
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and function of the silicifying peptide R5†
Laura
Senior
ab,
Matthew P.
Crump
c,
Christopher
Williams
c,
Paula J.
Booth‡
a,
Stephen
Mann
c,
Adam W.
Perriman
d and
Paul
Curnow
*a
aSchool of Biochemistry, University of Bristol, Medical Sciences Building, University Walk, BS8 1TD, UK. E-mail: p.curnow@bristol.ac.uk; Fax: +44(0) 17 331 2168; Tel: +44(0) 117 331 1837
bBristol Centre for Functional Nanomaterials, University of Bristol, BS8 1TD, UK
cSchool of Chemistry, University of Bristol, BS8 1TD, UK
dSchool of Cellular and Molecular Medicine, University of Bristol, BS8 1TD, UK
First published on 12th February 2015
Abstract
The 19-mer synthetic peptide known as R5 has been used widely in studies of peptide-driven silica condensation. Despite this, the structure and function of R5 have not yet been fully characterized. Here, we present a systematic study of R5 silicification focusing on three key variables: the concentration of the peptide, the concentration of the silica precursor silicic acid, and the solution pH. Additionally, we present the first study of R5 secondary structure in the presence and absence of silicic acid and introduce one-dimensional and two-dimensional solution NMR to probe both structure and higher-order peptide aggregation. We find that R5-directed silicification is linear with regard to silicic acid and H+ but, unexpectedly, that silicification appears to be cooperative with respect to peptide concentration. We also find that R5 is a random coil ensemble at subsaturating silicic acid concentrations and does not spontaneously self-assemble to form discrete aggregates in solution. These data contradict a model that invokes the functional micellization of R5 and provide a framework for future studies with the R5 peptide.
Introduction
The use of organic macromolecules for the controlled precipitation and deposition of inorganic materials is now an established area of modern chemistry.1–3 Such research is generally inspired by the composite biominerals found in the natural world. The assembly of many of these biominerals relies upon an organic component provided by macromolecules such as proteins and polysaccharides. The hybrid materials that result are often surprisingly ornate and complex with advantageous physical properties. In particular, hydrated silica can be synthesized in vitro from simple precursors using peptides,4 polymers,5 polyamines,6 and polysaccharides7 which mimic the macromolecules that are intimately involved in silicification processes in vivo.8,9 Such biomimetic approaches allow silica to be synthesized under environmentally sustainable conditions at near-neutral pH and room temperature, and may ultimately be scalable for industrial use.10,11Diatoms (Bacillariophyceae) are a group of abundant and diverse algae that are sheathed in a silicified external cell wall known as the frustule. The biomolecules responsible for diatom silicification8,9 include silicon-responsive12,13 silicifying polypeptides known as silaffins that were originally isolated as proteinaceous components of the frustule14 and have now been characterized in some detail.15–19 In Cylindrotheca fusiformis, a group of homologous silaffin isoforms known collectively as silaffin-1A1 are produced in vivo by endoproteolysis of the gene product Sil1p.12–14 Sequence analysis of silaffin-1A1 led to the identification of the repeat sequence motif SSKKSGSYSGSKGSKRR(I/N)L. Mature native silaffin-1A1, from which the C-terminal RRIL group is apparently removed, has unusual posttranslational modifications so that 5–11 repeats of N-methylpropylamine are introduced at the ε-amino group of K3 and K15, K4 is ε-N,N-dimethyllysine, K12 is a phosphorylated ε-N,N,N-trimethyl-δ-hydroxylysine, and all of the serine residues are phosphorylated. Both native silaffin-1A1 and a 19-mer synthetic peptide based upon the entire repeat motif, termed R5, were shown to induce rapid silica precipitation from silicic acid solutions in phosphate/citrate buffer. Although silaffin-1A1 could induce silicification down to pH 5 and was even partly active at pH 4, R5 was only active above pH 6.14 There is evidence that both silaffins and R5 become entrapped within the precipitated silica, giving rise to peptide–silica precipitates.14,20
There have since been numerous attempts to exploit this peptide sequence for biotechnology. For example, R5-associated silicification has been shown to be a generic strategy for bulk enzyme encapsulation,21 and covalent conjugation to R5 allows the encapsulation of small molecules22 and peptides.23 Additionally, R5 has been introduced to enzymes and other proteins as a silicifying motif by chemical and transcriptional fusion to enable the encapsulation of specific target proteins24–26 and for the formation of novel hybrid materials27,28 and biosensors.29,30 Modifications to the peptide sequence can also expand the utility of R5; for example to induce silicification at biomembranes.31 The dependence of silicification upon the R5 peptide sequence has been investigated and the C-terminal sequence RRIL appears to be essential for mineralization,20,32 with suggestions that this motif facilitates functionally-important, spontaneous peptide self-assembly either via micellization20,33 or through salt-bridging.32 These models are consistent with the electrostatically-driven aggregation that appears to be necessary for silicification by the zwitterionic native silaffins.16 Interestingly, scrambling the RRIL motif or moving it to the N-terminal or centre of R5 have a marginal effect on peptide activity but induce substantial changes to the resultant silica morphology.32
Significantly, the solution properties of R5 have not yet been fully characterized. To address this, we present here a study of the structure and function of R5 in physiologically-relevant buffers with a physiologically-relevant substrate, silicic acid (Si(OH)4). In particular, we examine whether R5 adopts any secondary structure in solution and study silicification as a function of peptide concentration, silicic acid concentration, and pH. We also determine whether the peptide spontaneously self-assembles in the absence and presence of silicic acid.
Materials and methods
Peptide sequences
The 19-mer R5 peptide with sequence SSKKSGSYSGSKGSKRRIL (MW, 2013 Da; pI, 11.2) was synthesized commercially (Generon). The peptide was confirmed as ≥95% pure by the vendor via LS-MS (Fig. S1†). Protamine sulphate salt from salmon, synonymously salmine, with sequence PRRRRSSSRPIRRRRPRRASRRRRRRGGRRRR (major component; MW, 4236 Da; pI, 13.3) was from Sigma-Aldrich. All other chemicals were supplied by Sigma-Aldrich at highest purity.Silica precipitation
Orthosilicic acid (referred to here as silicic acid) was prepared by incubating 4 ml of 0.2 M sodium silicate solution with 1.5 g Dowex 50WX4-50 cation exchange resin, prepared by acidification with 0.01 M sulfuric acid for 15 min at 4 °C.34 For precipitation studies, 60 μl of silicic acid solution was added to an equivalent volume of R5 or protamine in 50 mM sodium phosphate buffer and the final pH was adjusted as required. The samples were continuously mixed using a rotator for 15 min. To determine precipitate yield, reactions were carried out in a pre-weighed eppendorf tube and R5–silica and protamine–silica precipitates were pelleted at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 2 minutes. The supernatant was removed by pipetting and precipitates were dried for 16 h under vacuum. The tubes were then re-weighed to determine the mass of precipitate.
000 × g for 2 minutes. The supernatant was removed by pipetting and precipitates were dried for 16 h under vacuum. The tubes were then re-weighed to determine the mass of precipitate.
Analytical methods
Dynamic light scattering was performed using a Zetasizer Nano ZS (Malvern). Samples were 50 μL at concentrations up to 3.7 mM R5 in a low-volume quartz cuvette, measured in triplicate at 25 °C where each repeat was an average of 16 individual scans. Circular dichroism spectroscopy was performed using an AVIV model 410 spectrometer with a 0.01 mm path length cell. R5 was at 0.5 mM in 50 mM sodium phosphate buffer at the desired pH. Exceptions to this were experiments in the presence of 1 mM silicic acid, where the peptide concentration was 0.25 mM, and reactions at pH 4.5, where 50 mM MES buffer was used. The mean of two replicate scans was used in the data analysis.NMR spectra were recorded on a 600 MHz Varian VNMRS spectrometer equipped with a triple resonance cryogenically cooled probe-head. NMR samples contained 0.5 mM R5 in 50 mM sodium phosphate buffer and 90% H2O/10% D2O at pH 7.0 or 5.5. High-resolution one-dimensional proton spectra and two-dimensional nuclear Overhauser enhancement spectroscopy (NOESY), double-quantum filtered correlation spectroscopy (DQF-COSY) and total correlation spectroscopy (TOCSY) spectra were recorded. Mixing times were 150 ms and 65 ms for NOESY and TOCSY experiments respectively. Two-dimensional data were acquired with a spectral width of 8012 Hz, 4096 complex points in f2 and 800 complex points in f1 with sculpted excitation water suppression for both the NOESY and TOSCY spectra.35 Spectra were processed and plotted using VnmrJ 4.0 (Agilent).
For scanning electron microscopy, vacuum-dried R5–silica and protamine–silica precipitates were fixed onto aluminium stubs using adhesive tabs and were sputter-coated with silver. All samples were examined using a JSM 5600LV SEM (JEOL) fitted with the ISIS 300 X-ray analysis system (Oxford Instruments).
Results and discussion
Silicic acid concentration
We first assessed silica precipitation by both the lysine-rich peptide R5 and the arginine-rich comparator protamine as a function of silicic acid concentration. Both of these peptides are expected to facilitate silica formation from saturated solutions of silicic acid. In the early stages of silica formation, cationic peptides and small-molecule amines are thought to facilitate the local clustering of silicic acid by electrostatic interactions and hydrogen bonding and encourage dissociation to Si(OH)3−, which promotes the initial condensation events that lead to branched polysilicic acids. Additionally, it has been proposed that charged amines reduce the energy barrier to the growth of these condensed nuclei into spherical particles and induce the flocculation of these particles to form a three-dimensional network, largely by compensating for the build-up of negative charge at the particle surface.36–39As anticipated, the presence of either R5 or protamine resulted in rapid silica precipitation (within seconds) once silicic acid was raised above the saturating concentration of 2 mM at pH 7 (Fig. 1). No such rapid precipitation was observed in the absence of either peptide, confirming the roles of both R5 and protamine in promoting silica formation. The dry weight of these peptide–silica precipitates was found to be linear with silicic acid concentration, with the gradients of these linear curves being 0.36 ± 0.02 and 0.44 ± 0.02 mg product per ml per mM silicic acid for R5 and protamine, respectively (R2 = 0.96). Thus, under equivalent conditions, protamine generates approximately 1.2-fold greater dry weight of precipitate versus R5. This modest difference in activity presumably arises from the higher charge density carried by protamine (21 charged amino acids of 32 residues vs. 6 of 19 residues in R5). It was previously shown that the number of charged residues within the R5 sequence was a critical factor in controlling the rate and yield of silicification, and that the specific position of lysine and arginine groups within the peptide sequence also influenced silica morphology.20,32 The difference in activity between R5 and protamine may also reflect the side chain chemistries of the cationic amino acids within each peptide. The cation in protamine is exclusively arginine, and the arginine side-chain comprises a guanidino group with a geometry and charge distribution that are particularly favourable for the binding of tetrahedral oxoanions.40 This may make arginine-rich peptides more effective at silicification in vitro. We did not determine the contribution of any sequestered peptide to the weight of the peptide–silica products, but previous reports have suggested that R5 comprises about 25% of the precipitate by weight.20,33
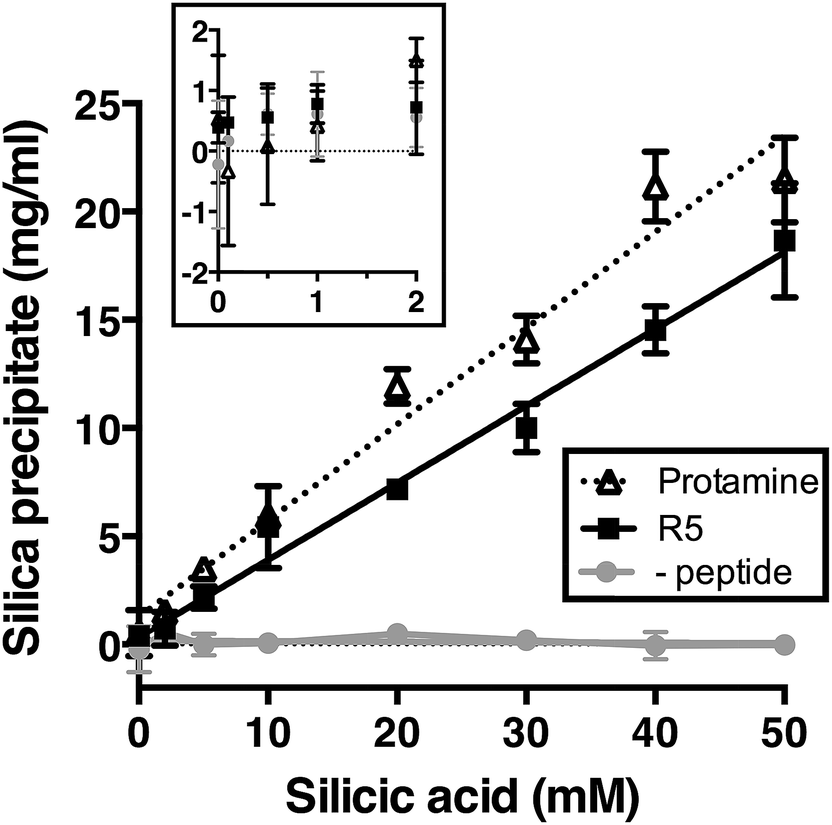 | ||
| Fig. 1 Rapid silica precipitation by R5 and protamine as a function of silicic acid at pH 7, 2.5 mM of R5 or protamine. Substantial rapid precipitation is only observed at saturating concentrations of silicic acid above 2 mM (inset; same axes as main figure). Rapid precipitation is approximately linear with silicic acid above 2 mM. Data are mean ± s.d. of at least two independent experiments with linear fits shown; R2 = 0.96. (-peptide), control sample without either R5 or protamine. | ||
Peptide concentration and pH dependence
Kröger and colleagues14,15 showed that while native silaffins were fully active over the range of pH 5 to pH 8, R5 was only able to precipitate silica above pH 6. To provide a more comprehensive assessment of both the pH-dependence and peptide concentration dependence of silica precipitation, we studied precipitation by 0–3.7 mM R5 and protamine in the presence of 50 mM silicic acid at pH 6, 7 and 8.The R5 results are shown in Fig. 2A–C. Fig. 2A shows that silica was only precipitated above 1 mM R5 at pH 7 and 8. No rapid precipitation by R5 was observed under any peptide concentrations tested at pH 6. The reaction appeared to be strongly cooperative and was well fit to the Hill equation with Hill slopes of 4.2 ± 1.0 and 3.5 ± 0.8 at pH 7 and 8, respectively (R2 ≥ 0.9). This is in contrast to previous experiments with native silaffins, where silica precipitation has a linear dependence on peptide concentration.14,16 From the curve-fitting shown in Fig. 2A, the maximal weight of precipitate that could be formed from 50 mM silicic acid was 19.3 ± 1.0 mg ml−1 at pH 7 and 22.5 ± 1.5 mg ml−1 at pH 8, in good agreement with Fig. 1. Half-maximal precipitation occurred at 1.3 ± 0.1 mM R5 at both pH 7 and 8. This R5 data could also be readily fit to a Boltzmann sigmoidal function. Similar behaviour was previously observed in a study of the peptide concentration dependence of R5-induced precipitation from tetramethoxysilane,20 confirming that this is not related to our preparation of the silicic acid precursor, reaction conditions or use of precipitate weight measurements. Although this prior study actually applied a linear fit to such data, we use here a wider R5 concentration range and so are able to confidently assign sigmoidal behaviour. SEM microscopy (Fig. 2B) confirmed that the presence of R5 resulted in the formation of R5–silica nanospheres with an average particle size (dav) of 540 ± 60 nm (N = 25) and EDX analysis of these precipitates (Fig. 2C) showed the expected elemental composition.
 | ||
| Fig. 2 Rapid silica precipitation from 50 mM silicic acid as a function of peptide concentration and pH. (A) R5 was active above 1 mM peptide at pH 7 and 8, with an apparently cooperative response, and was not active at pH 6. Data are mean ± s.d. from three independent repeats. Representative photographs of the peptide–silica precipitates are shown. Solid line, fit to the Hill equation; dashed line, fit to Boltzmann sigmoidal. See text for details. (B) R5–silica shows the expected spherical morphology, with an average measured diameter of these spheres (dav) being 540 ± 60. (C) These nanospheres were confirmed as biosilica by qualitative EDX. (D) Protamine was active at all pH values tested, with lower or absent cooperativity. Data are mean ± s.d. from three independent repeats. Representative photographs of precipitate are shown. Solid line, fit to hyperbolic equation. (E) Protamine–silica also has spherical morphology with slightly smaller dav of 380 ± 80 nm and (F) is confirmed as biosilica by qualitative EDX. | ||
Fig. 2D–F show the analogous experiments for protamine. Protamine was again found to be more active than R5 in silicification, generating substantial silica at 0.6 mM peptide (the lowest concentration tested). Protamine exhibits a less pronounced, though still discernible, pH dependence with the highest concentrations of protamine generating ∼1.5-fold more precipitate at pH 7 and 8 than the equivalent reaction at pH 6. In contrast to the results with R5, protamine reactions were not strongly cooperative with lower Hill coefficients of 1.5 ± 0.2 at all pH values. Fitting to a hyperbolic equation yielded maximal precipitation values of 22.5 ± 1.8 mg ml−1 at pH 6, 35.5 ± 1.7 mg ml−1 at pH 7 and 41.4 ± 2.0 mg ml−1 at pH 8, and gave half-maximal precipitation values of 0.8 ± 0.2 mM at pH 6, 1.2 ± 0.2 mM at pH 7 and 1.4 ± 0.2 mM at pH 8. SEM micrographs from the resulting precipitates (Fig. 2E) confirmed that the addition of protamine also resulted in the formation of silica nanospheres; EDX analysis (Fig. 2F) gave the anticipated elemental composition. The average diameter of the silica spheres formed (dav) was slightly smaller for protamine compared to R5, being 380 ± 80 nm (N = 25).
pH titration
Fig. 2 confirms that pH is clearly an important parameter in rapid silica precipitation by R5. To further investigate the pH dependence of the R5 reaction we undertook a more detailed pH titration. Fig. 3 shows that silica precipitation is linear with H+ concentration over the pH range 6.4–8.0 at 2.5 mM R5 and 50 mM silicic acid. This pH dependence may arise from pH-induced changes to silicic acid, peptide or buffer components individually or in combination. We consider each of these below.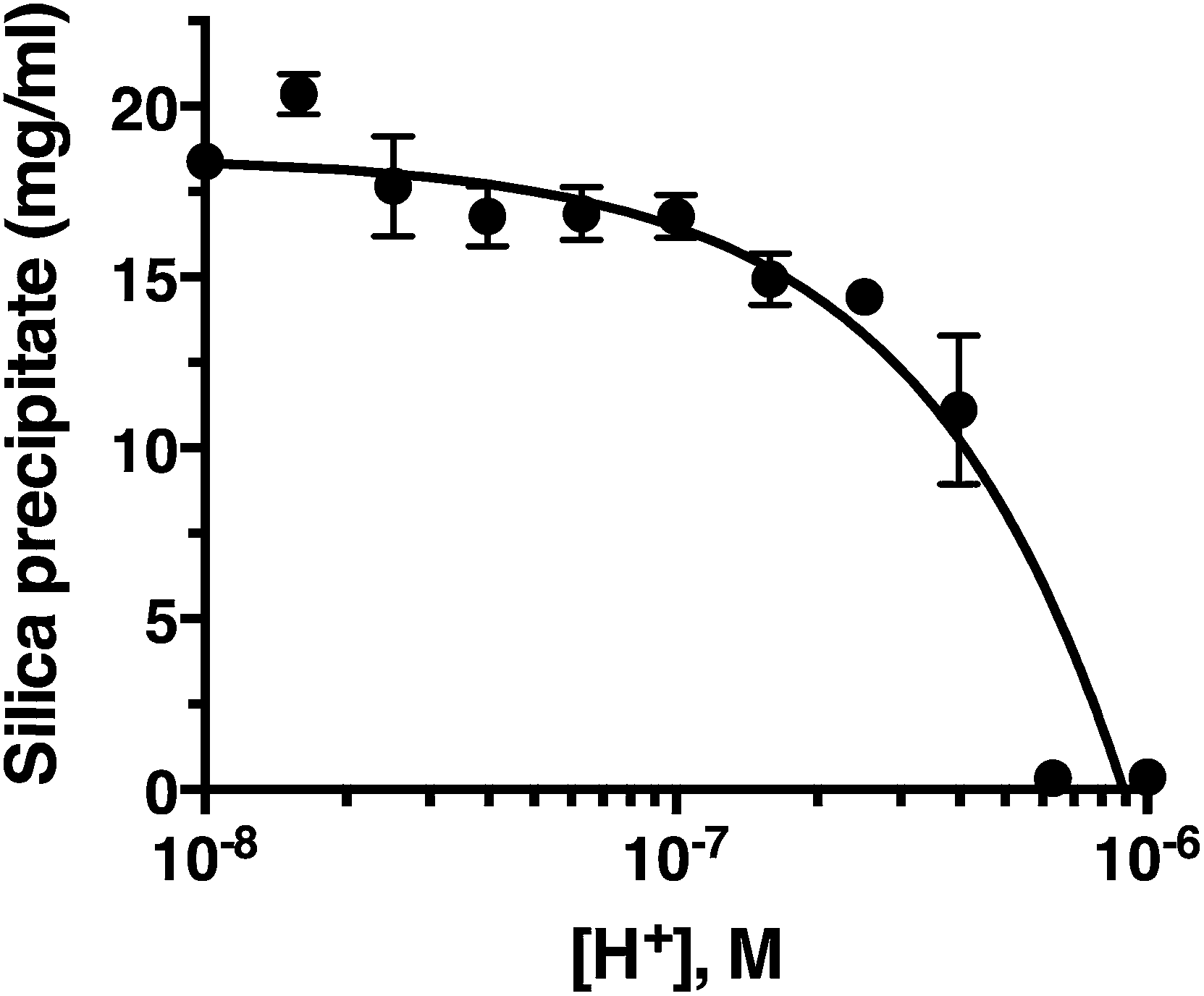 | ||
| Fig. 3 Silica precipitation is linear with respect to proton concentration. Data are mean ± s.d. from three independent experiments, shown on a log scale for clarity with a linear fit (R2 = 0.9). All reactions were 50 mM silicic acid, 2.5 mM R5. | ||
The reactive species for silicification is Si(OH)3O−, giving a linked equilibrium between the ionization of silicic acid and silica precipitation. It seems plausible that increased silicification at higher pH (lower [H+]) arises from a linear increase in Si(OH)3O−. Silicification only occurs above pH 6.4 and given that the pKa for the first ionization state of silicic acid is 9.8, this equates to a ratio of ∼2500:1 Si(OH)4:Si(OH)3O− being the critical threshold condition for rapid silicification. Silica formation by native silaffins and polyamines depends upon the presence of phosphate groups, occurring either as phosphorylated serines introduced via post-translational modifications or through phosphate ions being present in the reaction buffer.16,41,42 This requirement for phosphate does not appear to extend to R5, where introducing phosphoserine residues into the peptide has the opposite effect of inhibiting silicification at neutral pH;22 nonetheless, buffer phosphate may still be required to promote peptide self-assembly by bridging between cationic sidechains. Since the pH range explored in this study does encompass the equilibrium between H2PO4− and HPO42− (pKa = 7.2) it is possible that changes in the protonation state of buffer phosphate could influence silicification across this pH range.
It is also possible that the pH-dependence of silicification arises from changes in peptide charge at increasing pH. However, the pKa values of the lysine and arginine sidechains are expected to be close to the literature values of ∼10 and ∼12 in the absence of significant secondary structure (see below) and will remain fully protonated over the pH range tested here. The terminal amino and carboxyl groups are expected to have pKa values close to 8 and 3, respectively, but the N-terminal amino group alone is not sufficient to induce silica precipitation.32 Thus although we cannot exclude changes in peptide ionization playing a role in silicification, the overall peptide charge is unlikely to change significantly over the range pH 6.0–8.0.
Structure and aggregation of R5
Positively-charged peptides have been shown to adopt secondary structures that can influence silicification.43–47 In some cases e.g. polylysine47 it appears that the presence of silicic acid can actually induce structure formation. Using 31P NMR, native silaffins were shown to form a spontaneous aggregate of at least 700 peptide molecules in solution, and this assembly was suggested to be important in silicification.16 Additionally, it was previously suggested that long-chain polyamines self-assemble in the presence of bridging anions and that this is central to their ability to synthesize silica in vitro.36,39 Specifically with regard to R5, solid-state 13C two-dimensional NMR was previously used to study the structure of lyophilized R5 powders and R5–silica precipitates.33 This study observed significant changes in peptide backbone and side-chain chemical shifts that were apparently induced by silicification. These peturbations to the local environment were interpreted as arising from changes in peptide structure and from peptide–silica interactions, and the pattern of the chemical shift variations was consistent with a model in which the peptide forms a micelle-like structure. Within this micelle the C-terminal region is buried, and participates in peptide–peptide interactions, and the N-terminal is exposed. Inspired by these studies, we sought to use solution methods to determine whether R5 adopts secondary structure in solution and whether the peptide can spontaneously assemble into higher-order micelle-like structures or other aggregates.A computational prediction of the secondary structure of R5 was made by submitting the peptide sequence to the PSIPRED server.48 The results, shown in Fig. 4, suggest that R5 is largely unstructured, with the surprising possibility of a short length of β-strand at the C-terminal residues KRR. The confidence in this prediction is low, and it seems unlikely to be correct since independent β-strands are unstable. We extended our computational analysis by using the PEP-FOLD software49 to build multiple de novo models of the three-dimensional structure of R5, and representative models are shown in Fig. 4. The models did not converge upon any stable secondary structure in any part of the peptide sequence and this analysis suggests that the peptide is a diverse random coil ensemble in solution in agreement with experimental circular dichroism (CD) and NMR data presented here. In order to compare these random coils with experimental dynamic light scattering measurements (Fig. 7) we measured the distance between the two furthest separated main chain carbons for each of the models as shown. This measurement gives an approximate cross-section for the random coil of 1.4–2 nm, in good agreement with the hydrodynamic diameter of 2 nm determined using DLS (Fig. 7).
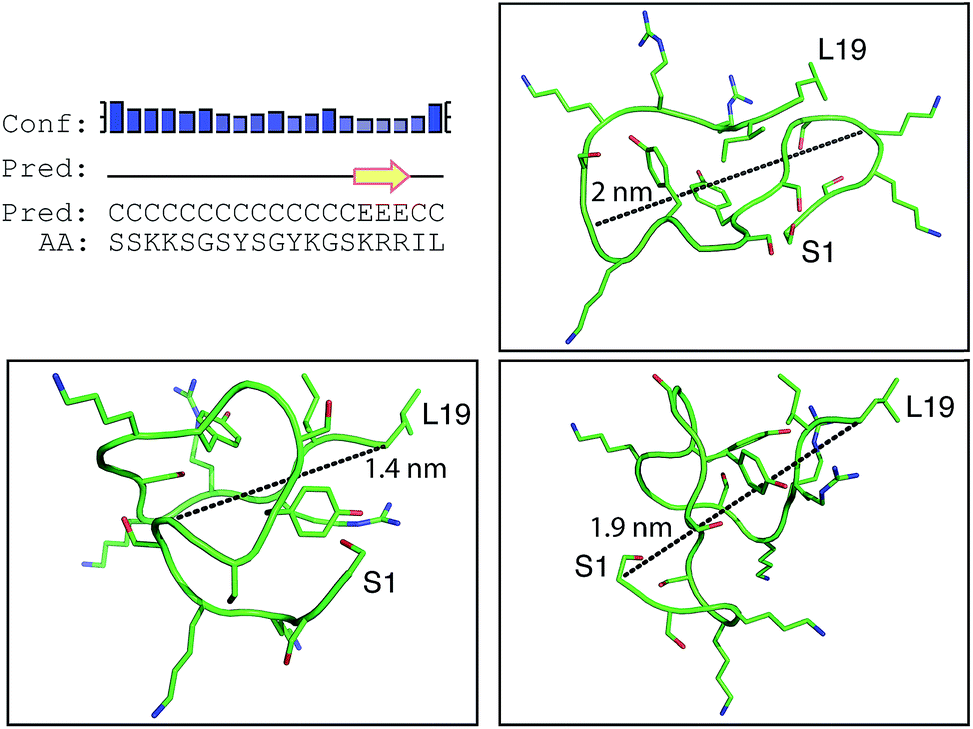 | ||
| Fig. 4 Bioinformatic predictions of R5 secondary structure. Sequence-based prediction by the PSIPRED server48 gives a low-confidence prediction of a short region of β-strand at the C-terminal. However, de novo structure predictions using PEP-FOLD49 suggest that R5 is a disparate random coil ensemble in solution. Three representative models from the PEP-FOLD output are shown with N-terminal serine and C-terminal leucine indicated as S1 and L19, respectively. Black dotted lines show distances measured in Pymol,52 which was used to construct the figures. | ||
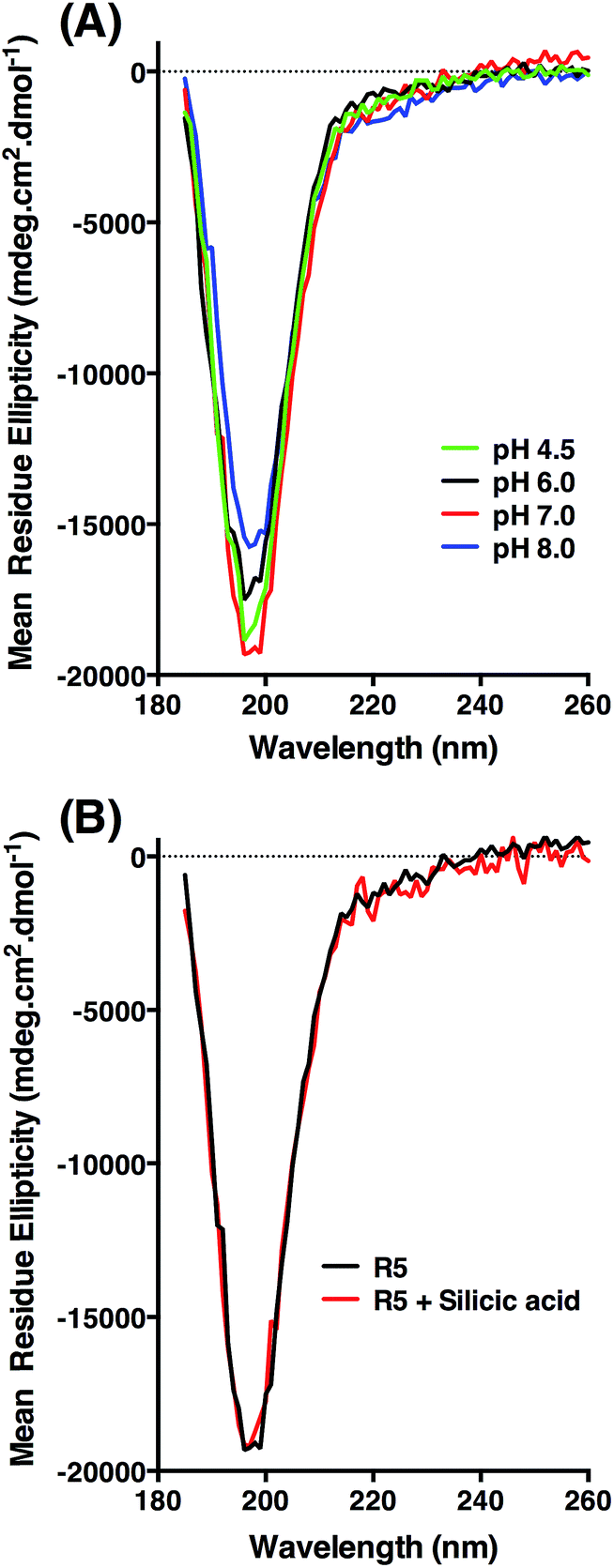 | ||
| Fig. 5 Circular dichroism spectra of R5. (A) Peptide ellipticity is characteristic of random coil at all pH values tested. (B) Peptide ellipticity is unchanged in the presence of silicic acid at pH 7.0. | ||
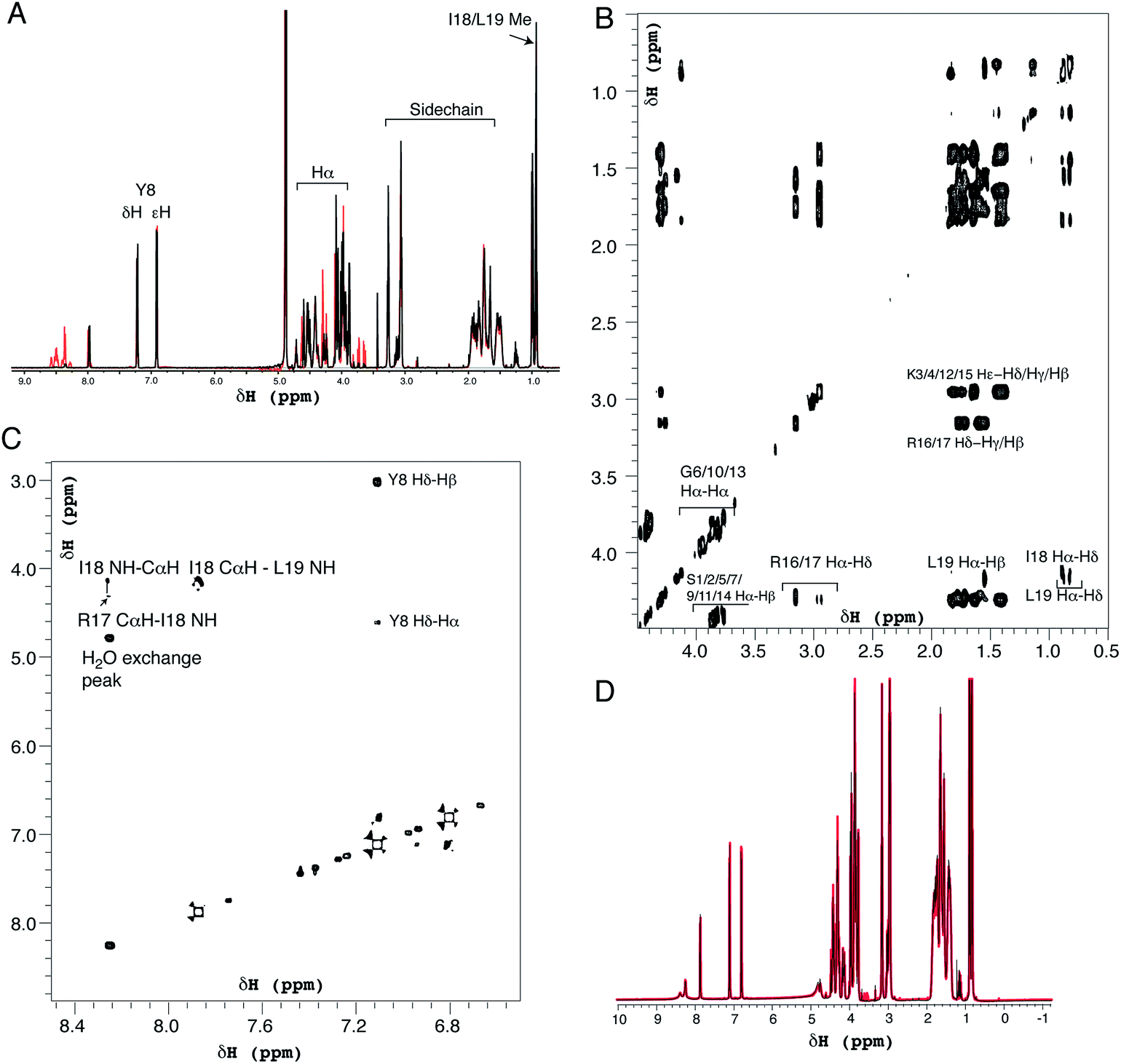 | ||
| Fig. 6 Solution 1H NMR spectra of R5 collected as (A) one-dimensional 1H spectrum at pH 7.0 (black) and pH 5.5 (red), (B) two-dimensional TOCSY spectrum at pH 7.0 expanded over the aliphatic region and (C) two-dimensional NOESY spectrum showing the fingerprint region at pH 7.0. (D) Comparison of one-dimensional spectra of R5 collected as in (A) (black) and in the presence of 50 μM silicic acid (red). Peptide concentration was 0.5 mM for all spectra. | ||
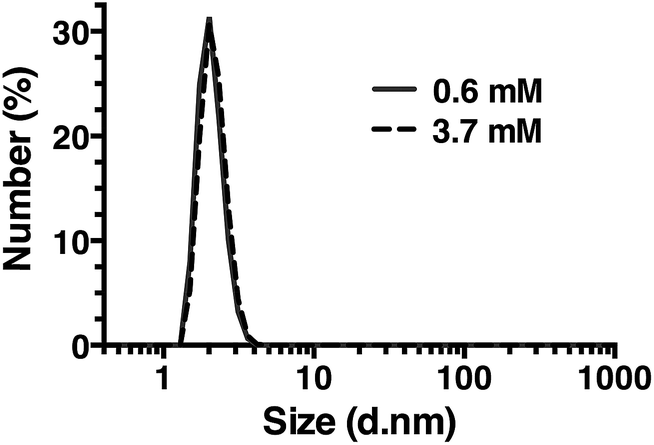 | ||
| Fig. 7 Dynamic light scattering of R5 solutions. R5 is monodisperse at all concentrations tested with a single peak centered at a hydrodynamic diameter of 2 nm. Representative results at the lowest and highest concentrations tested are shown. | ||
CD was used to assess whether R5 contained any substantial secondary structure, or whether such structure was induced upon interacting with subsaturating concentrations of silicic acid. The results are shown in Fig. 5. R5 exhibited a classical random coil signature at all pH values tested between 4.5–8.0 (Fig. 5A). Although the signal intensities were slightly varied between samples the shape of the spectra were identical with a single deflection at 198 nm characteristic of random coil.50 The presence of 2 mM silicic acid at pH 7 did not induce any structural change (Fig. 5B). It was not possible to collect data at higher silicic acid concentrations because of turbidity from precipitated silica.
1H solution NMR spectra were also collected to ascertain the secondary structure and aggregation state of R5. The 1D spectrum (Fig. 6A) showed sharp peaks in the aliphatic region (0–5 ppm) of the spectrum as well as clear signals for the aromatic protons of the single tyrosine residue present. At pH 7.0 the amide signals for all but the C-terminal isoleucine (I18) and leucine (L19) were broadened out to the baseline by solvent exchange effects. By comparison, sixteen of the nineteen amide signals were observable at pH 5.5. The aliphatic region of spectra collected at pH 7.0 and pH 5.5 were very similar with the exception of several intensity changes for side-chain βH protons of serine residues that sharpened at low pH due to reduced exchange effects from the side-chain (Oγ) hydroxyl group proton. At pH 7.0, all of the expected aliphatic signals could be identified in the two-dimensional TOCSY spectra (Fig. 6B), confirming solvent exchange as the source of line broadening and ruling out association or dynamic effects. The TOCSY experiment showed CαH and sidechain proton correlations for each amino acid type (Ser, Gly, Tyr, Lys, Arg, Leu and Ile) that were clustered around the expected values for a random coil peptide and did not show the dispersion associated with structured sequences. This apparent lack of secondary structure was also suggested by NOESY experiments (Fig. 6C) that showed no significant short-range or long-range NOEs at pH 5.5 or pH 7.0. At pH 5.5 two short stretches from G6 to Y8 and I18 to L19 could be assigned using sequential assignment methods but severe overlap and lack of dispersion prevented any further assignment.
CαH chemical shifts are strong indicators of secondary structure and the chemical shift index51 predicted random coil structure along the length of the peptide; the lack of variation of CαH chemical shift for each residue type with sequence position meant a chemical shift index prediction could be made without complete assignments.
Identical NMR spectra were recorded in the presence of 50 μM silicic acid and Fig. 6D shows the one-dimensional spectra in the absence and presence of silicic acid for comparison. Because of the long time-scale of the NMR experiments, we empirically determined that 50 μM silicic acid was the highest concentration that could be used without samples gelling or precipitating. Collectively, the NMR and CD data both suggest that R5 is a monomeric random coil in solution and that soluble silicic acid does not induce the formation of secondary structure at the concentrations and pH values examined here. Our data thus confirm that primary amino acid sequence, rather than any peptide structure, is the critical factor in R5-driven silicification.
Dynamic light scattering
Dynamic light scattering (DLS) was also used to assess whether R5 spontaneously aggregated in solution. The hydrodynamic diameter of R5 solutions was determined across a range of peptide concentrations up to 3.7 mM. A single population of particles with ∼2 nm diameter (Fig. 7) was observed at all peptide concentrations, which was similar to the approximate sizes of the simulated peptide monomers (Fig. 4). This further supports the results from NMR (Fig. 6) which showed that R5 did not spontaneously aggregate in solution. These observations are in contrast to a previous study20 that detected R5 aggregates in DLS with a diameter of 825 nm. Interestingly, the introduction of even 50 μM silicic acid to the R5 solution gave rise to a dramatic change in the DLS readings, with the peptide peak at ∼2 nm being abolished and replaced with either a single peak at ∼1500 nm diameter (when calculating by particle volume) or a bimodal distribution with peaks at ∼200 and ∼800 nm diameter (when calculating by particle number). However, given that the NMR spectra were identical in the presence and absence of silicic acid, it seems likely that this size shift was either a DLS artefact caused by changes in solvent viscosity or that these large aggregates involve only a small fraction of the total peptide in solution. While this minor population would dominate the DLS signal, which scales with particle radius to the sixth power, they would be effectively invisible to NMR.Conclusions
Studies of R5-dependent silicification have previously been described at lower resolution, but to our knowledge this is the first detailed study of R5 structure and function. We find that R5 has no ordered secondary structure in solution and does not appear to assemble into discrete peptide aggregates. The structure and aggregation of the peptide appear to be unaffected by the addition of soluble silicic acid. In terms of promoting mineralization, we find that silica formation is saturable and cooperative with regard to peptide concentration above 0.5 mM R5. Silicification is linear with [H+] above a strict threshold value of ≥pH 6.4 that corresponds to a ratio of ∼2500:1 Si(OH)4:Si(OH3)O−. Our findings should inform the further applications of this peptide in vitro.
Acknowledgements
We thank Hannah Maple for collection of 600 MHz NMR spectra. This work was supported by European Research Council Starting Grant 282101 under the European Union's Seventh Framework Programme (FP7/2007–2013) to PC. LS is supported by Engineering and Physical Sciences Research Council Doctoral Training Centre grant EP/G036780/1.References
- C. L. Chen and N. L. Rosi, Angew. Chem., Int. Ed., 2010, 49(11), 1924–1942 CrossRef CAS PubMed.
- M. Sarikaya, C. Temerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS PubMed.
- R. R. Naik, P. W. Whitlock, F. Rodriguez, L. L. Brott, D. D. Glawe, S. J. Clarson and M. O. Stone, Chem. Commun., 2003, 238–239 RSC.
- M. R. Knecht and D. W. Wright, Langmuir, 2004, 20(11), 4728–4732 CrossRef CAS.
- D. J. Belton, S. V. Patwardhan, V. V. Annenkov, E. N. Danilovtseva and C. C. Perry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(16), 5963–5968 CrossRef CAS PubMed.
- Y. A. Shchipunov and T. Y. Karpenko, Langmuir, 2004, 20(10), 3882–3887 CrossRef CAS.
- M. Hildebrand, Chem. Rev., 2008, 108, 4855–4874 CrossRef CAS PubMed.
- N. Kröger and N. Poulsen, Annu. Rev. Genet., 2008, 42, 83–107 CrossRef PubMed.
- C. Drummond, R. McCann and S. V. Patwardhan, Chem. Eng. J., 2014, 244, 483–492 CrossRef CAS PubMed.
- S. V. Patwardhan, Chem. Commun., 2011, 47, 7567–7582 RSC.
- T. Mock, M. P. Samanta, V. Iverson, C. Berthiaume, M. Robison, K. Holtermann, C. Durkin, S. S. BonDurant, K. Richmond, M. Rodesch, T. Kallas, E. L. Huttlin, F. Cerrina, M. R. Sussman and E. V. Armbrust, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(5), 1579–1584 CrossRef CAS PubMed.
- R. P. Shrestha, B. Tesson, T. Norden-Krichmar, S. Federowicz, M. Hildebrand and A. E. Allen, BMC Genomics, 2012, 13, 499 CrossRef CAS PubMed.
- N. Kröger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129–1132 CrossRef.
- N. Kröger, R. Deutzmann and M. Sumper, J. Biol. Chem., 2001, 276(28), 26066–26070 CrossRef PubMed.
- N. Kröger, S. Lorenz, E. Brunner and M. Sumper, Science, 2002, 298(5593), 584–586 CrossRef PubMed.
- N. Poulsen, M. Sumper and N. Kröger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(21), 12075–12080 CrossRef CAS PubMed.
- N. Poulsen and N. Kröger, J. Biol. Chem., 2004, 279, 42993–42999 CrossRef CAS PubMed.
- N. Poulsen, A. Scheffel, V. C. Sheppard, P. M. Chesley and N. Kröger, J. Biol. Chem., 2013, 288(28), 20100–20109 CrossRef CAS PubMed.
- M. R. Knecht and D. W. Wright, Chem. Commun., 2003, 3038–3039 RSC.
- H. R. Luckarift, J. C. Spain, R. R. Naik and M. O. Stone, Nat. Biotechnol., 2004, 22, 211–213 CrossRef CAS PubMed.
- C. C. Lechner and C. F. W. Becker, Chem. Sci., 2012, 3, 3500 RSC.
- C. C. Lechner and C. F. W. Becker, Bioorg. Med. Chem., 2013, 21(12), 3533–3541 CrossRef CAS PubMed.
- W. D. Marner, A. S. Shaikh, S. J. Muller and J. D. Keasling, Biotechnol. Prog., 2009, 25(2), 417–423 CrossRef CAS PubMed.
- W. D. Marner, A. S. Shaikh, S. J. Muller and J. D. Keasling, Biomacromolecules, 2008, 9(1), 1–5 CrossRef CAS PubMed.
- D. H. Nam, K. Won, Y. H. Kim and B. I. Sang, Biotechnol. Prog., 2009, 25(6), 1643–1649 CAS.
- C. Wong Po Foo, S. V. Patwardhan, D. J. Belton, B. Kitchel, D. Anastasiades, J. Huang, R. R. Naik, C. C. Perry and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(25), 9428–9433 CrossRef PubMed.
- A. J. Mieszawska, L. D. Nadkami, C. C. Perry and D. L. Kaplan, Chem. Mater., 2010, 22(20), 5780–5785 CrossRef CAS PubMed.
- O. Choi, B. C. Kim, J. H. An, K. Min, Y. H. Kim, Y. Um, M. K. Oh and B. I. Sang, Enzyme Microb. Technol., 2011, 49(5), 441–445 CrossRef CAS PubMed.
- D. H. Nam, J. O. Lee, B. I. Sang, K. Won and Y. H. Kim, Appl. Biochem. Biotechnol., 2013, 170(1), 25–31 CrossRef CAS PubMed.
- M. S. Kent, J. K. Murton, F. J. Zendejas, H. Tran, B. A. Simmons, S. Satija and I. Kuzmenko, Langmuir, 2009, 25(1), 305–310 CrossRef CAS PubMed.
- C. C. Lechner and C. F. W. Becker, J. Pept. Sci., 2014, 20, 152–158 CrossRef CAS.
- A. Roehrich and G. Drobny, Acc. Chem. Res., 2013, 46(9), 2136–2144 CrossRef CAS PubMed.
- G. B. Alexander, J. Am. Chem. Soc., 1953, 75(12), 2887–2888 CrossRef CAS.
- T.-L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112 Search PubMed.
- M. Sumper and N. Kröger, J. Mater. Chem., 2004, 14, 2059–2065 RSC.
- F. Rodriguez, D. D. Glawe, R. Naik, K. P. Hallinan and M. O. Stone, Biomacromolecules, 2004, 5, 261–265 CrossRef CAS PubMed.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS PubMed.
- M. Sumper and E. Brunner, Adv. Funct. Mater., 2006, 16, 17–26 CrossRef CAS.
- P. Blondeau, M. Segura, R. Pérez-Fernández and J. de Mendoza, Chem. Soc. Rev., 2006, 36, 198–210 RSC.
- M. Sumper, S. Lorenz and E. Brunner, Angew. Chem., Int. Ed., 2003, 42, 5192–5195 CrossRef CAS PubMed.
- A. Bernecker, R. Wieneke, R. Riedel, M. Seibt, A. Geyer and C. Steinem, J. Am. Chem. Soc., 2010, 132, 1023–1031 CrossRef CAS PubMed.
- K. M. Hawkins, S. S.-S. Wang, D. M. Ford and D. F. Shantz, J. Am. Chem. Soc., 2004, 126(29), 9112–9119 CrossRef CAS PubMed.
- S. C. Holmström, P. J. King, M. G. Ryadnov, M. F. Butler, S. Mann and D. N. Woolfson, Langmuir, 2008, 24(20), 11778–11783 CrossRef PubMed.
- V. M. Yuwono and J. D. Hartgerink, Langmuir, 2007, 23(9), 5033–5038 CrossRef CAS PubMed.
- H. Xu, Y. Wang, X. Ge, S. Han, S. Wang, P. Zhou, H. Shan, X. Zhao and J. R. Lu, Chem. Mater., 2010, 22(18), 5165–5173 CrossRef CAS.
- S. V. Patwardhan, R. Maheshwari, N. Mukherjee, K. L. Kiick and S. J. Clarson, Biomacromolecules, 2006, 7, 491–497 CrossRef CAS PubMed.
- J. T. Jones, J. Mol. Biol., 1999, 292, 195–202 CrossRef PubMed.
- J. Maupetit, P. Derreumaux and P. Tuffery, Nucleic Acids Res., 2009, 37, W498–W503 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- N. E. Hafsa and D. S. Wishart, J. Biomol. NMR, 2014, 60(2–3), 131–146 CrossRef CAS PubMed.
- W. L. DeLano, http://www.pymol.org, 2002.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01679c |
| ‡ Current address: Department of Chemistry, King's College, London, SE1 1DB, UK. |
| This journal is © The Royal Society of Chemistry 2015 |