
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced uptake and siRNA-mediated knockdown of a biologically relevant gene using cyclodextrin polyrotaxane†
P.
Dandekar‡
*a,
R.
Jain§
*a,
M.
Keil
b,
B.
Loretz
a,
M.
Koch
d,
G.
Wenz
b and
C.-M.
Lehr
ac
aDepartment of Drug Delivery (DDEL), Helmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Campus A4 1, Saarland University, Saarbrücken, 66123, Germany. E-mail: pd.jain@ictmumbai.edu.in; rd.jain@ictmumbai.edu.in
bOrganic Macromolecular Chemistry, Campus C4 2, Saarland University, Saarbrücken, 66123, Germany
cDepartment of Biopharmaceutics and Pharmaceutical Technology, Campus A4 1, Saarland University, Saarbrücken, 66123, Germany
dInnovative Electron Microscopy, INM-Leibniz Institute for New Materials, Campus D2 2, Saarland University, Saarbrücken, 66123, Germany
First published on 5th February 2015
Abstract
Ideal cationic polymers for siRNA delivery could result in its enhanced cellular internalization, escape from endosomal degradation, and rapid release in cell cytoplasm, to facilitate knockdown of the target gene. In this study, we have investigated the ability of an in-house synthesized cationic polyrotaxane to bind siRNA into nanometric complexes. This polymer, which had earlier shown improved transfection of model siRNA (luciferase), was used to improve the cellular internalization of the siRNA molecule with therapeutic implications. In cellular assays, the polymer enhanced the knockdown of a gene involved in the pathogenesis of tuberculosis, when the nanocomplexes were compared with free siRNA. The efficacy and cellular non-toxicity of this polymer encourage its further exploitation in animal models of tuberculosis and other intracellular bacterial infections.
1. Introduction
Cationic cyclodextrin (CD)-based polymers are well documented for delivering nucleic acids due to their ability to condense them into nanometric structures, their low toxicity, and their ability to transfect mammalian cells.1 Amongst the cationic CD polymers, CD polyrotaxanes have rendered substantial utility, typically in the delivery of plasmid DNA (pDNA).2–5 The CD polyrotaxanes possess a supramolecular architecture in which mobile CD rings are non-covalently threaded over linear polymer backbones, which bear bulky substituents (stoppers) at both ends, thus hindering the rapid dethreading of the CD rings. The nature of the stoppers may be varied to facilitate programmed dissociation of the CD rings and hence the release of complexed pDNA, which offers an added advantage as compared to the other polymers which have been explored for intracellular delivery of nucleic acids. Also alternative CD containing polymers have been known to self-assemble into nanoparticles and have shown efficacy for delivery of nucleic acids in humans and have been explored in clinical studies.1–3,6 This further suggests the potential of CD polyrotaxanes for clinical investigations pertaining to nucleic acid delivery.In recent times, CD polyrotaxanes have also demonstrated success in intracellular delivery of siRNA (small interfering RNA) and have facilitated knockdown of reporter genes like luciferase and green fluorescence protein.7–9 However, none of these recent studies report the ability of CD polyrotaxanes to knock down genes of therapeutic relevance.
Recently, we have reported a CD-based cationic polyrotaxane, which can condense siRNA into nanometric complexes (nanoplexes) along with a cargo pDNA, the latter being non-functional in mammalian cells. As compared to the previously reported CD polyrotaxanes, based on CDs threaded over neutral polymers (polyethylene glycol), the CD polyrotaxane discussed in our investigation involves thermally activated threading of cationic CDs onto suitable cationic polymers, obviating the need for attaching bulky stoppers or post-derivatization of the polyrotaxane. Thus, our method avoids tedious multi-step synthesis or introduction of stoppers via cleavable linkages, to assist intracellular dissociation of the polymer and associated nucleotides, as observed with other CD polyrotaxanes.10,11 Cellular investigations have proved that our polymer could successfully overcome the cellular barriers and resulted in intra-cellular siRNA delivery in a biologically active state, which was proven by the luciferase knockdown assay in A 549 human lung adenocarcinoma epithelial cell line. We also studied the intracellular uptake and release of siRNA in live cells, using confocal microscopy.11 With success in these investigations, we furthered our research to conduct cellular assays to prove the cellular safety of our CD polyrotaxane (Fig. 1), which we have discussed subsequently. Thereafter, we also investigated its ability to deliver siRNA, capable of down-regulating a gene relevant to the pathogenesis of tuberculosis. Ours is the first investigation that describes the potential of CD polyrotaxanes for therapeutic applications, to the best of our knowledge. With tuberculosis as the first therapeutic target, we foresee that our polymer may be employed for the RNA interference (RNAi) therapy of various lethal intracellular infections.
 | ||
| Fig. 1 Structure of CD Polyrotaxane. Alpha-cyclodextrin (α) and Heptakis-[6-deoxy-6-cysteaminyl)]-beta-cyclodextrin (β) rings were threaded over ionene-6,10 backbone (i). 9% of the polymer repeats are covered by αCD and 20% by the βCD derivative. Total molecular weight is ∼125 kDa, molecular weight per nitrogen of 243 Da. | ||
2. Materials and methods
2.1. Materials for preparation of nanoplexes and cellular evaluations
Dulbecco's modified Eagle's medium (DMEM) and fetal calf serum (FCS) were purchased from GIBCO (Karlsruhe, Germany). Dimethyl sulfoxide (DMSO) for MTT assay was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Fluorescein wheat germ agglutinin (FITC-WGA) was procured from Vector (Vector Laboratories, Inc., Burlingame, CA, USA) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) was from Fluka Chemie GmbH (Buchs, Switzerland). All the other chemicals and solvents employed were of analytical grade. Deionized, double-distilled water (Milli Q Plus system, Millipore, Bedford, MA, USA) was used whenever required.2.2. siRNA
The cellular uptake studies were conducted by formulating the nanoplexes with Silencer® Cy™3 labeled negative control siRNA purchased from Ambion, Inc. (TX, USA). The knockdown of the tuberculosis relevant Bfl1/A1 gene was investigated by formulating the nanoplexes with Bfl1/A1 siRNA and scrambled siRNA (Santa Cruz Biotechnology, CA, USA) at the concentration specified in the transfection protocol recommended by the manufacturer.2.3. Synthesis of CD polyrotaxanes
We have previously reported the synthesis of cationic CD polyrotaxane comprising ionene-6,10; heptakis-[6-deoxy-(2-amino-ethylsulfanyl)]-βCD (HAESβCD) and αCD,11 which was used in the present investigation.Briefly, 51.4 mg (0.134 mmol) of ionene-6,10 and 483.6 mg (0.268 mmol) of HAESβCD were dissolved together in 5.14 mL of 0.1 M NaCl solution. After stirring the resulting solution for 8 h at 25 °C and successively adding a solution of 514 mg αCD (0.528 mmol) in 5 mL water, the solution was further stirred for 16 h at 70 °C. After cooling to room temperature, the solution was subjected to ultrafiltration over a cellulose membrane (UC010, cut-off 10 kDa, MICRODYN-NADIR GmbH, Wiesbaden, Germany). The retentate was lyophilized to obtain 93 mg (85% based on ionene-6,10) of CD polyrotaxane, 1H NMR δ/ppm: 5.08 (1.35 H, HAESβCD), 5.02 (0.58 H, αCD), 3.79, 3.66, 3.44, 3.20, 3.05, 2.96, 2.83, 1.70 (8.0 H, methylene), 1.27–1.34 (16 H, methylene). The coverage of individual CD molecules per polymer repeat unit was estimated to be 9% for αCD and 20% for HAESβCD.
2.4. Formulation of the nanoplexes of CD polyrotaxane and nucleic acids
The nanoplexes were formulated in HEPES buffered glucose (10 mM, pH 7.0 containing 5% glucose; w/v), as described previously.11 The nucleotide combination employed consisted of pUC 18 control plasmid DNA (E. coli XL-1 cells were transfected with pUC 18 for propagation and the pDNA was purified using QIAGEN Plasmid Mini Kit, Qiagen Inc., CA, USA) along with the respective siRNAs in the ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w. Nanoplexes were formulated by mixing fixed volumes of CD polyrotaxane solution, (3.6–29.4 μg mL−1, depending on N/P) and of pDNA (8.25 μg mL−1) and siRNA (2.75 μg mL−1). The N/P ratios (ratio of nitrogen atoms of polyrotaxane per phosphate of nucleotides) were varied between 0.5 and 4 to identify the concentration of polymer required for completely binding the nucleic acids. This was observed at the N/P ratio of 2.0 as verified by agarose gel electrophoresis (described in the ESI†).
:1 w/w. Nanoplexes were formulated by mixing fixed volumes of CD polyrotaxane solution, (3.6–29.4 μg mL−1, depending on N/P) and of pDNA (8.25 μg mL−1) and siRNA (2.75 μg mL−1). The N/P ratios (ratio of nitrogen atoms of polyrotaxane per phosphate of nucleotides) were varied between 0.5 and 4 to identify the concentration of polymer required for completely binding the nucleic acids. This was observed at the N/P ratio of 2.0 as verified by agarose gel electrophoresis (described in the ESI†).
2.5. Characterization of nanoplexes
WET-STEM was performed on a FEI Quanta 400 ESEM with a field emission gun (FEG) operating at an energy of 30 keV. The procedure was conducted at room temperature (T = 293 K) using transmitted electrons. 100 μL of the freshly prepared nanoplex suspension (containing 14.74 and 22.12 μg mL−1 CD polyrotaxane, 8.25 μg mL−1 pDNA and 2.75 μg mL−1 siRNA) was placed on a carbon coated copper grid (300 #), which in turn was mounted on a WET-STEM sample holder. Initially, the sample was investigated in low-vacuum conditions (p = 100 Pa; T = 276 K) to maintain a thin liquid film of the sample over the substrate. Eventually, the applied vacuum was gradually increased (p = 400 Pa) to evaporate the liquid and the drying was investigated in situ in transmission mode (STEM) by detecting the transmitted electrons using a gaseous secondary electron detector (GSED).
AFM studies were conducted employing a Bioscope SPM and a Nanoscope IV controller (Veeco Instruments, CA, USA) in a vibration-free environment. A drop of the freshly prepared nanoplex suspension was placed on a silicon wafer and air-dried. Measurements were performed in tapping mode, using a commercially available pyramidal tip (silicon, Ultrasharp, MikroMasch, Tallinn, EST) on a cantilever with a length of 230 μm. The resonance frequency of the tip was approximately 170 kHz, scanning probe of the force constant was ∼40 N m−1, and the scan frequency between 0.1 and 1 Hz.
2.6. Cell viability: MTT assay
Cellular safety of the nanoplexes at various N/P ratios (CD polyrotaxane concentration: 3.6–29.4 μg mL−1) was determined using the standard MTT assay.14 Murine macrophage cell line, RAW 264.7 (ATCC Cat. no. TIB-71, VA, USA) was utilized for this assay since the same cells were intended to be subsequently employed for studying the cellular uptake and biological efficacy of the nanoplexes. Another important reason for choosing macrophages was that these cells serve as reservoirs of mycobacteria, wherein up-regulation of certain anti-apoptotic proteins by the mycobacteria facilitates their intracellular survival. However, we have also studied the transfection efficacy of this polymer in human lung adenocarcinoma epithelial cells (A 549).14 The cells were cultivated at 37 °C, 85% relative humidity, and 5% CO2, in DMEM supplemented with 10% FCS. Cells were seeded into 96-well plates at a density of 20000 cells per well in 200 μL of growth medium. Once confluent, they were incubated with the nanoplexes, at individual N/P ratios, for 24 h in DMEM supplemented with FCS (final volume of 100 μL per well). Nanoplexes formulated with polyethylene imine (PEI) at the same N/P ratios (PEI concentration: 0.83–6.6 μg mL−1), were maintained as a control. The cellular treatment (ESI†) was identical for both types of nanoplexes.
2.7. FACS
Cellular association of nanoplexes containing Cy3-labeled siRNA was investigated using Fluorescent Activated Cell Sorting (FACS). Nanoplexes with N/P ratios of 1.0 (negative zeta potential; anionic surface charge) and 2.0 (positive zeta potential; cationic surface charge) were used for this study and for all the other cellular investigations, to compare whether the cationic nanoplexes had any distinct advantage over the anionic ones. Processed untreated cells and cells treated with naked siRNA, in a manner similar to the nanoplexes, were used as controls during this study. All the experiments were conducted in triplicate and values of the percent mean fluorescence intensity were presented as the mean ± SEM. Studies were conducted exactly as reported by our research group previously11 and the associated details have been provided in the ESI† of this manuscript.2.8. Confocal laser scanning microscopy (CLSM)
The precise cellular uptake pathway of the nanoplexes in RAW 264.7 cells was conducted using CLSM, in the presence of inhibitors of the specific pathways. The inhibitors used were 10 μg mL−1 chlorpromazine (CHL; C8138, Sigma-Aldrich, Taufkirchen, Germany), 10 μg mL−1 nystatin (NYS; N3503, Sigma-Aldrich, Taufkirchen, Germany), and 5 μg mL−1 5-(N-ethyl-N-isopropyl)amiloride (EIPA) (A3085, Sigma-Aldrich, Taufkirchen, Germany). Literature reports have already indicated the cellular non-toxicity of the respective agents at the concentrations employed in the present study.15 The procedural details of this study (see the ESI†) were identical to those reported by our research group previously.11Conclusions were drawn by visually analyzing images corresponding to a total of 100 cells for each inhibitor and quantification of the number of cells positive for nanoparticle uptake (nanoparticles inside the cells). Results of manual counting were verified by a combined multiphoton pixel analysis method reported by Labouta et al.16
2.9. Biological efficacy of nanoplexes in cellular assays
2.9.2.1. Inducing Bfl1/A1 gene expression in RAW 264.7 cells. RAW 264.7 cells were plated in a 24-well plate at a density of 50
000 cells per well, in 1 mL of the growth medium. After 24 h, lipopolysaccharide (LPS) from E. coli (solution in sterile pyrogen free water, L 8274, Sigma-Aldrich, Taufkirchen, Germany) was added to the cells (10 μg mL−1) to induce the expression of endogenous Bfl1/A1 gene. The concentration of LPS was chosen according to a reported procedure. LPS induced expression of NF-κB-regulated, Bfl1/A1 gene expression in RAW 264.7 cells has already been reported in literature.17,18 After a further 24 h, the expression of Bfl1/A1 gene was confirmed by western blot analysis according to a reported procedure, using identical buffers and solutions.19 The primary antibody used was mouse monoclonal IgG1 (A1 (B-3) Antibody: sc-166943; Santa Cruz Biotechnology, CA, USA) at a dilution of 1:200 in blocking buffer. The secondary antibody employed was chicken polyclonal antibody to Mouse IgG-H & L (HRP) (ab6814; Abcam, MA, USA) at a dilution of 1:3000 in blocking buffer. The blots were developed using Amersham™ ECL™ Prime Western blotting chemiluminescence kit (GE Healthcare Europe GmbH, Freiburg, Germany) as per the manufacturer's instructions. Imaging was carried out using FUSION FX7™ Advance documentation system (PEQLAB Biotechnologie GMBH, Erlangen, Germany) equipped with Acquisition Data Interface and Fusion-CAPT-Software.
2.9.2.2. Nanoplex mediated silencing of Bfl1/A1 gene. RAW 264.7 cells were plated in a 24-well plate at a density of 50
000 cells per well, in 1 mL of the growth medium and Bfl1/A1 gene expression was induced in the cells as previously described. After 24 h of LPS activation, the medium containing LPS was replaced by serum-free medium containing nanoplexes formulated with anti-Bfl1/A1 siRNA (Santa Cruz Biotechnology, CA, USA) at a concentration of 80 pmol per well. Untreated cells, cells treated with naked siRNA, siRNA formulated with jetPRIME™ reagent, and nanoplexes formulated with scrambled siRNA, employing the same siRNA concentrations in each case, were maintained as controls during this study. The medium containing the formulations or free siRNA was removed after 4 h and the cells were incubated with a normal growth medium, for a further 65 h, with intermittent replacement of medium every 24 h. Subsequent to the transfection, the gene silencing efficacy of the nanoplexes, in comparison to the controls, was determined by estimating the expression of Bfl1/A1 by RT-PCR and western blot analysis (as in Section 2.9.2.1.).
RT-PCR was conducted using a reported procedure with slight modifications (details provided in the ESI†). The cycling protocol employed was: initial denaturation at 94 °C for 5 min, 35 cycles of 1 min at 94 °C, 1 min at 60 °C, 1.5 min at 72 °C and a final amplification at 72 °C for 5 min, followed by a melt curve analysis (from 72 °C to 98 °C in 0.5 °C steps for 5 s).20 The primer sequences for Bfl1/A1 (Santa Cruz Biotechnology, CA, USA) were: primer sense sequence 5′-TAC AGG CTG GCT CAG GAC TAT C-3′ and primer antisense sequence 5′-GGT ATC CAC ATC CGG GGC AAT-3′. β-Actin was included as an internal standard for each set of RNA samples analyzed and non-template controls, replacing cDNA with water, were maintained to control DNA contamination of the reagents. Sequences of β-actin primer pairs used were: primer sense sequence 5′-TGC GTG ACA TTA AGG AGA AG-3′ and primer antisense sequence 5′-GTC AGG CAG CTC GTA GCT CT-3′.
3. Results and discussion
3.1. Formulation of the nanoplexes of CD polyrotaxane and nucleic acids
Complexation of CD polyrotaxane with anti-Bfl1/A1 siRNA or Cy™3 labeled siRNA (and anti-luciferase siRNA) was conducted using a mixture of the respective siRNA along with pUC 18 control plasmid DNA (pDNA:siRNA = 3:1 w/w). The complex size and molecular topography of pDNA were exploited for efficient complexation with cationic polymer and siRNA into nanometric structures. RNA molecules have been reported to be more rigid as compared to dsDNA (ds: double-stranded). One persistence length for RNA, quantifying the stiffness of a polymer, is about 260 bp (basepairs); therefore, siRNA molecules, being only about 21 bp in length, fundamentally behave as rigid rods. The minimum length of nucleotides required to condense cationic materials has been reported to be between 80 and 140 bp. Thus siRNA molecules, by themselves, can result in a disorderly complexation with cationic materials, resulting in poor loading or formation of large complexes, poor cellular internalization, and hence, inefficient gene knockdown.21 Furthermore, siRNA alone requires more cationic material to form nanometric complexes, thus increasing the probable toxicity of the resulting system. Other researchers have also reported these observations previously.21,22 pUC 18 control plasmid DNA was employed to ensure that the pDNA would not have its own action, once inside the mammalian cells, and hence, any biological efficacy observed could be attributed entirely to the specific condensed siRNA.
3.2. Particle size, zeta potential, and morphology of the nanoplexes
The particle size varied between 219.9 ± 1.6 nm (polydispersity index; PI: 0.28 ± 0.024) and 154.8 ± 8.7 nm (PI: 0.15 ± 0.031) for N/P ratios ranging from 0.5 to 4.0, in the case of nanoplexes formulated with Cy™3 siRNA. In this case, the lowest particle size (140.9 ± 2.7 nm) and polydispersity index (PI; 0.14 ± 0.04) was observed at an N/P ratio of 2. Similarly for nanoplexes formulated with anti-Bfl1/A1 siRNA, the particle size varied between 259.9 ± 1.3 nm (polydispersity index; PI: 0.22 ± 0.024) and 194.8 ± 0.87 nm (PI: 0.19 ± 0.031) for N/P ratios ranging from 0.5 to 4.0. Again, the lowest particle size (147.9 ± 2.5 nm) and polydispersity index (PI; 0.15 ± 0.01) was observed at an N/P ratio of 2. With an increase in N/P ratio beyond 2, the size of the nanoplexes slightly increased. This may be attributed to an excess of free polymer leading to electrostatic interactions between the nanoplexes and their aggregation. The same is also reflected through an increase in the PI of the nanoparticles.23,24The surface charge of nanoplexes governs their cellular interactions and uptake. When the zeta potential values of the nanoplexes were recorded at various N/P ratios, a significant change in zeta potential from a negative to positive value was observed at an N/P ratio of 2.0 (−11.7 ± 2.7 mV at N/P ratio of 1.0 to +24.3 ± 2.4 mV for nanoplexes formulated with Cy™3 siRNA and −21.8 ± 0.6 mV at N/P ratio of 1.0 to +27.8 ± 2 mV for nanoplexes formulated with anti-Bfl1/A1; differences in the zeta potential may be attributed to the presence of dye on Cy™3 siRNA). Increase in N/P ratio beyond 2 further increased the surface charge of the nanoplexes. However, nanoplexes with very high cationic charge were considered to be unsuitable for cellular evaluations as very stable complexation (indicated by high zeta potential values) was predicted to hamper an efficient cytosolic release of the condensed siRNA and hence their knockdown efficiency. Also, highly cationic particles could increase the probability of cellular toxicity. Thus, an N/P ratio of 2 was finalized as the working ratio for further cellular evaluations.
The morphology of nanoplexes (N/P = 2.0) was studied by WET-STEM and AFM to procure detailed information about the shape and homogeneity of the nanoplexes.
The AFM image of the nanoplexes is depicted in Fig. 2A. Though the image revealed a homogenous distribution of the nanoplexes, the size of the nanoplexes recorded by this method was slightly less (117 ± 0.2 nm) than that obtained by Zetasizer Nano-ZS (based on the principle of dynamic light scattering; DLS). The vertical height of the nanoplexes by AFM was observed to be 2.1 nm. Moreover, AFM measurements involve drying of the nanoplexes, resulting in detachment of their hydration shell, which may be responsible for the observed smaller diameters and aggregation of nanoplexes located within close vicinity of each other.23 Furthermore, the low vertical height of nanoplexes may again be attributed to the drying procedure resulting in their horizontal spreading on the AFM substrate.
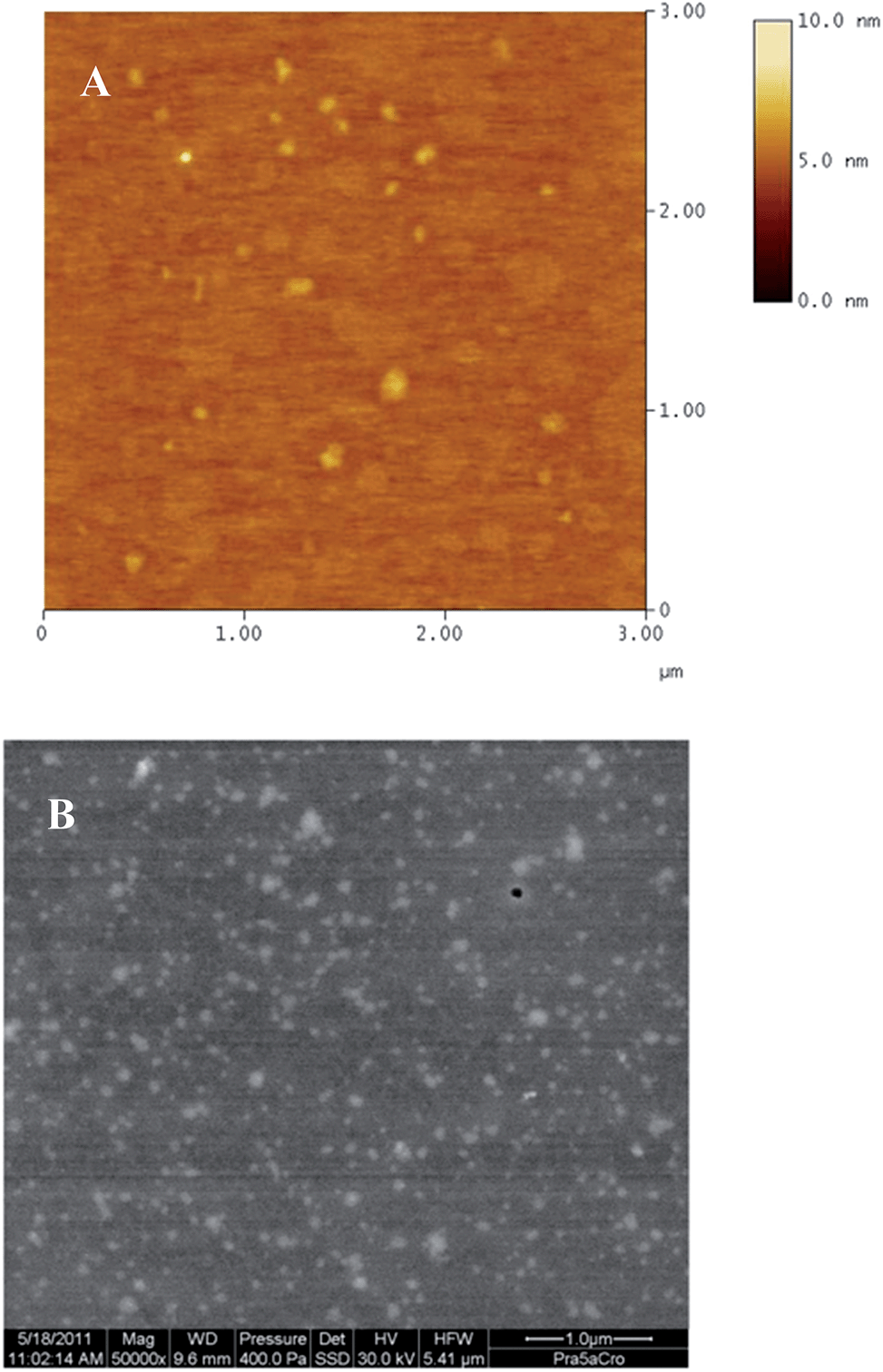 | ||
| Fig. 2 (A) AFM image of the nanoplexes depicting a diameter smaller than that measured by DLS and some aggregation of nanoplexes. (B) WET-STEM image of the nanoplexes depicting their homogenous distribution and size in compliance with DLS results. | ||
Thus, to assess the morphology of nanoplexes in their natural aqueous environment, WET-STEM was performed. Analysis in a wet state avoids deformation or collapsing of polymeric structures and enables an accurate estimation of their size and morphology.25 As seen from Fig. 2B, the WET-STEM image revealed a homogenous distribution of the nanoplexes, with sizes fairly in compliance with DLS results.
3.3. Cell viability: MTT assay
Toxicity of cationic polymers is one of the prime hurdles for their application in effective siRNA delivery. Many such cationic polymers have been reported to cause cellular damage, such as membrane destabilization, cell shrinkage, reduction in frequency of mitosis, and formation of vacuoles in the cell cytoplasm. It is also known that the cationic polymers used for siRNA delivery adversely affect certain cellular proteins.26 Thus, toxicological assessment of nanoplexes is important for validating their efficacy at the sub-cellular level. This is even more critical for newly synthesized polymers due to the unavailability of a toxicological profile. The cellular cytotoxicity assays provide the quickest way of assessing the toxicity of nanoplexes as cell death is considered a direct indicator of the toxicity potential of the test materials. The MTT assay, used in the present investigation, is an indicator of mitochondrial toxicity induced by test materials. This assay is based on the ability of the mitochondrial dehydrogenases of viable cells to cause reductive conversion of the yellow tetrazolium dye (MTT) to blue formazan crystals.27 The results of the MTT assay have been depicted in Fig. 3. | ||
| Fig. 3 MTT assay of nanoplexes in RAW 264.7 cells. Cells were incubated with nanoplexes for 24 h in DMEM supplemented with FCS and washed twice with PBS before incubation with MTT (5 mg mL−1) for 4 h. After removing the dye solution, the formazan crystals were dissolved in DMSO and absorbance was measured at 550 nm. The nanoplexes formulated with polyrotaxane resulted in almost complete cell viability at the working N/P ratio of 2.0. At the same N/P ratio PEI (70 kDa, branched) was found to be severely toxic to the cells. | ||
As evident from the results, the nanoplexes did not affect the cell viability at the working N/P ratio of 2. Even at a higher N/P ratio of 4, the viability of the cells was around 75%. The results indicated the suitability of CD polyrotaxane nanoplexes for cellular evaluations concerning their uptake and biological efficacy. On the other hand, PEI, which is the “standard” polymer for gene delivery, resulted in severe toxicity to the RAW 264.7 cells at all the evaluated concentrations.
3.4. FACS
Insufficient cellular internalization constitutes one of the major barriers to effective siRNA delivery. Free nucleic acids cannot passively diffuse across the cell membranes due to high molecular weight, hydrophilicity, and an overall negative charge. In such cases, cationic nanoplexes facilitate their enhanced association with anionic cell membranes and subsequent internalization by one or more of the internalization mechanisms (endocytosis, phagocytosis, or macropinocytosis).20,28 The results of cellular association of nanoplexes are depicted in Fig. 4.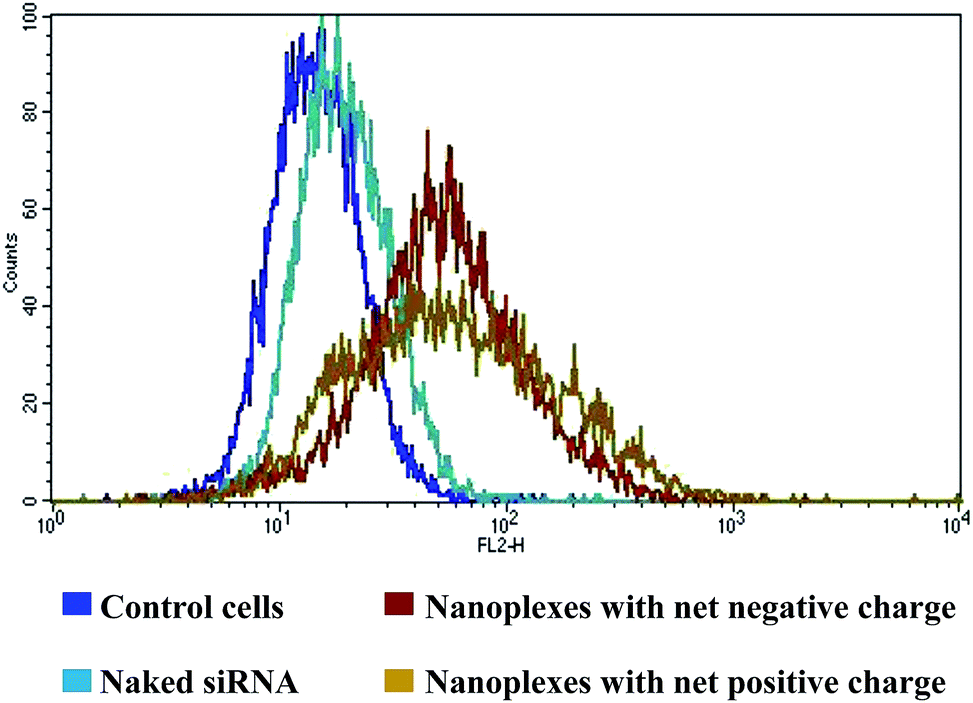 | ||
| Fig. 4 FACS analysis of nanoplexes formulated with Cy3 siRNA. The cellular association was in the order: nanoplexes with net positive charge > nanoplexes with net negative charge > naked siRNA as inferred from the mean fluorescent intensity (%) values. Nanoplexes dispersed in Krebs–Ringer's solution were incubated with confluent RAW 264.7 cells for 4 h and thereafter with DMEM supplemented with FCS for a further 18 h. The cells were then washed twice with PBS, centrifuged and dispersion of the cell pellet was analyzed by FACS. | ||
There was only a small shift in the mean fluorescent intensity (MFI) peak of free siRNA as compared to the control cell, which correlated well with the failure of naked siRNA in efficient cellular internalization. In the case of nanoplexes, there was a distinct shift in the MFI peaks as compared to either the control cell or naked siRNA, indicating their higher cellular association and hence possibility of higher cellular uptake. The results confirm the ability of CD polyrotaxane to enhance the cellular uptake of complexed siRNA. Further, the peak of cationic nanoplexes was broader than that of the anionic nanoplexes, indicating a higher cellular association of the former. This was confirmed from the MFI (%) value, which was 81.6 ± 0.87 for the cationic nanoplexes as against 67.9 ± 2.5 for the anionic nanoplexes. The MFI (%) value for the naked siRNA was only 21.2 ± 0.2 as compared to 15.62 ± 0.01 for cell control, which may be attributed to the auto-fluorescence of cells.
3.5. CLSM
The precise uptake pathway of nanoplexes was evaluated using specific inhibitors of various endocytic pathways. Chlorpromazine (CHL) inhibits clathrin-mediated endocytosis through loss of clathrin and the adaptor protein, AP2, from the cell surface and their artificial assembly on endosomal membranes. Nystatin (NYS), the polyene antibiotic, acts as a selective inhibitor of caveolin mediated endocytosis due to its ability to interact with the cholesterol rich domains in the cell membrane and alter their properties. This agent sequesters cholesterol from the cell membrane by forming large aggregates within the membrane, and thereby results in aberration of the caveolar structure and function, these being cholesterol-rich membrane domains. Finally, EIPA has been reported to block macropinocytosis and phagocytosis by inhibiting the Na+/H+ exchange required for both these endocytic mechanisms.15The 3D CLSM images depicted distinct intracellular localization of nanoplexes (red spots) with respect to the green cell membranes and blue nuclei, when the nanoplexes were incubated in the presence of either CHL or NYS, thus precluding the possibility of clathrin or caveolin mediated endocytosis as the possible uptake mechanisms for the nanoplexes. However, the majority of the nanoplexes remained outside the cells in case of EIPA, suggesting macropinocytosis and phagocytosis as the uptake mechanisms. The representative CLSM images, in the presence of each inhibitor, have been depicted in Fig. 5(I).
 | ||
| Fig. 5 (I) Mechanism of uptake of nanoplexes: nanoplexes were internalized in the presence of CHL (A) and NYS (B), indicating that the nanoparticles did not enter the cells by clathrin or caveolin mediated endocytosis. Nanoplexes were not internalized in the presence of EIPA (C), suggesting macropinocytosis and phagocytosis as their possible uptake pathways. Green fluorescence: cell membranes stained with FITC-WGA; blue fluorescence: cell nuclei stained with DAPI; red fluorescence: nanoplexes formulated with Cy3 siRNA. RAW cells were incubated with inhibitors for 3 h, further with nanoplexes in the presence of inhibitors for 4 h. The nanoplex solution was then replaced by inhibitor solutions in cell culture medium and the cells were incubated for further 18 h before subjecting them to fixation and staining. (II) Uptake of nanoplexes analyzed by multiphoton-pixel analysis method. The weighed number of nanoplexes in the selected region of interest for CHL and NYS were significantly higher than EIPA, thus indicating that the nanoplexes were uptaken by macropinocytosis/phagocytosis. (III) Manual counting of nanoplexes; the percentage of cells with distinct intracellular localization of nanoparticles: 73 ± 13% for CHL; 80 ± 15% for NYS and 21 ± 1.5% for EIPA; macropinocytosis and phagocytosis possible uptake pathways of nanoplexes. | ||
This visual observation was confirmed by determining the weighed number of nanoplexes in CLSM images by a combined multiphoton pixel analysis method reported by Labouta et al.16 The advantage of this method over manual counting of the fluorescent spots per field in confocal images is that the method avoids human error and considers the area of the fluorescent spots that are larger than the resolution limit. This method, based on pixel analysis, considers the number of events and not their intensity. The weighed number of nanoplexes in the selected region of interest, in the presence of all the three inhibitors, has been represented in Fig. 5(II).
As seen in Fig. 5(II), the weighed number of nanoplexes was significantly lower for EIPA, clearly indicating that EIPA blocked the cellular uptake. This confirmed the involvement of macropinocytosis and phagocytosis in the cellular uptake of nanoplexes.
The 3D CLSM images were also manually evaluated for estimating the number of cells with distinct intracellular localization of nanoplexes, for verifying the above observations. The results of manual counting were in compliance with visual observation and those obtained with the multiphoton pixel analysis method. These results have been presented in Fig. 5(III).
The percentage of cells with distinct intracellular localization of nanoparticles was 73 ± 13% in the presence of CHL, 80 ± 15% in the presence of NYS, and only 21 ± 1.5% when the nanoplexes were incubated in the presence of EIPA. The experimental findings thus suggested macropinocytosis and phagocytosis as the possible uptake pathways for the nanoplexes.
With reference to polycation mediated nucleotide delivery, using PEI as the “gold” standard, it has been reported that larger polycation–nucleotide complexes (>200 nm) are generally taken up by macropinocytosis, whereas intermediate nanoplexes (100–200 nm) are taken up by receptor-mediated endocytosis.29,30 However, in the present investigation, we speculate that the nanoplexes were internalized by macropinocytosis, despite their small size, due to their mechanical properties. The nanoplexes employed herein are based on spontaneous electrostatic interactions between the polymer and nucleic acids and are soft nanoparticles. This fact was also demonstrated by a decrease in the vertical height of the nanoplexes while drying during AFM, since soft nanoparticles are prone to deformation upon drying. Although the exact effect of the mechanical strength of nanoparticles on their intracellular uptake is unknown, the dynamics of the membrane–nanoparticle interface deformation has been anticipated to govern the nanoparticle uptake. Thermal fluctuations of the cell membrane–nanoparticle interfaces, upon contact with the nanoparticles, are expected to distort soft nanoparticles and hence influence the stability of contacts formed and subsequent receptor involvement. Hence cellular uptake of soft nanoparticles has been hypothesized to take place by receptor independent mechanisms such as macropinocytosis. Other researchers have reported a similar influence of mechanical properties of hydrogel nanoparticles on their cellular uptake in RAW 264.7 cells.30 In the present study, we speculate the same to happen during the cellular uptake of our nanoplexes. However size-independent phagocytosis of nanoplexes, as has been observed for alternative nanoparticle systems, or their uptake after aggregation upon contact with cellular plasma membrane cannot be completely overruled.31,32
3.6. Biological efficacy of nanoplexes in cellular assays
Thus, targeting of Bfl-1/A1 using nanoplexes formulated with anti-Bfl-1/A1 siRNA was hypothesized to be a rational therapeutic intervention for tuberculosis. Bfl-1/A1 expression was induced in RAW 264.7 cells taking advantage of the fact that this protein is a direct transcriptional target of NF-κB. Thus, up-regulation of Bfl-1/A1 expression was achieved by stimulating the endogenous NF-κB activity in RAW 264.7 cells. Numerous studies have already proved the potential of bacterial LPS to stimulate NF-κB signaling in macrophage cell lines including RAW 264.7.17,18,35 Thus, LPS from E. coli was used for inducing the expression of Bfl-1/A1 in the present study.
The induction of Bfl-1/A1 expression in RAW 264.7 cells was confirmed by western blot analysis of the protein. Further, Bfl-1/A1 protein and mRNA levels, after treatment with nanoplexes formulated using anti-Bfl-1/A1 siRNA, naked siRNA, and anti-Bfl-1/A1 siRNA-jetPRIME™ reagent combination, were analyzed using western blot and RT-PCR. The results were compared with untreated cells induced with LPS and cells without any treatment. These have been represented in Fig. 6A and B, respectively.
 | ||
| Fig. 6 (A) Western blot analysis of Bfl-1/A1 protein expression after transfection of RAW 264.7 cells with nanoplexes formulated using anti-Bfl-1/A1 siRNA (80 pmol) for 72 h; (B) Bfl-1/A1 gene silencing monitored using RT-PCR. Bfl-1/A1 mRNA levels were normalized to β-actin mRNA expression. Red bars indicate treatment with anti-Bfl-1/A1 siRNA whereas black bars indicate treatment with scrambled siRNA. Endogenous Bfl1/A1 gene expression in RAW 264.7 cells was stimulated using lipopolysaccharide from E. coli for 24 h. Bfl-1/A1 expression was analyzed after treatment with naked siRNA and nanoplexes for 4 h, followed by incubation with normal growth medium for further 65 h, with intermittent replacement of medium every 24 h. | ||
As seen from Fig. 6A, there was only a slight endogenous expression of Bfl-1/A1 in RAW 264.7 cells in the absence of LPS stimulation whereas LPS resulted in up-regulation of Bfl-1/A1. Further, the nanoplexes were able to reduce the expression of Bfl-1/A1, when compared with its expression after LPS stimulation and after treatment with naked siRNA. A single treatment of the cells with 80 pmol of anti-Bfl-1/A1 siRNA formulated with CD polyrotaxane was able to significantly reduce the protein levels in comparison with naked siRNA. Also, protein levels after treatment with cationic nanoplexes was lower as compared to treatment with anionic nanoplexes, indicating their superior efficacy.
These results were further reinforced by the reduction in Bfl-1/A1 mRNA expression as observed in RT-PCR (Fig. 6B). Bfl-1/A1 mRNA levels were normalized to β-actin mRNA expression (endogenous gene control). This was done to eliminate any possibility of reduced Bfl-1/A1 expression due to altered metabolic activity of the cells arising from polymer mediated cellular stress.
As seen from Fig. 6B, the cationic nanoplexes resulted in almost 50% reduction in Bfl-1/A1 mRNA levels when compared to those after LPS stimulation. Decrease in mRNA levels after treatment with naked siRNA was negligible, indicating the inability of naked anti-Bfl-1/A1 siRNA to effectively traverse the cell barriers. The Bfl-1/A1 mRNA levels after treatment with anionic nanoplexes were higher as compared to those after treatment with cationic nanoplexes, once again suggesting the effect of the overall positive charge of CD polyrotaxane nanoplexes in mediating better intracellular uptake, release, and hence, biological competence of anti-Bfl-1/A1 siRNA. Moreover, the gene expression was analyzed 65 h after single treatment with nanoplexes or naked siRNA, thus indicating a persistent knockdown with nanoplexes in the cellular assay. The protein and mRNA levels of Bfl-1/A1 were not affected when the cells were treated with nanoplexes formulated with scrambled siRNA (Fig. 6B, black bars). The experiments were performed in triplicate to confirm their reproducibility. Thus the polymer was indeed able to release the siRNA within the cells to knockdown the targeted protein. We have earlier proved the ability of the polymer to release complexed nucleic acids,11 which corroborated well with the biological efficacy assay in the present investigation. The results confirmed the potential of CD polyrotaxane mediated siRNA delivery for the treatment of intracellular bacterial infections such as tuberculosis.
4. Conclusion
Our findings validate the potential of CD polyrotaxane to mediate efficient siRNA delivery. The synthesized polymer condensed siRNA molecules into nanometric complexes, bearing an overall positive charge. The resulting nanoplexes facilitated an enhanced knockdown of the targeted gene, as compared to naked siRNA. These results suggested the potential of the polymer to improve cellular internalization of the fragile siRNA and prevent its degradation in endosomes. Prevention of endosomal degradation of siRNA may also be speculated to occur due to possible macropinocytic uptake of the nanoplexes resulting from their soft mechanical properties. More comprehensive studies in this direction will be required to provide concrete proof of our projections. Nevertheless, the investigations proved the potential of CD polyrotaxane to downregulate the expression of a gene relevant in the pathogenesis of lethal intracellular infections such as tuberculosis. Due to the observed efficacy and cellular safety of the polymer at the working concentrations, we foresee its broad implications for the treatment of other intracellular infections as well. In vitro cellular assays suggest a clear potential of CD polyrotaxane for delivering siRNA in animal models of tuberculosis and other intracellular bacterial infections. This accompanied by in vivo toxicity assays will assist to further investigate their clinical potential.Acknowledgements
Dr Prajakta Dandekar is a recipient of a European Respiratory Society/Marie Curie Joint Research Fellowship – Number MC [1634]-2010. The research leading to these results has received funding from the European Respiratory Society and the European Community's Seventh Framework Programme FP7/2007-2013-Marie Curie Actions under grant agreement RESPIRE, PCOFUND-GA-2008-229571. Dr Ratnesh Jain is recipient of Alexander von Humboldt Foundation's postdoctoral fellowship and thankful to the foundation for the funding.References
- S. Y. Wong, J. M. Pelet and D. A. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS PubMed.
- T. Albuzat, M. Keil, J. Ellis, C. Alexander and G. Wenz, J. Mater. Chem., 2012, 22, 8558–8565 RSC.
- J. J. Li, F. Zhao and J. Li, Appl. Microbiol. Biotechnol., 2011, 90, 427–443 CrossRef CAS PubMed.
- K. Tokuhisa, E. Hamada, R. Karinaga, N. Shimada, Y. Takeda, S. Kawasaki and K. Sakurai, Macromolecules, 2006, 39, 9480–9485 CrossRef CAS.
- T. Ooya, H. S. Choi, A. Yamashita, N. Yui, Y. Sugaya, A. Kano, A. Maruyama, H. Akita, R. Ito, K. Kogur and H. Harashima, J. Am. Chem. Soc., 2006, 128, 3852–3853 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- A. Kulkarni, K. DeFrees, R. A. Schuldt, S. H. Hyun, K. J. Wright, C. K. Yerneni, R. VerHeul and D. H. Thompson, Mol. Pharm., 2013, 10, 1299–1305 CrossRef CAS PubMed.
- A. Tamura and N. Yui, Biomaterials, 2013, 34, 2480–2491 CrossRef CAS PubMed.
- Y. Yamada, M. Hashida, T. Nomura, H. Harashima, Y. Yamasaki, K. Kataoka, A. Yamashita, R. Katoono and N. Yui, ChemPhysChem, 2012, 13, 1161–1165 CrossRef CAS PubMed.
- G. Wenz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6333–6341 CrossRef CAS.
- P. Dandekar, R. Jain, M. Keil, B. Loretz, L. Muijs, M. Schneider, D. Auerbach, G. Jung, C. M. Lehr and G. Wenz, J. Controlled Release, 2012, 164, 387–393 CrossRef CAS PubMed.
- K. Moh, U. Werner, M. Koch and M. Veith, Adv. Eng. Mater., 2010, 12, 368–373 CrossRef CAS.
- B. Weiss, M. Schneider, L. Muys, S. Taetz, D. Neumann, U. F. Schaefer and C. M. Lehr, Bioconjugate Chem., 2007, 18, 1087–1094 CrossRef CAS PubMed.
- P. Dandekar, R. Jain, T. Stauner, B. Loretz, M. Koch, G. Wenz and C. M. Lehr, Macromol. Biosci., 2012, 12, 184–194 CrossRef CAS PubMed.
- A. I. Ivanov, Methods Mol. Biol., 2008, 440, 15–33 CAS.
- H. I. Labouta, T. Kraus, L. K. El-Khordagui and M. Schneider, Int. J. Pharm., 2011, 413, 279–282 CrossRef CAS PubMed.
- W. X. Zong, L. C. Edelstein, C. Chen, J. Bash and C. Gélinas, Genes Dev., 1999, 13, 382–387 CrossRef CAS.
- M. Fukui, R. Imamura, M. Umemura, T. Kawabe and T. Suda, J. Immunol., 2003, 171, 1868–1874 CrossRef CAS.
- C. Schulze, U. F. Schaefer, C. A. Ruge, W. Wohlleben and C. M. Lehr, Eur. J. Pharm. Biopharm., 2011, 77, 376–383 CrossRef CAS PubMed.
- M. Dominska and D. M. Dykxhoorn, J. Cell Sci., 2010, 123, 1183–1189 CrossRef CAS PubMed.
- H. Rhinn, C. Largeau, P. Bigey, R. L. Kuen, M. Richard, D. Scherman and V. Escriou, Biochim. Biophys. Acta, 2009, 1790, 219–230 CrossRef CAS PubMed.
- D. J. Gary, N. Puri and Y. Y. Won, J. Controlled Release, 2007, 121, 64–73 CrossRef CAS PubMed.
- J. Nguyen, R. Reul, S. Roesler, E. Dayyoub, T. Schmehl, T. Gessler, W. Seeger and T. H. Kissel, Pharm. Res., 2010, 27, 2670–2682 CrossRef CAS PubMed.
- N. D. Weber, O. M. Merkel, T. Kissel and M. Á. Muñoz-Fernández, J. Controlled Release, 2012, 157, 55–63 CrossRef CAS PubMed.
- A. Bogner, G. Thollet, D. Basset, P. H. Jouneau and C. Gauthier, Ultramicroscopy, 2005, 104, 290–301 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- P. Dandekar, R. Jain, C. Kumar, S. Subramanian, G. Samuel, M. Venkatesh and V. Patravale, J. Biomed. Nanotechnol., 2009, 5, 445–455 CrossRef PubMed.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. Au, AAPS J., 2010, 12, 492–503 CrossRef CAS PubMed.
- S. Grosse, Y. Aron, G. Thévenot, D. François, M. Monsigny and I. Fajac, J. Gene Med., 2005, 7, 1275–1286 CrossRef CAS PubMed.
- X. Banquy, F. Suarez, A. Argaw, J.-M. Rabanel, P. Grutter, J.-F. Bouchard, P. Hildgen and S. Giasson, Soft Matter, 2009, 5, 3984–3991 RSC.
- N. Oh and J.-H. Park, Int. J. Nanomed., 2014, 9, 51–63 Search PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5–15 CrossRef PubMed.
- R. Dhiman, M. Kathania, M. Raje and S. Majumdar, Biochim. Biophys. Acta, 2008, 1780, 733–742 CrossRef PubMed.
- M. Kathania, C. I. Raje, M. Raje, R. K. Dutta and S. Majumdar, Int. J. Biochem. Cell Biol., 2011, 43, 573–585 CrossRef CAS PubMed.
- E. Jones, I. M. Adcock, B. Y. Ahmed and N. A. Punchard, J. Inflammation, 2007, 4, 23 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01821d |
| ‡ Present address: Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Matunga, Mumbai 400019, India. E-mail: E-mail: pd.jain@ictmumbai.edu.in; Fax: +91 22 33611020; Tel: +91 22 33612221 |
| § Present address: Department of Chemical Engineering, Institute of Chemical Technology, Matunga, Mumbai 400019, India. E-mail: E-mail: rd.jain@ictmumbai.edu.in; Fax: +91 22 33611020; Tel: +91 22 33612221 |
| This journal is © The Royal Society of Chemistry 2015 |