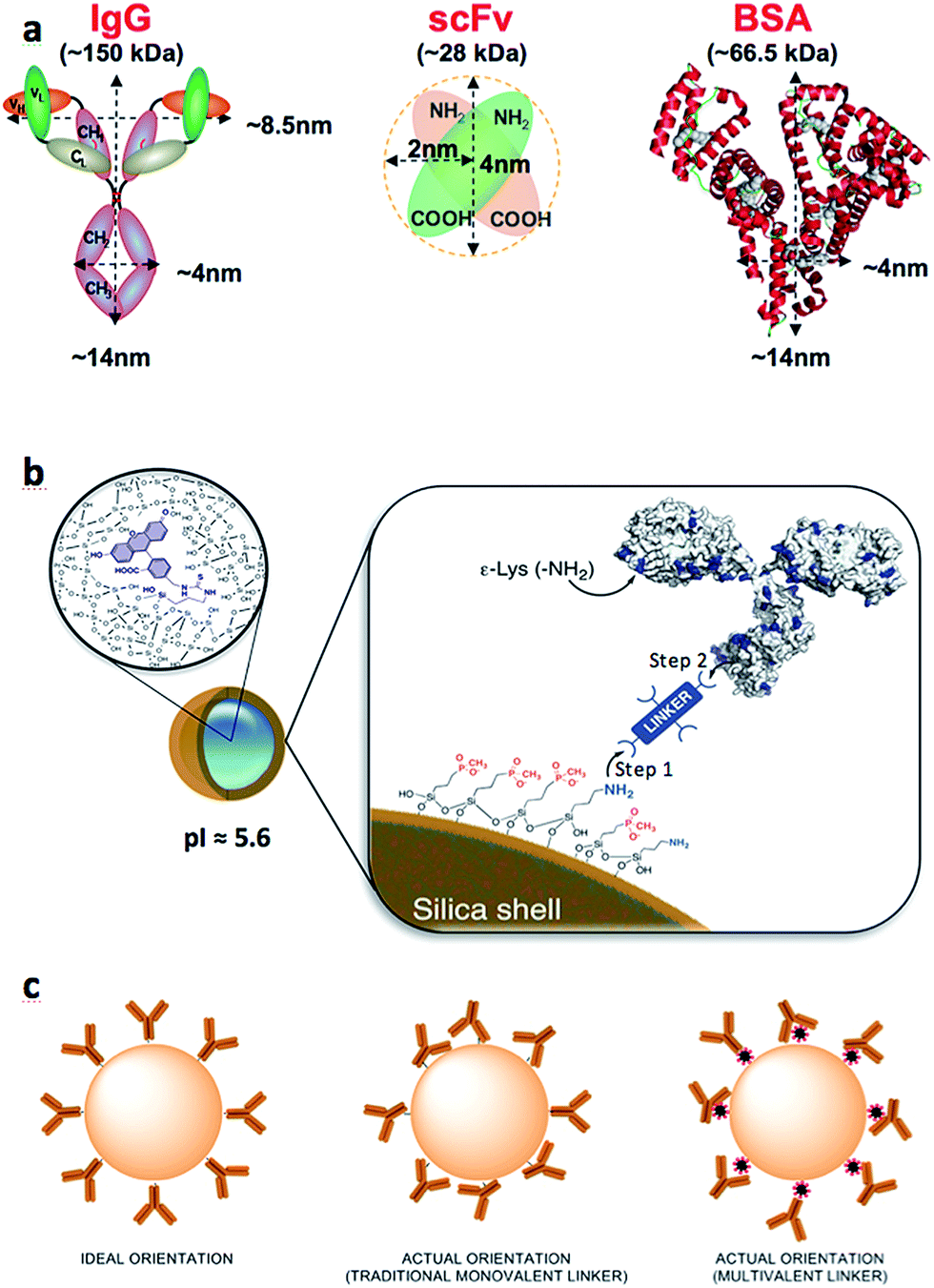 | ||
| Fig. 1 (a) Cartoons of proteins used in this study, their corresponding size and known isoelectric points (pI); (b) magnification of the NP-core showing FITC molecule covalently bound inside the silica matrix. The two steps required to tether antibodies to the NP surface: (Step 1) NP functionalization with crosslinker followed by (Step 2) the attachment of the biomolecule to the linker-coated NP; (c) cartoon representation of idealized and commonly drawn antibodies on NP surface compared to a more realistic scenarios. | ||
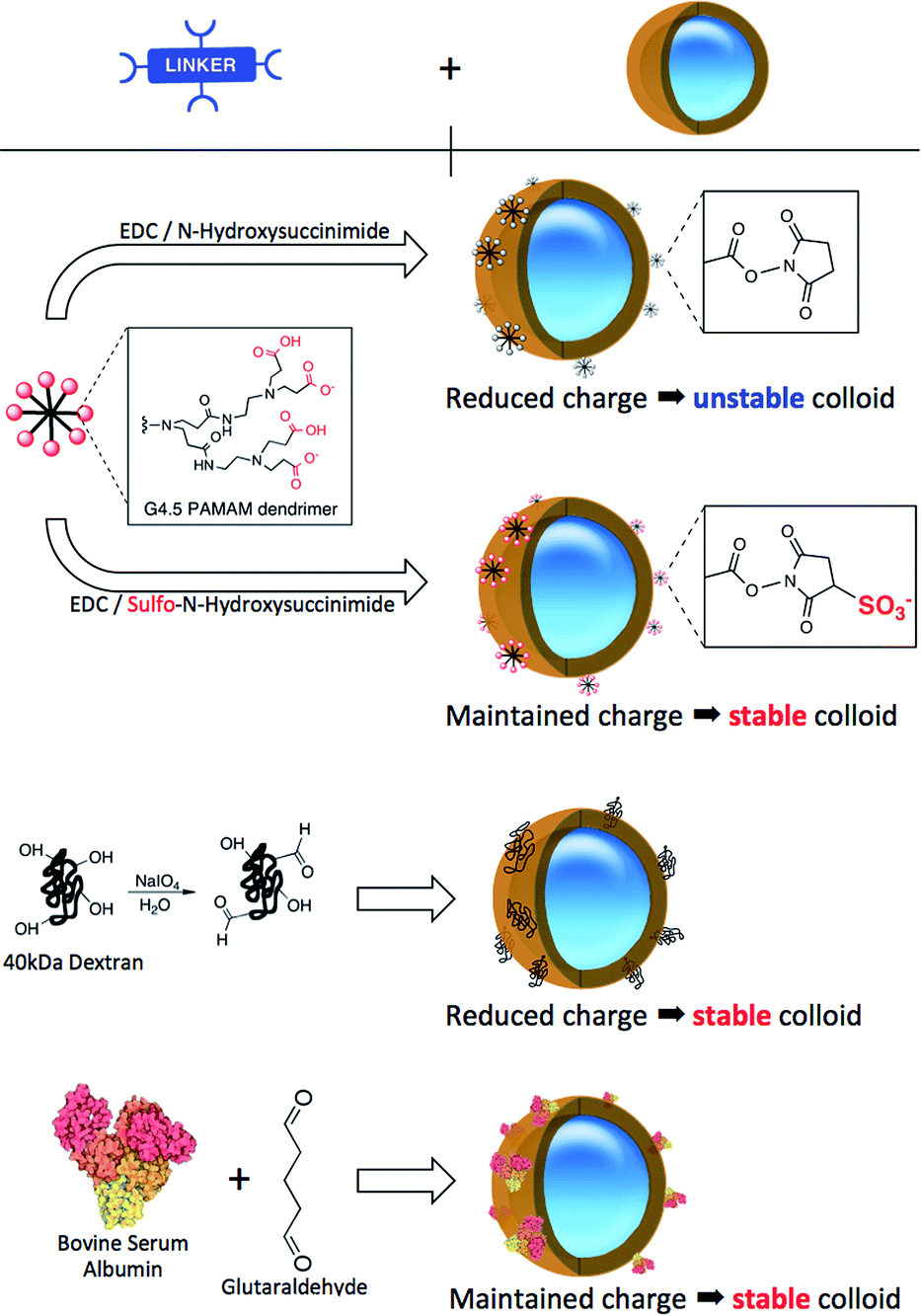 | ||
| Scheme 1 Crosslinkers used in the bioconjugation. | ||
Results and discussion
Using G4.5 PAMAM for bioconjugation optimization
Dye-doped silica nanoparticles were prepared using a well-known, reverse microemulsion method. The synthesis was adapted from Bagwe et al.10 The synthetic protocol includes a nanoparticle surface modification step using siloxane derivatives with both ‘inert’ phosphonate groups and ‘reactive’ amino groups in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 1b). Such specific composition of the NP-shell displays unique electrostatic properties (strongly negative zeta potential and relatively low isoelectric point), due to which the colloid maintains its stability in aqueous solutions. Because of the limited amount of the reactive amino groups on the NP surface, multivalent linkers are employed to increase the number of potential binding sites for biomolecule attachment. We have previously reported on the effects of different half-generations of PAMAM dendrimer with COOH surface groups to couple proteins onto silica nanoparticles.16,18 To successfully perform this reaction, the COOH groups of the crosslinker must be activated. This activation is routinely accomplished using a mixture of NHS/EDC to convert the carboxylic acids to amine-reactive NHS-esters. Such activated dendrimer is added in large excess (∼106 PAMAM:1 NP) to the NP solution to avoid unwanted cross-linking of individual particles. As illustrated in Scheme 1, NHS-esters are neutral, uncharged species. We hypothesized that the activation of PAMAM dendrimers (containing 128 terminal carboxy groups) with EDC/NHS renders it neutral, and upon attachment to the plain NP surface its neutrality is inherited by the negatively charged NPs, thus lowering the stability of the resultant NP-linker colloidal system. This could in turn lead to nanoparticle polydispersity during antibody attachment (Fig. 1b, Step 1 and 2). However, a second form of succinimide is available for COOH activation: sulfo-NHS, which is known to be more water soluble and stable against hydrolysis than NHS.19 The presence of the negatively charged sulfonates (–SO3−) (Fig. 2, bottom) on the activated dendrimer could maintain the necessary electrostatic repulsion to keep the particles highly monodispersed during NP – linker attachment (Fig. 1b, Step 1). This was a prerequisite for the subsequent, successful protein coupling. We tested this hypothesis using dynamic light scattering (DLS) to monitor the particle size evolution during the reaction between NPs and the activated dendrimers (Fig. 2A) and the succeeding protein reaction with the dendrimer-coated NPs (Fig. 2B and C). Prior to the addition of the activated dendrimer, the NP solutions were monodisperse, as shown by TEM (Fig. S.1A†) and DLS (Fig. S.4†). Upon mixing the NPs with the activated dendrimers (using either EDC/NHS or EDC/sulfo-NHS), significant difference in particle aggregation was observed between the two samples (Fig. 2A). Substantial clusters of NPs were detected in the case of NHS-ester activation during the course of 30 minutes (Fig. 2A, top). This broadening in size distribution is attributed to the attachment of the uncharged PAMAM dendrimer and increase in colloidal instability. This was confirmed by monitoring NP zeta potential. Plain NPs exhibited a zeta potential of −42.2 mV and upon binding PAMAM activated by EDC/NHS the resultant linker-coated NP had a zeta potential of −22.5 mV (Fig. S.1B†). In this case, this drop in zeta potential of the colloid, often described as ‘charge’, was insufficient for retaining particle stability in solution and therefore led to partial sample aggregation (Fig. 2A, top, Scan 3). On the contrary, by activating PAMAM with EDC/sulfo-NHS and reacting it with NPs, the resultant NP-linker colloidal system retained its stability (Fig. 2A, bottom). We attribute this to the fact that the dendrimer activation strategy generates a negatively charged macromolecule, and its physicochemical properties are inherited by the NP. This ultimately benefits the NP-linker colloidal system in terms of its overall stability and size population distribution. Unreacted dendrimer was removed from the NP-linker mixture and the colloids were incubated with the protein (Fig. 2B). As a model, we have used Cy5-labeled BSA, with an isolelectric pH of 4.7.20 The isoelectric point of the dye-doped silica nanoparticles depends on the fluorophore and it is estimated between 2.0–6.3 (data not shown). Notably, the hydrolysis of both NHS-esters and sulfo-NHS esters is much slower at lower pH. We have therefore chosen 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES), pH = 4.2 as the optimal buffered solution for the protein conjugation. Aggregation occurred rapidly during the protein conjugation reaction with the NHS-ester activated dendrimer-NPs (Fig. 2B, top). A very broad NP size distribution evolved over the course of the reaction, which carried through to the purified NP product (Fig. 2C, top). However, the sulfo-NHS activation preserved the stability of the colloid during conjugation (Fig. 2B and C, bottom). We attribute this to the state of the linker-coated NPs before being reacted with the protein. Importantly, the differences in the colloidal stability of the samples were visible to the naked eye after only 12 hours at 4 °C (Fig. S.1D†). The monodisperse sulfo-NHS activated dendrimer-NPs were 40% more efficient in the coupling reaction with proteins compared to that of its neutral analogue, as determined by fluorescence measurements (Fig. S.1C†). Such increased binding efficiency is presumably due to the charged nature of the overall linker-coated NPs when reacted with proteins. By incorporating the anionic character of the EDC/sulfo-NHS activated PAMAM dendrimers, the linker-coated NPs displayed a zeta potential of −33.5 mV (Fig. 3) and were therefore highly electrostatically attractive for the positively charged BSA molecules (at pH 4.2). Due to the high probability of BSA being proximal to the linker-coated NPs in solution, there was therefore an increased likelihood of covalent bond formation to generate Ab-NPs. It is also important to note that the use sulfo-NHS-ester activated NPs in this bioconjugation reaction with proteins allowed for the formation of Ab-NPs of narrow size distribution. For example, in nanomedicine, it is important to develop nanomaterials within a given size range (typically <200 nm) such that they can avoid clearance by the reticuloendothelial system, reduce toxicity and successfully locate their target site of interest via the EPR effect.11 In our case, this strategy generated Ab-NPs with an average size of 135 ± 11 nm in diameter (Fig. 3).
:1 ratio (Fig. 1b). Such specific composition of the NP-shell displays unique electrostatic properties (strongly negative zeta potential and relatively low isoelectric point), due to which the colloid maintains its stability in aqueous solutions. Because of the limited amount of the reactive amino groups on the NP surface, multivalent linkers are employed to increase the number of potential binding sites for biomolecule attachment. We have previously reported on the effects of different half-generations of PAMAM dendrimer with COOH surface groups to couple proteins onto silica nanoparticles.16,18 To successfully perform this reaction, the COOH groups of the crosslinker must be activated. This activation is routinely accomplished using a mixture of NHS/EDC to convert the carboxylic acids to amine-reactive NHS-esters. Such activated dendrimer is added in large excess (∼106 PAMAM:1 NP) to the NP solution to avoid unwanted cross-linking of individual particles. As illustrated in Scheme 1, NHS-esters are neutral, uncharged species. We hypothesized that the activation of PAMAM dendrimers (containing 128 terminal carboxy groups) with EDC/NHS renders it neutral, and upon attachment to the plain NP surface its neutrality is inherited by the negatively charged NPs, thus lowering the stability of the resultant NP-linker colloidal system. This could in turn lead to nanoparticle polydispersity during antibody attachment (Fig. 1b, Step 1 and 2). However, a second form of succinimide is available for COOH activation: sulfo-NHS, which is known to be more water soluble and stable against hydrolysis than NHS.19 The presence of the negatively charged sulfonates (–SO3−) (Fig. 2, bottom) on the activated dendrimer could maintain the necessary electrostatic repulsion to keep the particles highly monodispersed during NP – linker attachment (Fig. 1b, Step 1). This was a prerequisite for the subsequent, successful protein coupling. We tested this hypothesis using dynamic light scattering (DLS) to monitor the particle size evolution during the reaction between NPs and the activated dendrimers (Fig. 2A) and the succeeding protein reaction with the dendrimer-coated NPs (Fig. 2B and C). Prior to the addition of the activated dendrimer, the NP solutions were monodisperse, as shown by TEM (Fig. S.1A†) and DLS (Fig. S.4†). Upon mixing the NPs with the activated dendrimers (using either EDC/NHS or EDC/sulfo-NHS), significant difference in particle aggregation was observed between the two samples (Fig. 2A). Substantial clusters of NPs were detected in the case of NHS-ester activation during the course of 30 minutes (Fig. 2A, top). This broadening in size distribution is attributed to the attachment of the uncharged PAMAM dendrimer and increase in colloidal instability. This was confirmed by monitoring NP zeta potential. Plain NPs exhibited a zeta potential of −42.2 mV and upon binding PAMAM activated by EDC/NHS the resultant linker-coated NP had a zeta potential of −22.5 mV (Fig. S.1B†). In this case, this drop in zeta potential of the colloid, often described as ‘charge’, was insufficient for retaining particle stability in solution and therefore led to partial sample aggregation (Fig. 2A, top, Scan 3). On the contrary, by activating PAMAM with EDC/sulfo-NHS and reacting it with NPs, the resultant NP-linker colloidal system retained its stability (Fig. 2A, bottom). We attribute this to the fact that the dendrimer activation strategy generates a negatively charged macromolecule, and its physicochemical properties are inherited by the NP. This ultimately benefits the NP-linker colloidal system in terms of its overall stability and size population distribution. Unreacted dendrimer was removed from the NP-linker mixture and the colloids were incubated with the protein (Fig. 2B). As a model, we have used Cy5-labeled BSA, with an isolelectric pH of 4.7.20 The isoelectric point of the dye-doped silica nanoparticles depends on the fluorophore and it is estimated between 2.0–6.3 (data not shown). Notably, the hydrolysis of both NHS-esters and sulfo-NHS esters is much slower at lower pH. We have therefore chosen 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES), pH = 4.2 as the optimal buffered solution for the protein conjugation. Aggregation occurred rapidly during the protein conjugation reaction with the NHS-ester activated dendrimer-NPs (Fig. 2B, top). A very broad NP size distribution evolved over the course of the reaction, which carried through to the purified NP product (Fig. 2C, top). However, the sulfo-NHS activation preserved the stability of the colloid during conjugation (Fig. 2B and C, bottom). We attribute this to the state of the linker-coated NPs before being reacted with the protein. Importantly, the differences in the colloidal stability of the samples were visible to the naked eye after only 12 hours at 4 °C (Fig. S.1D†). The monodisperse sulfo-NHS activated dendrimer-NPs were 40% more efficient in the coupling reaction with proteins compared to that of its neutral analogue, as determined by fluorescence measurements (Fig. S.1C†). Such increased binding efficiency is presumably due to the charged nature of the overall linker-coated NPs when reacted with proteins. By incorporating the anionic character of the EDC/sulfo-NHS activated PAMAM dendrimers, the linker-coated NPs displayed a zeta potential of −33.5 mV (Fig. 3) and were therefore highly electrostatically attractive for the positively charged BSA molecules (at pH 4.2). Due to the high probability of BSA being proximal to the linker-coated NPs in solution, there was therefore an increased likelihood of covalent bond formation to generate Ab-NPs. It is also important to note that the use sulfo-NHS-ester activated NPs in this bioconjugation reaction with proteins allowed for the formation of Ab-NPs of narrow size distribution. For example, in nanomedicine, it is important to develop nanomaterials within a given size range (typically <200 nm) such that they can avoid clearance by the reticuloendothelial system, reduce toxicity and successfully locate their target site of interest via the EPR effect.11 In our case, this strategy generated Ab-NPs with an average size of 135 ± 11 nm in diameter (Fig. 3).
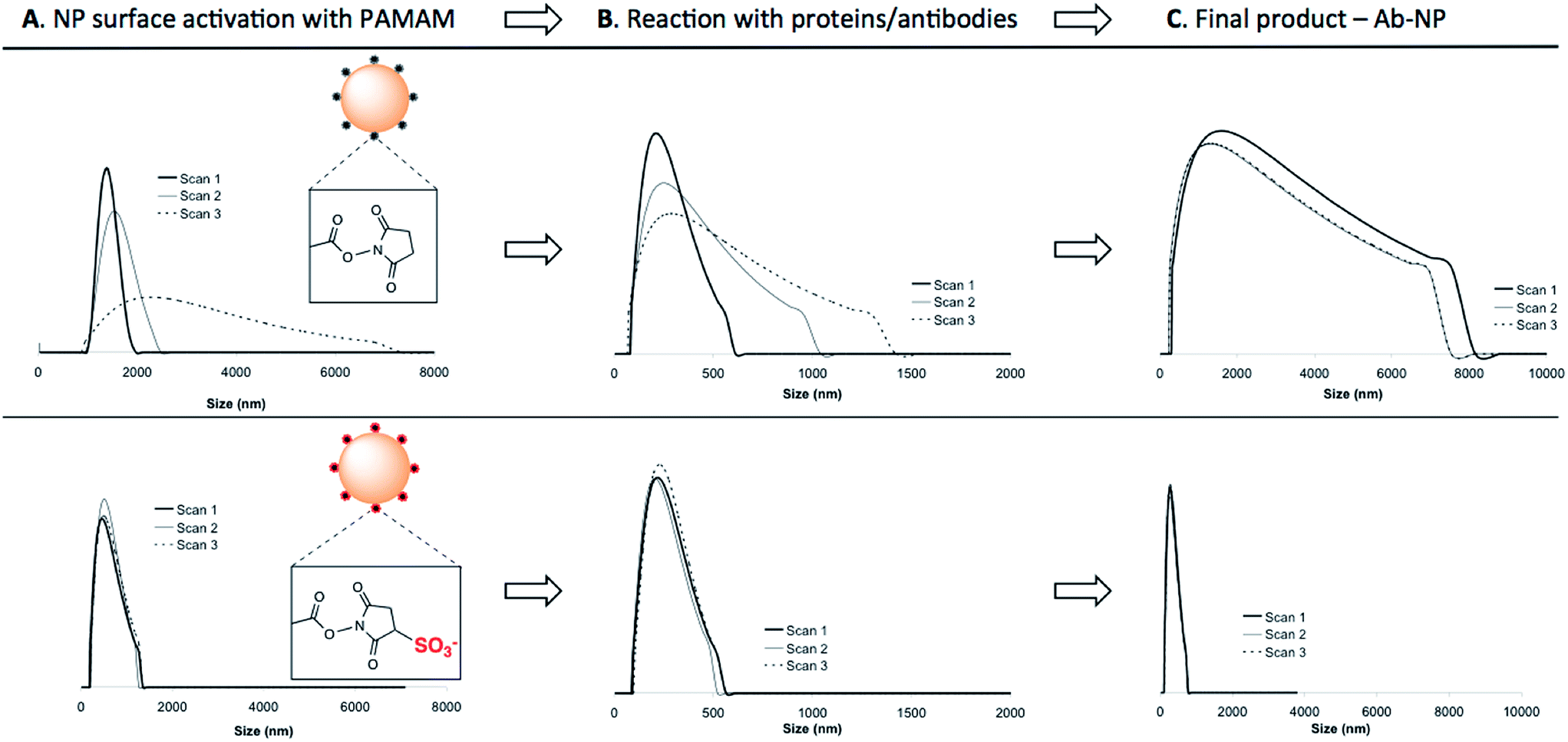 | ||
| Fig. 2 Progress of protein (BSA) conjugation reactions with NPs monitored by dynamic light scattering (DLS). The evolution of the size was measured at different time intervals upon mixing the reagents. In the Step 1: (A) the NPs were reacted with PAMAM dendrimers activated with either EDC/NHS (top) or EDC/sulfo-NHS (bottom). When bound to the NP surface, the negatively charged, EDC/sulfo-NHS activated dendrimer supported NP stability in solution (unlike EDC/NHS activated PAMAM). Measured time intervals: Scan 1 – 0 min, Scan 2 – 15 min and Scan 3 – 30 min; Step 2: (B) corresponds to the reaction of activated NPs with a Cy5-labeled BSA performed in MES 0.1 M pH 4.2. Measured time intervals: Scan 1 – 0 min, Scan 2 – 60 min and Scan 3 – 120 min; and (C) size distributions of the final Ab-NPs following the removal of non-covalently bound protein (measured in triplicate). The use of EDC/sulfo-NHS activated dendrimer resulted in a narrow protein-coated NP size distribution. | ||
 | ||
| Fig. 3 DLS data showing the size and zeta potential evolution of anti-IgG coated NPs prepared using different multivalent linkers. The addition of linker molecules and antibodies to the NP surface induced an increase in particle size. PAMAM and dextran attachment induced NP stability and particle size increases upon linker and antibody attachment were seen. Zeta potential analysis of the NPs showed that PAMAM retained a high negative charge on the NP surface, whilst uncharged dextran produced a more ‘neutral’ colloid. The attachment of BSA led to undesirable ‘growth’ in particle size, attributed initially to the use of glutaraldehyde. | ||
Conjugation of anti-CRP, a single chain variable fragment (scFv): a demonstration of ‘active surface area’ of NPs
To demonstrate the robustness of the sulfo-NHS conjugation strategy and to qualitatively assess that the protein on the NP surface is in fact immobilized in its active, native form, we conjugated small antibody fragments to NPs and show their retained function in a simple aggregation-induced experiment (Fig. S.2A†). These fragments, usually just the active domains of the full proteins, are of <30 kDa in size (Fig. 1a) and can suffer from relatively fast aggregation and denaturation.21 Stripped of the glycosylated and other heavy parts of the typical full size antibody, the fragments tend to be immobilized onto surfaces in conformations that compromise their ability to specifically recognize their corresponding antigens. We speculate that the 3-dimensional shape, size and the flexibility of the G4.5 PAMAM dendrimer introduces a small degree of roughness on the NP surface. As a result, the antibody fragments are ‘anchored’ on the surface through fewer points, thus are able to retain their specific activity and binding capacity. In our experiment, the dendrimer-activated NPs (via sulfo-NHS-ester) were incubated with anti-CRP scFv to produce anti-CRP coated NPs. The colloidal stability of such antibody-conjugated NPs, and their antigen-reactivity were observed by DLS. Fig. S.2B-left† shows that the anti-CRP-NPs were stable over the course of 3 hours of the experiment in the absence of the penta-valent CRP-antigen, although later measurement confirmed stability over several months. The ‘active surface area’ of the NPs (surface of the NPs covered by active anti-CRP) was qualitatively tested by measuring the evolution of particle size over time after addition of the CRP. Upon addition of the CRP-antigen, aggregation of the particles was induced (Fig. S.2B-right†). The resulting, time-evolving cluster size distribution increased exponentially with time (not shown), as expected for reaction-limited aggregation. To support this interpretation, the anti-CRP-NP sample showed negligible aggregation when random, non-specific protein, human chorionic gonadotropin (hCG) was added into the sample instead of CRP (Fig. S.2C†).Alternative multivalent crosslinkers: dextran and BSA
Despite the robustness of G4.5 PAMAM dendrimer in bioconjugation reactions, the cost of this well-defined macromolecule is considerable. Therefore, we searched for alternative hyperbranched molecules that are readily available and inexpensive, but with reasonably predictable chemistries. Dextran and BSA were chosen as appropriate models. Dextran is a polysaccharide already used in medicine, with well-documented chemistry. It is neutral, water-soluble and possesses some favorable properties such as biocompatibility and biodegradability. The use of 40 kDa dextran molecules to attach antibodies onto a flat surface of biosensor devices has been well described in the literature.22–24 Jonsson et al. presented an interesting approach to graft antibodies onto plastic substrates using dextran ‘activated’ by oxidation using NaIO4.25 We have adapted this method for the silica NPs. Oxidation of dextran using NaIO4 generates polyaldehyde dextran that can then act as a crosslinker with the primary amines of the NPs, and those of proteins, via Schiff base formation. The detailed optimization of this reaction including the effects of reaction variables on the reaction efficiency and colloidal stability of the NPs are summarized in the ESI.† BSA and other albumins are also widely studied supramolecules and are the most abundant proteins in plasma.26,27 Albumins are characterized by high content of charged amino acids, aspartic and glutamic acids, lysine, and arginine.28 Activation of the surface carboxylic acids with EDC/sulfo-NHS leads to rapid gelation of a BSA solution (not shown). Alternatively, additional steps employing glutaraldehyde were introduced to obtain the BSA-coated NPs, onto which antibodies could be immobilized. To compare the bioconjugation efficiency of the various multivalent linkers and the effect of the reaction conditions, we have grafted full size antibodies (Alexa Fluor 647 goat anti-human IgG) onto the surface of silica NPs as illustrated in Scheme 1. The number of antibodies immobilized on the NPs was quantified by using fluorescence measurements and described in Table 1. The effect of the surface chemistries on the NP charge and size was evaluated by DLS and shown in Fig. 3 and S.4.†| Linkers | Number of Ab's per NP | NP coverage by Ab's |
|---|---|---|
| PAMAM | 120 ± 9 | 60 ± 5% |
| Dextran | 47 ± 14 | 24 ± 7% |
| BSA | 78 ± 6 | 39 ± 3% |
The incremental increase in the size of the NPs using the PAMAM and dextran method corresponded well to the size of the material that was being immobilized in each reaction step. PAMAM and dextran both have individual sizes of approximately 4–5 nm. The changes in the zeta potential (as an indicator of charge) of the NPs were as expected for both PAMAM and dextran-coated NPs. The zeta potential of the dextran-coated NPs changed to −22.5 mV, which confirmed that such neutral hyperbranched glucan was firmly immobilized on the NP surface. The value rose to −32.5 V after the reaction with antibodies, indicative of successful conjugation. Though the dextran-coated NPs exhibited a zeta potential similar to that of PAMAM-coated NPs activated by EDC/NHS, dextran acts as a steric barrier to the aggregation of adjacent NPs and they ultimately remain stable in solution during the bioconjugation reaction. Importantly for both PAMAM and dextran, no visible or measurable aggregation of the NPs was detected during any of these steps. On the other hand, the BSA procedure showed signs of a small degree of aggregation. The original size of the plain NPs was 106 nm; the BSA-coated NPs ‘grew’ to ∼186 nm and the size of the final Ab-NPs for the BSA method reached nearly 228 nm. This undesired effect was attributed to the introduction of the additional steps, in which glutaraldehyde (monovalent, homo-functional crosslinker) was employed to produce the BSA-coated NP intermediate. We have reported previously on the negative effect of monovalent linkers in bioconjugation reactions.18 To this end, all three methods yielded in Ab-NPs with different surface coverage of antibodies (Table 1).
The robust and reliable conditions for the PAMAM dendrimer reaction resulted in the highest yield of antibodies per NP. The yield for the reaction using multivalent dextran, on the other hand, was the lowest. This is presumably because PAMAM-coated NPs are more negatively charged than dextran-coated NPs, thus the former NPs electrostatically attract more antibodies in solution (which in turn leads to higher probability of covalent bond formation). This is in agreement with our previously published data on the use of different generations of PAMAM dendrimer (antibody-coated surface area of NPs decreases with decreasing generation of PAMAM dendrimer16). On the whole, PAMAM and dextran crosslinking strategies appear to be very robust in that they generate stable, monodisperse Ab-NPs. Although the BSA method bound a relatively high number of antibodies per NP (78 per NP), the fact that the overall NP size was in excess of 200 nm suggests that this method of grafting antibodies to NPs results in a small degree of NP aggregation. However, high antibody surface-coverage does not necessarily equate to high antibody activity. In the last section of this manuscript, the performance of Ab-NPs prepared by these methods will be evaluated in a simple fluorescence-linked immunoassay.
Evaluation of NP stability upon storage and long distance transportation through interactions studies with in vitro cell cultures
Nanomaterials are usually prepared by expert material scientists who often rely on biologists and clinicians to apply such prepared materials in many in vitro and/or in vivo experiments. It is therefore imperative to control the conditions of the nanoparticle storage so that the samples retain their unique properties and function over a long period of time. Crucially, the storage method should also enable simple means of transportation of the nanomaterial. The most convenient methods to preserve the nanomaterial and extend its shelf-life include storing the samples in solution at either +4 °C or −20 °C or in a dried state by a method known as lyophilisation (or freeze-drying). Lyophilisation might be the preferred method because it reduces the cost and simplifies long distance transportation. However, the freeze-drying process can add extensive strain on the nanomaterial, which often leads to particle aggregation. This phenomenon is particularly notable with antibody-coated nanoparticles. Before lyophilisation, it is therefore a common practice to suspend the NPs in formulations containing ‘cryoprotectants’, which are typically glycols or sugar-containing solutions. Common examples of the dessication-stress protectants that are widely used in the industry are trehalose, glucose, sucrose, fructose, dextran, mannitol and sorbitol.29 The choice of the cryoprotectant will significantly influence the nanoparticle aggregation, which in turn presents important consequences on the interactions between the NPs and a biological material, such as cells. In the initial experiment, we have qualitatively investigated the effects of trehalose (2% (w/v)), a commonly used cryoprotectant, on the colloidal stability of silica NPs (plain FITC-doped and human IgG-coated) and their interactions with HeLa cells.The results obtained using CLSM and SEM, shown on Fig. 4, provided very important information. Firstly, the optical properties of NPs were not affected by the storage procedure and due to their green fluorescence emission they were clearly visible under a CLSM (Fig. 4a). NPs can be observed either internalized in the cytoplasm or associated to the cell surface (arrows in Fig. 4a). Secondly, upon reconstitution in the cell culture medium and 60 minutes incubation with the cells, the IgG-coated NPs were observed to form aggregates at the micrometer scale and adhered to the surface of the cell (Fig. 4b and a magnified area containing NP aggregates is shown in Fig. 4c). Presumably, such large NP clusters were formed as a result of structural changes of the NP-bound proteins during the freeze-drying process, during the subsequent sample-reconstitution method, imbalanced charge effects of the Ab-NPs in the high-salt concentration aqueous medium or a combination of these effects. By contrast, the unmodified NPs, freeze-dried from the same solution, appeared monodisperse. Due to their small size (∼100 nm), they were unable to be seen (Fig. 4d) at the same magnification, at which Ab-NP aggregates were observed. Moreover, these NPs were homogenously distributed and adhered over the cell surface (Fig. 4e). These simple, qualitative experiments critically highlighted the need for a simple screening protocol that would allow for the identification of the most reliable cryoprotectant and storage conditions, especially for protein coated-NPs. Without a storage method that does not influence the Ab-NP performance, it is unrealistic to envisage a successful translation of such nanomaterials into a biological setting.
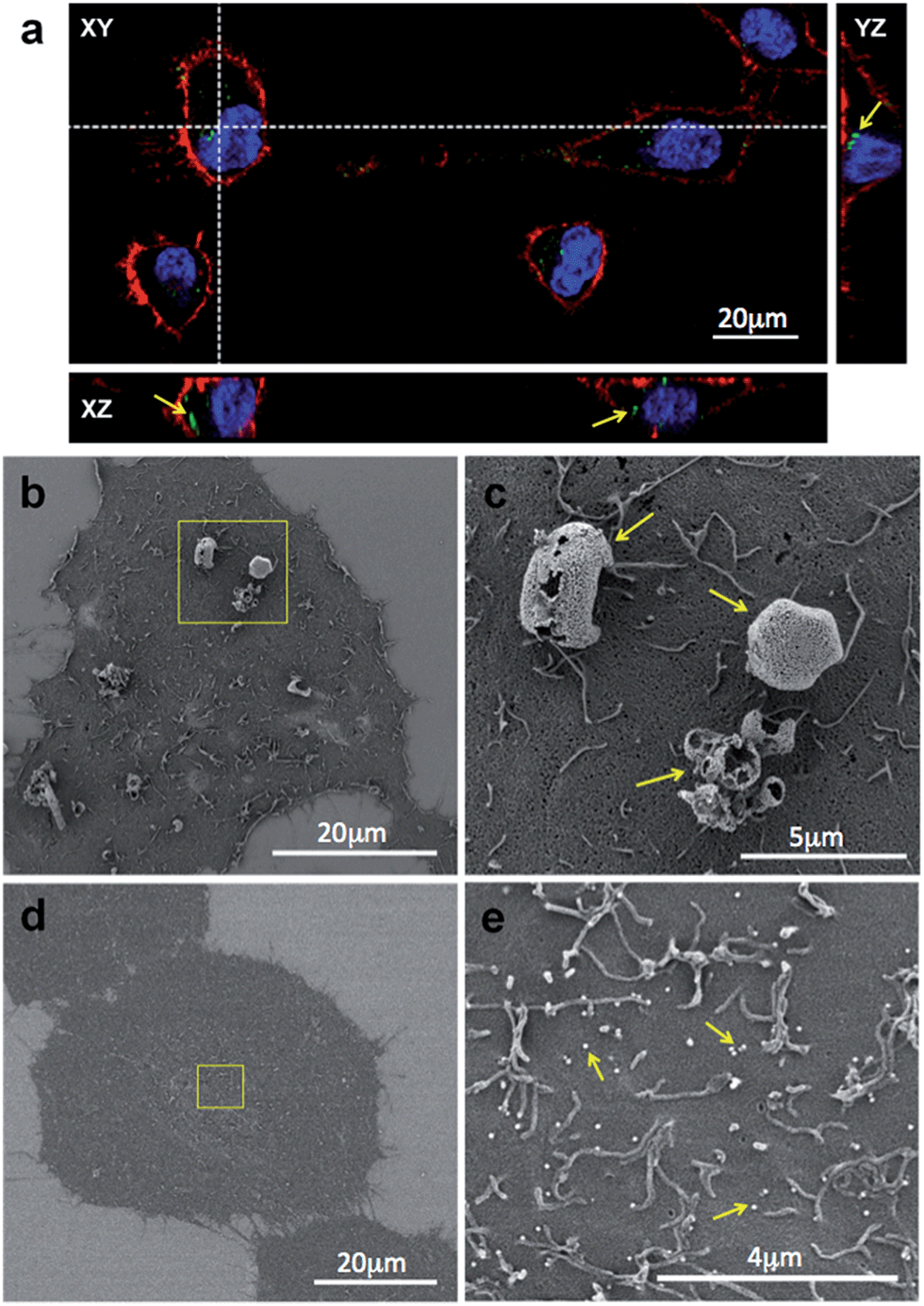 | ||
| Fig. 4 HeLa cells interacting with NPs (plain and human IgG-coated, freeze-dried in 2% trehalose solution). (a) After storage and shipment, NPs do not lose their optical properties (green fluorescence) and are clearly visible under CLSM. Cells were further stained to make their membranes (red) and nuclei (blue) visible. NPs are observed either internalized or interacting at the cell surface level (arrows). (b) Following reconstitution in MEM and incubation of IgG-coated NPs with the cells, micron sized colloidal aggregates were visualised by SEM. (c) A 5000× magnified image of (b). (d) In contrast, plain NPs remained monodisperse (∼100 nm) and were homogenously distributed over the cell surface (e). | ||
The effect of cryoprotectants on the long-term storage and manipulation of Ab-NPs
In light of the cellular uptake experiments, we considered it absolutely necessary to prepare the Ab-NPs under conditions that allowed for long-term storage without any damage to the expensive antibodies on the NP surface. Equally important, the storage method must enable cost-effective transport of the nanomaterial and easy reconstitution of the Ab-NPs in the desired medium. The specific condition for the latter is that the end-user should be able to re-dissolve the Ab-NPs without excessive manipulation with the sample (e.g. ultrasonication, vortexing etc.).In an ideal scenario, a solid formulation of the Ab-NP sample should be reconstituted in the desired solvent by simply shaking it by hand. Such reformed Ab-NP solution should maintain excellent colloidal stability for the minimum of 2 hours, which was arbitrarily chosen as a sufficiently long working-window for the end-user to manipulate with the NPs (i.e. for sample dilution, partition, sterilization, etc.). We believe this to be a frequently overlooked facet of nanomaterial preparation and it is in fact of paramount importance. Addressing these storage issues means scientists could then confidently know that the re-dispersed material is as close to identical as its pre-storage form as possible.
Several useful articles review the freeze-drying process.17,30,31 This simple method is based on removing water from a frozen sample by sublimation and desorption under vacuum. It is argued though that this process generates various stresses during freezing and drying steps. Therefore, cryoprotectants are usually added to the formulation to protect the nanoparticles from freezing and desiccation stresses. The most popular cryoprotectants encountered in the literature for freeze-drying nanoparticles are sugars: trehalose, sucrose, glucose and mannitol.17 In the cell-interaction experiment, trehalose was identified as an inefficient protectant for the Ab-NPs system. We have therefore investigated the effect of another common stabilizer – sucrose and one unusual protectant – BSA on the colloidal stability of the reconstituted NPs and Ab-NPs. The experimental design is illustrated on Fig. S.5.† Initially, considering the protocols published by industrial laboratories, plain NPs were freeze-dried in solutions containing either sucrose (20% w/v) or mixtures of BSA and sucrose (1–6% w/v BSA + 20% w/v sucrose). The samples were freeze-dried and stored for several days before they were reconstituted in 0.01 M PBS solution and also in a commonly used cell culture medium called Minimum Essential Medium (MEM). MEM is an enriched basal medium that enhances growth of sensitive or difficult to culture mammalian cells. The only user manipulation involved the addition of the desired medium to the solid-form nanoparticles and shaking the nanoparticles into solution by hand (see Movie† at: https://www.youtube.com/watch?v=HczrDJCaC38). The size of the NPs upon re-dispersion was measured by DLS, as was the evolution of the NP size over the course of 4 hours. The results, summarized in Fig. 5 show that the addition of 20% w/v of sucrose had a destabilizing effect on the colloid in both PBS and MEM. However, the addition of BSA had a stabilizing influence in both media (Fig. 5A left & centre, S5A and S5B†). When BSA (1–6% w/v) was solely used as the cryoprotectant the colloid reconstituted in MEM retained its excellent stability for a minimum of 2 hours (Fig. 5A right). 1% w/v BSA solution was identified as the optimal concentration to protect the solid formulation of the NPs. Addition of the MEM medium to the NP pellet containing 1% w/v BSA stabilized the NP solution for 4 hours (Fig. 5A right). In addition, a similar experiment was performed upon re-dissolving the NP pellet in Dulbecco's Modified Eagle's Medium (DMEM). DMEM is an example of an even more complex cell culture medium that contains approximately four times more vitamins and amino acids and two to four times more glucose than the MEM formula used. As seen in Fig. S.6B,† the NP pellet containing 1% w/v BSA retained excellent colloidal stability upon re-dispersion for up to 2.5 hours. However, in order to demonstrate the real practicality of the freeze-drying approach employing cryoprotectants, we examined the colloidal stability of antibody-coated NPs. Firstly, goat anti-human IgG was conjugated to the NPs using the three multivalent-linkers strategies described in this study. All prepared Ab-NP samples were tested by DLS and showed well-defined sizes (Fig. 3) before they were lyophilized from a solution containing 0, 1, 3 and 6% w/v BSA. After several days of storage, they were reconstituted both in water and PBS. PBS usually has a destabilizing effect on Ab-NPs colloidal stability due to the high content of NaCl (0.137 M), and also it is a good example of an isotonic medium widely used both in immunodiagnostics and nanomedicine applications. Fig. 5B illustrates that yet again, 1% w/v BSA concentration as the cryoprotectant has a stabilizing effect on the Ab-NP solution in both water and PBS. In the absence of the stabilizers (i.e. freeze dried in DI water), it was not possible to re-dissolve the Ab-NPs in aqueous solvent by shaking it by hand without a high degree of aggregation (0% w/v BSA). Such samples would be practically unusable without ultrasonication. Overall, we demonstrated that it is possible to precisely control the freeze-drying of colloidal systems in order to reach a long shelf-life of the Ab-NPs and to empower the end-user to exploit the full potential of the nanomaterial without unnecessary manipulation of the samples that could potentially damage proteins bound to the surface of the NPs.
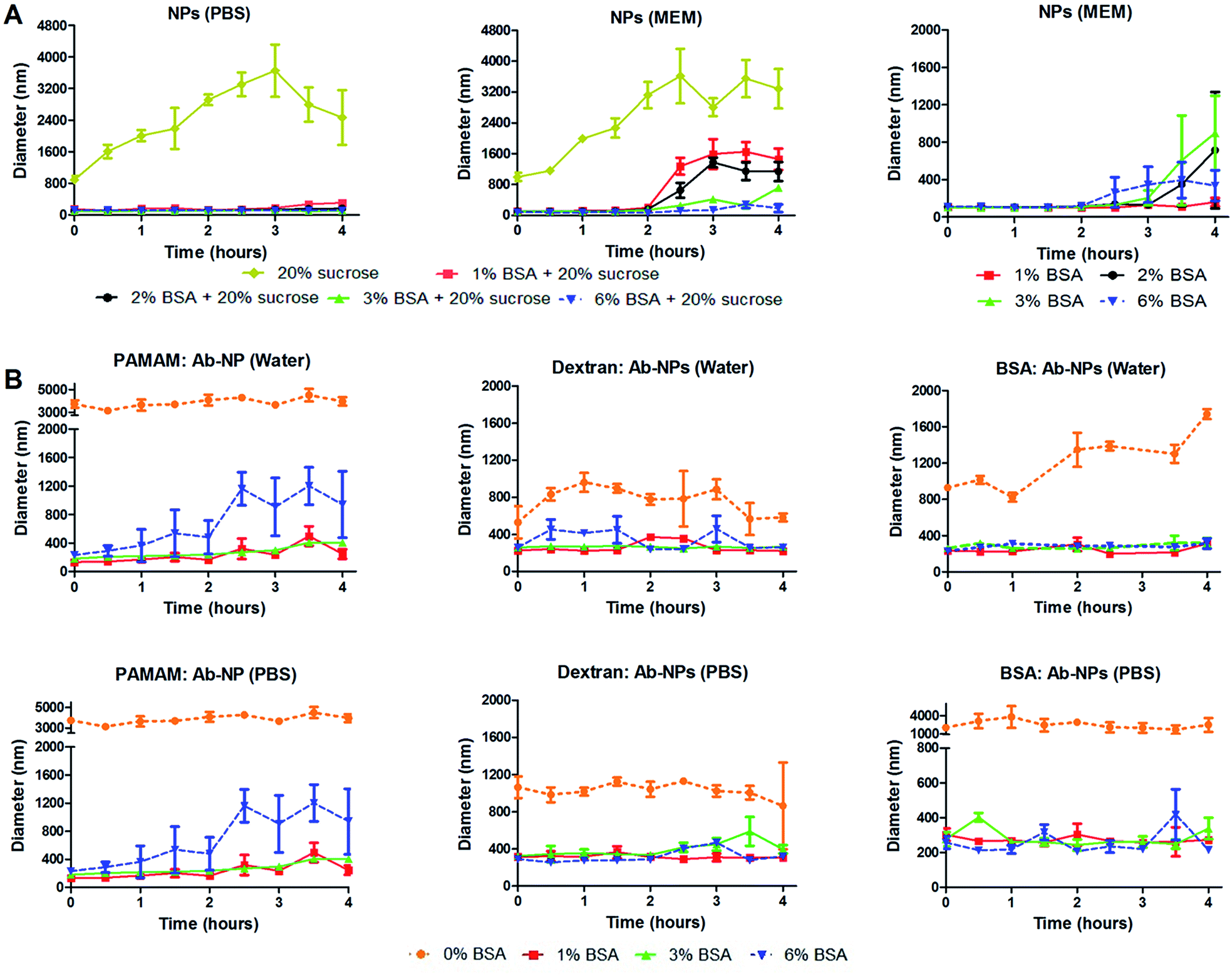 | ||
| Fig. 5 Nanoparticles were freeze-dried from solutions containing various cryoprotectants (described in legends) and redispersed in DI water, PBS or MEM. Redispersion was performed by adding the appropriate medium to the solid form NPs and shaking the dried samples (by hand) back into solution (A) unmodified NPs were freeze-dried in different concentrations of BSA (right) or BSA solutions containing 20% sucrose (left and centre). The solid form of unmodified NPs was reconstituted in PBS and MEM; (B) with BSA clearly demonstrating a stabilising effect during freeze-drying, Ab-NPs prepared with different linkers were redispersed in water and PBS after being freeze-dried in a 0% (i.e. only DI water), 1%, 3% or 6% BSA solution. Following redispersion, the evolution of particle size over time was monitored by DLS. | ||
The function of Ab-NPs measured by immunoassay
High surface coverage of antibodies in Ab-NPs does not automatically translate to better performance of the Ab-NPs in applications such as biomedical sensing and drug delivery. Specific to these practices, one critical parameter that measures the function of the Ab-NPs is their signal-to-noise ratio (S/N). In diagnostics, this relates to the ‘positive’ signal generated by the specific Ab-NPs/antigen binding compared to the signal obtained in the absence of the antigen (i.e. non-specific binding). For targeted drug delivery and in vivo imaging, the S/N denotes the selective binding of the nanocarrier (e.g. Ab-NPs) to the desired cells/tissues with minimal collateral exposure to other cells. The function of the Ab-NPs prepared by the methods presented in this article was evaluated in a simple fluorescence-linked immunoassay. The schematic of the assay format is illustrated on Fig. S.7.† All Ab-NPs were freeze-dried from a solution containing 1% w/v BSA as the cryoprotectant. The S/N value for Ab-NPs is underpinned by the ‘active surface area’ (ASA), i.e. surface area of NPs covered by antibodies in their active, native state, available to bind to their target. We hypothesized that the higher the ASA, the higher is the S/N. The ASA for our Ab-NPs was modulated by the different multivalent linkers and the underlying conjugation chemistries. However, in nanomedicine, the ASA can also be impeded by the protein adsorption layer (known as a “protein corona”) that forms on the surface of colloidal nanoparticles in biological media such as serum or plasma.32 The Dawson group has shown that this corona effect plays a key role at the nanoparticle-tissue/cell interface. A corona of adsorbed proteins on the nanoparticle surface is in fact what interacts with the biological machinery of cells rather than the carefully engineered particle surfaces created by materials scientists.33 We have therefore probed the performance of Ab-NPs in immunoassays using PBS, 10% fetal bovine serum (FBS) and whole FBS as reaction media. As before, the solid formulations of Ab-NPs used in the immunoassays, were re-dispersed in these media by shaking by hand. To determine the specific vs. non-specific binding phenomenon, goat anti-human IgG (hAb-NPs) and goat anti-rabbit IgG (rAb-NPs) antibody-coated NPs were prepared using the three multivalent linkers. Both hAb-NPs and rAb-NPs were incubated in 96 well plates modified with either human IgG or BSA respectively. Only the hAb-NPs were expected to bind to the human IgG antigen present in the wells. We have also compared the S/N ratios of these Ab-NPs to that of free dye-labelled goat anti-human and goat anti-rabbit IgG to gauge the reporting capacity of the FITC-NPs versus free dye. The fluorescence signal in each well was recorded using a fluorescence plate reader; the immunoassay results are displayed in Fig. 6. The results suggest that all of the Ab-NPs outperformed the free-antibody. Although this is in agreement with our previous published work,18 such results were not so easily predicted for the assays performed in 10% and whole serum. What is evident though, is that the ‘protein corona’ effect has played a significant role in the performance of the Ab-NPs. While the specific signal (Fig. 6, red stripy bars) was relatively high in PBS, it significantly dropped when the buffer was being replaced by FBS (red striped bars in 10% and whole serum). We presume that a corona occurs on the Ab-NPs and therefore leads to a decrease in ‘positive’ binding. The effect of the different media played little role for Ab-NPs using PAMAM, which justifies the use of such linkers and the reliability of the method. Also, the PAMAM hAb-NPs showed the highest specific signal (red stripy bars) across all tested media. Interestingly though, the non-specific binding (grey bars) of PAMAM hAb-NPs was relatively high in PBS but improved when introduced to complex serum conditions: S/N of 5.0 in PBS compared to 5.8 and 6.1 in 10% and whole serum respectively. The S/N ratio of the Ab-NPs in PBS decreased in the following order: dextran > BSA > PAMAM. This was surprising finding for the dextran method. As presented earlier in this article, the Ab-NPs prepared by the dextran-mediated method yielded the lowest quantity of antibodies per one NP (Table 1). However, because of its low non-specific binding (grey bars), the dextran hAb-NPs showed the highest S/N ratio in PBS. This outcome is attributed to the combination of the following two parameters: (1) high active surface area – high % of active antibodies on the NP surface and (2) the consequences of dextran's hydration and swelling ability, which helps to suppress non-specific binding of molecules on its surface. The S/N ratio of dextran hAb-NPs dropped in serum but it was still comparable to the PAMAM nanoparticles, which is remarkable considering the differences in cost between the two hyperbranched linkers. On average, the BSA molecule proved to be the least effective crosslinker for the conjugation of antibodies onto the NP surface. Its S/N ratio was the lowest in serum, which was ascribed to the issues related to the low degree of aggregation specific to this sample. All things considered, we have identified an inexpensive alternative to the robust PAMAM-conjugation approach to immobilize antibodies on the NP surface. The use of readily available dextran proved to be straightforward with evident benefits for the nanoparticle's colloidal stability and its function.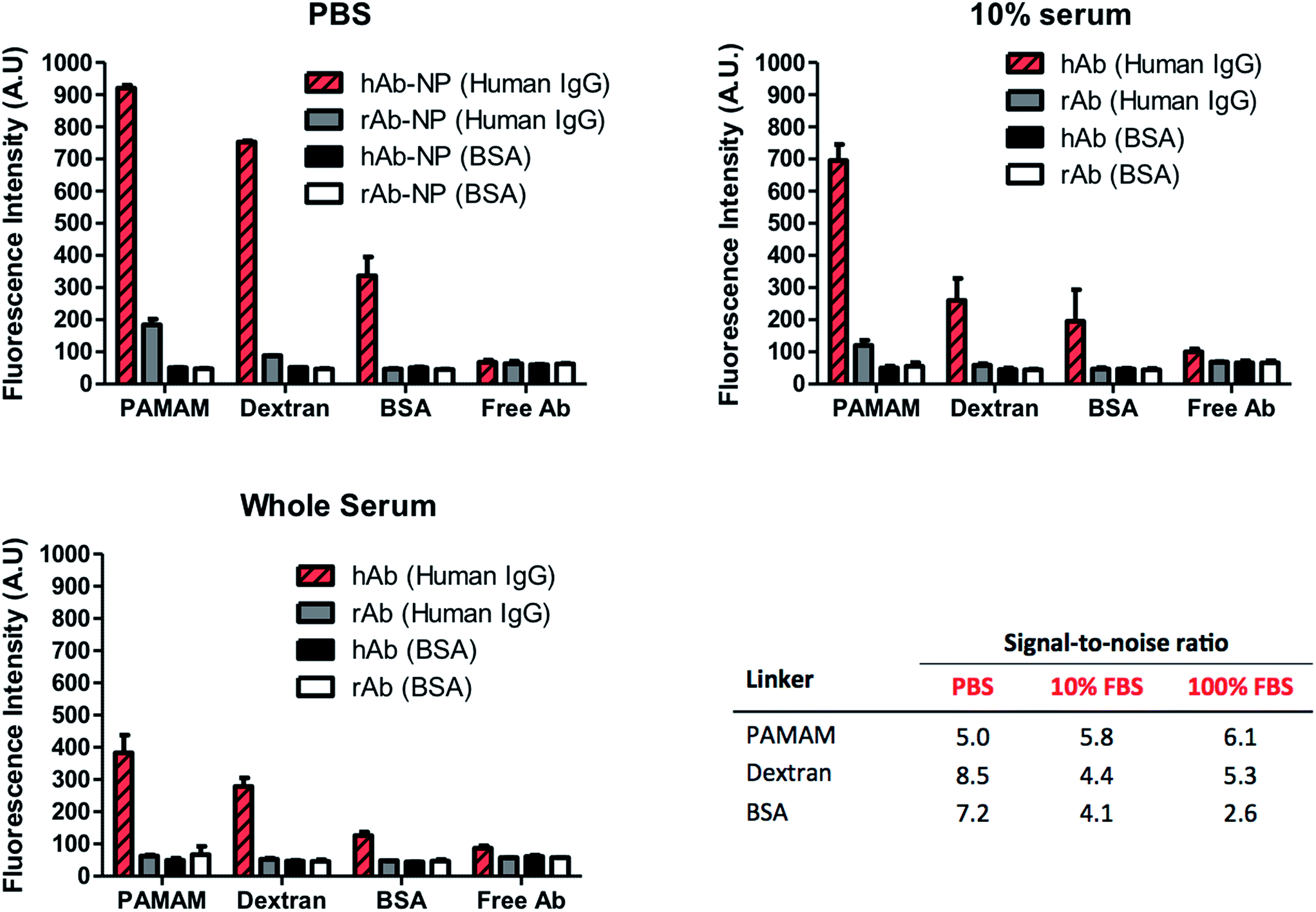 | ||
| Fig. 6 Immunoassay results and the corresponding signal-to-noise (S/N) ratios for goat anti-human (hAb) and anti-rabbit (rAb)-coated NPs when introduced to either human IgG or BSA coated surfaces. Increasing serum concentration saw a decrease binding due to the presence of a protein corona. The high active surface area of Ab-NPs using PAMAM showed the best performance in whole serum. Though Ab-NPs using dextran were coated with less antibodies, the ability of the sugar to suppress non-specific binding allowed for a S/N comparable to PAMAM. Dextran is therefore a viable alternative multivalent linker to costly PAMAM. | ||
Conclusions
It can be argued that the surface chemistry strategies presented in this article are not universal and adaptable to every type of known nanomaterial. In this work however, we aimed to empower scientists with a robust and reliable ‘recipe’ or reference point for producing protein-coated nanomaterial that will enable the translation of the “on-bench potential” towards commercially attractive products or applications. We tried to clarify some currently used misconceptions and formulate some of the unaddressed challenges related to the active surface area of antibody-coated nanoparticles and the most appropriate means of preserving and handling them. We have investigated the optimal conditions for bioconjugation strategies to immobilize three different proteins on the surface of silica nanoparticles. We have probed the function of the protein-coated nanoparticles using mainly two analytical tools, usually readily available to material scientists: DLS and fluorescence spectrophotometry. We have shown that the physicochemical properties of the chosen crosslinker play a fundamental role in nanoparticle colloidal stability and antibody binding. In particular, we have shown that when activated using EDC/sulfo-NHS that the PAMAM mediated immobilization method is very robust and reliable for both full size antibodies and also their single chain variable fragments. Significantly though, the dextran method represents a very good and cost effective alternative to PAMAM. It is quite clear that to translate the many formulations of nanomaterials, such as drug delivery systems or signal reporters into the clinical market, expertise from a multitude of disciplines including chemistry, materials science, engineering, biology, pharmaceutical sciences, and clinical research is required. Therefore, we have crucially established the most adequate protocols for storage and reconstitution of the nanomaterial with minimum end-user manipulation. To this end, freeze-drying as the optimal method and BSA (1% w/v) as the optimal example of cryoprotectant were identified to preserve the colloidal stability of our Ab-NPs in biologically relevant media. However, a number of challenges remain before such nanomaterials can be produced industrially; for example, would the conjugation reactions be as efficient if performed on a larger scale? We believe that the work presented in this article will help to advance the multidisciplinary efforts for the common objective of clinical benefits of nanotechnology.Experimental
Materials
Cyclohexane (anhydrous, 99.5%), 1-hexanol (anhydrous, ≥99%), Triton® X-100, aminopropyl trimethoxysilane [APTMS] (97%), tetraethyl orthosilicate [TEOS] (99.99%), ammonium hydroxide solution (28% w/v in water, ≥99.99%), 3-(trihydroxysilyl)propyl methylphosphonate monosodium salt (42 w/v in water) [THPMP], poly(amidoamine) [PAMAM] dendrimer (ethylenediamine core, generation 4.5 solution, 5% w/v in methanol), albumin from bovine serum (lyophilised powder, ≥98%), glutaraldehyde (50% w/v in water), sucrose (for microbiology, ≥99.0%), 6-aminohexanoic acid (≥98.5%), dextran (from Leuconostoc spp, Mr ∼ 40000), sodium periodate (99.8%), sodium borohydride (≥99%), N,N-dimethylformamide (anhydrous, 99.8%) [DMF], IgG from human serum (reagent grade, ≥95%), 4-morpholoneethanesulfonic acid [MES] (≥99%), Tween®20, trehalose dehydrate (≥99.0%), fetal bovine serum, N-hydroxysuccinimide (98%), fetal bovine serum and Dulbecco's Modified Eagle Medium – high glucose were purchased from Sigma Aldrich. Absolute ethanol, 1-ethyl-3(3-dimethylaminopropyl) carbodiimide HCl [EDC], phosphate buffer saline tablets (one tablet dissolved in 200 mL DI water yields 0.01 M phosphate buffer, pH 7.4) were purchased from Fisher Scientific. Here after, the use of ‘PBS’ refers to 0.01 M PBS, pH 7.4. N-Hydroxysulfosuccinimide sodium salt (>98%) was purchased from TCI. AlexaFluor®647 labelled goat anti-human and goat anti-rabbit IgG (H + L, 2 mg mL−1), minimum essential medium were purchased from Life Technologies. Transparent Nunc Maxisorb 96 well plates used in the immunoassay were purchased from Fisher Scientific. Deionised water (<18 MΩ) was obtained from a Milli-Q system from Millipore. The dye used in this work, fluorescein isothiocyanate isomer (95%) was purchased from ABCR.
Synthetic procedures
000 rpm, 7 min, ×3). The centrifugation steps led to the formation of a NP pellet. The supernatant was removed and the pellet was re-suspended in fresh ethanol, ultrasonicated and then re-centrifuged. After purification the nanoparticles were stored in ethanol solution at 4 °C.
000 rpm, 8 min). The supernatant contained unbound dendrimer and was discarded. The pellet contained dendrimer-coated NPs and was redispersed (via ultrasonication) in 968.5 μL MES buffer (pH 4.2, 0.1 M). 31.5 μL of 2 mg mL−1 AlexaFluor647 goat anti-human or goat anti-rabbit was added to the dendrimer-coated NP solution and allowed to shake for 4 hours at 600 rpm. Goat anti-human and anti-rabbit IgG contained 4 moles of dye per antibody. The concentration of protein added was calculated using Bangs Lab Technical Note 205. The amount of proteins needed to achieve high nanoparticle surface coverage was determined from the formula:where S = amount of protein required to achieve surface saturation (mg protein per g nanoparticle), ρs is solid sphere density (2.4 g cm−3 for silica), d is the mean particle diameter (which, from TEM, was calculated to be 80 nm) and C is the capacity of sphere surface for a given protein (mg protein per m2 of sphere surface), which is approximately 2.5 mg m−2 for IgG molecules and 3 mg m−2 for BSA. For purification, the sample was centrifuged for 8 min at 14
 000 rpm, sonicated and redissolved in 1 mL PBS (×4).
000 rpm, 8 min, ×2). The NP pellet was dispersed (via ultrasonication) in DI water after the first centrifugation. This procedure was shown to be most effective way of conjugating dextran to the NP surface (Fig. S.3†). After the second centrifugation, the pellet was redissolved in 500 μL of 40 mM of sodium periodate and shaken for 30 min (600 rpm), and then centrifuged (14000 rpm, 8 min, ×2). After the second centrifugation, 484 μL 0.1 M MES pH 4.2 was added to the pellet and sonicated. 16 μL of AlexaFluor647 antibody was added and shaken for 4 h (600 rpm). 500 μL of 40 mM sodium borohydride (in DMF) was added directly into the tube and allowed shake for another 40 min. The sample was purified as per the PAMAM section.
000 rpm, 5 min) and the pellet was redispersed in 1 mL 10% glutaraldehyde in 0.01 M PBS pH 7.4 and allowed to shake for 75 min (800 rpm). The sample was centrifuged (14000 rpm, 5 min) and the pellet was redispersed in 968 μL 0.01 M PBS, pH 7.4. 32 μL of AlexaFluor647 antibody (2 mg mL−1) was added to the sample and allowed to shake for 2 h. After 2 h, the sample was centrifuged (14000 rpm, 5 min) and the pellet was redispersed in 1 mL of 1% BSA, 40 mM aminohexanoic acid in 0.01 M PBS pH 7.4 for 15 min. The sample was purified as per the PAMAM section.
000 rpm, sonicated and redissolved in 1 mL PBS (×4).
000 rpm, 8 min, ×2). The NP pellet was dispersed (via ultrasonication) in DI water after the first centrifugation. This procedure was shown to be most effective way of conjugating dextran to the NP surface (Fig. S.3†). After the second centrifugation, the pellet was redissolved in 500 μL of 40 mM of sodium periodate and shaken for 30 min (600 rpm), and then centrifuged (14000 rpm, 8 min, ×2). After the second centrifugation, 484 μL 0.1 M MES pH 4.2 was added to the pellet and sonicated. 16 μL of AlexaFluor647 antibody was added and shaken for 4 h (600 rpm). 500 μL of 40 mM sodium borohydride (in DMF) was added directly into the tube and allowed shake for another 40 min. The sample was purified as per the PAMAM section.
000 rpm, 5 min) and the pellet was redispersed in 1 mL 10% glutaraldehyde in 0.01 M PBS pH 7.4 and allowed to shake for 75 min (800 rpm). The sample was centrifuged (14000 rpm, 5 min) and the pellet was redispersed in 968 μL 0.01 M PBS, pH 7.4. 32 μL of AlexaFluor647 antibody (2 mg mL−1) was added to the sample and allowed to shake for 2 h. After 2 h, the sample was centrifuged (14000 rpm, 5 min) and the pellet was redispersed in 1 mL of 1% BSA, 40 mM aminohexanoic acid in 0.01 M PBS pH 7.4 for 15 min. The sample was purified as per the PAMAM section.
Freeze-drying
NP samples (plain or protein-coated) were suspended in DI water such that their concentration was 2 mg mL−1 100 μL of NP sample was added to 100 μL of particular cryoprotectant solution. The aliquots were then frozen using liquid nitrogen and lyophilised. For example, 100 μL of NPs was added to 100 μL of 2% (w/v) BSA solution (i.e. each aliquot contained 200 μg of NPs in a 1% BSA solution). The aliquots were then frozen using liquid nitrogen and lyophilised using a ScanVac Coolsafe freeze drier connected to an Edwards RV12 pump. Following freeze drying, the various solid form NPs were stored at −20 °C until further use.Nanoparticles characterization
Cell cultures
Acknowledgements
We gratefully acknowledge the Medway School of Pharmacy, University of Kent and Ministerio de Ciencia e Innovación, Spain (TEC2011-29140-C03) for the financial support of this project. C.J.M. thanks the University of Kent for providing his postgraduate studentship.Notes and references
- C. N. R. Rao, A. Muller and A. K. Cheetham, Nanomaterials Chemistry: Recent Developments and New Directions, Wiley-VCH, 2007 Search PubMed.
- C. N. R. Rao and A. K. Cheetham, The Chemistry of Nanomaterials: Synthesis, Properties and Applications, Wiley-VCH, 2006 Search PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- R. Paull, J. Wolfe, P. Hebert and M. Sinkula, Nat. Biotechnol., 2003, 21, 1144–1147 CrossRef CAS PubMed.
- D. W. Hobson, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 189–202 CrossRef PubMed.
- T. Tsuzuki, Int. J. Nanotechnol., 2009, 6, 567–578 CrossRef CAS.
- R. Juliano, Nat. Rev. Drug Discovery, 2013, 12, 171–172 CrossRef CAS PubMed.
- V. J. Venditto and F. C. Szoka Jr, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed.
- H. L. Nie, X. X. He, X. Q. Huo, L. Hai, X. Wu, J. Ge and W. H. Tan, Chem. Res. Chin. Univ., 2008, 29, 477–481 CAS.
- R. P. Bagwe, L. R. Hilliard and W. H. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- S. W. Bae, W. Tan and J.-I. Hong, Chem. Commun., 2012, 48, 2270–2282 RSC.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- D. Knopp, D. P. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14–30 CrossRef CAS PubMed.
- V. Gubala, C. Crean, R. Nooney, S. Hearty, B. McDonnell, K. Heydon, R. O'Kennedy, B. D. MacCraith and D. E. Williams, Analyst, 2011, 136, 2533–2541 RSC.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- V. Gubala, X. Le Guevel, R. Nooney, D. E. Williams and B. MacCraith, Talanta, 2010, 81, 1833–1839 CrossRef CAS PubMed.
- G. T. Hermanson and G. T. Hermanson, Bioconjugate Techniques, 2nd edn, 2008 Search PubMed.
- S. R. Ge, K. Kojio, A. Takahara and T. Kajiyama, J. Biomater. Sci., Polym. Ed., 1998, 9, 131–150 CrossRef CAS PubMed.
- P. Leonard, P. Safsten, S. Hearty, B. McDonnell, W. Finlay and R. O'Kennedy, J. Immunol. Methods, 2007, 323, 172–179 CrossRef CAS PubMed.
- D. J. Oshannessy, M. Brighamburke and K. Peck, Anal. Biochem., 1992, 205, 132–136 CrossRef CAS.
- A. W. Drake, M. L. Tang, G. A. Papalia, G. Landes, M. Haak-Frendscho and S. L. Klakamp, Anal. Biochem., 2012, 429, 58–69 CrossRef CAS PubMed.
- D. A. Edwards, Bull. Math. Biol., 2006, 68, 627–654 CrossRef CAS PubMed.
- C. Jonsson, M. Aronsson, G. Rundstrom, C. Pettersson, I. Mendel-Hartvig, J. Bakker, E. Martinsson, B. Liedberg, B. MacCraith, O. Ohman and J. Melin, Lab Chip, 2008, 8, 1191–1197 RSC.
- G. Li, R. Stewart, B. Conlan, A. Gilbert, P. Roeth and H. Nair, Vox Sang., 2002, 83, 332–338 CrossRef CAS.
- O. J. M. Bos, J. F. A. Labro, M. J. E. Fischer, J. Wilting and L. H. M. Janssen, J. Biol. Chem., 1989, 264, 953–959 CAS.
- K. Hirayama, S. Akashi, M. Furuya and K. Fukuhara, Biochem. Biophys. Res. Commun., 1990, 173, 639–646 CrossRef CAS.
- P. Fonte, S. Soares, F. Sousa, A. Costa, V. Seabra, S. Reis and B. Sarmento, Biomacromolecules, 2014, 15, 3753–3765 CrossRef CAS PubMed.
- D. Quintanar-Guerrero, A. Ganem-Quintanar, E. Allemann, H. Fessi and E. Doelker, J. Microencapsulation, 1998, 15, 107–119 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert and H. Fessi, Eur. J. Pharm. Biopharm., 2006, 63, 87–94 CrossRef CAS PubMed.
- P. D. Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- R. I. Nooney, E. McCormack and C. McDonagh, Anal. Bioanal. Chem., 2012, 404, 2807–2818 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data on the bioconjugation protocols, optimization of the reaction parameters for the Dextran reaction and details about the immunoassay design. Short movie demonstrating the Ab-NPs redispersion is available at https://www.youtube.com/watch?v=HczrDJCaC38. See DOI: 10.1039/c4tb01915f |
| This journal is © The Royal Society of Chemistry 2015 |