
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of protein imprinted polymer beads by Pickering emulsion polymerization†
Tongchang
Zhou
ac,
Ka
Zhang
a,
Tripta
Kamra
ab,
Leif
Bülow
a and
Lei
Ye
*a
aDivision of Pure and Applied Biochemistry, Lund University, Box 124, 221 00 Lund, Sweden. E-mail: lei.ye@tbiokem.lth.se; Fax: +46 46 2224611; Tel: +46 46 2229560
bDivision of Synchrotron Radiation Research, Lund University, Box 118, 221 00 Lund, Sweden
cGuangzhou Pharmaceutical Holdings, Ltd., Guangzhou 510130, China
First published on 17th December 2014
Abstract
We present a new method for preparation of protein-specific polymer beads based on surface molecular imprinting in Pickering emulsion. In the first step, adult human hemoglobin (Hb) was adsorbed on silica nanoparticles. The protein-coated silica particles were then used to stabilize an oil-in-water emulsion (Pickering emulsion) composed of cross-linking monomer in the oil phase. After free radical polymerization of the oil phase, the protein-silica particles were removed to leave Hb-imprinted sites on the polymer surface. The protein-imprinted polymer microspheres were characterized by scanning electron microscopy and their selectivity was investigated by protein binding analysis. The new synthetic method based on Pickering emulsion polymerization produced easily accessible Hb binding sites on the surface of spherical polymer particles, which are useful for protein separation, purification and analysis.
Introduction
Molecular imprinting is a versatile technique for preparing synthetic polymers with specific recognition property for molecular targets.1,2 Due to their low cost and high stability, molecularly imprinted polymers (MIPs) can be used as substitutes for biological macromolecules in applications covering affinity separations, enzyme catalysis and protein crystallization.3–7 To date, molecular imprinting has been successful mainly for small molecules.8–10 In the case of biological macromolecules such as proteins, polysaccharides and nucleic acids, the use of molecular imprinting to create recognition materials for these biomacromolecules has been more challenging. This is largely due to the large molecular size and conformational diversity of these entities. Furthermore, biomacromolecules are often sensitive to both pH and temperature changes, and can be easily denatured under the harsh polymerization conditions.Because of the enormous demand for protein-selective materials in the areas of biology and life science, there have been some successful attempts to prepare protein imprinted polymers via different strategies, such as through the sol–gel process,11 free-radical polymerization,12 and other methods.13 Most of these studies have used a low crosslinking density to enable protein diffusion,14,15 in particular in hydrophilic polyacrylamide gels.16,17 One technical issue, however, is that MIPs prepared by these approaches are soft and have low mechanical strength, making their application limited and the MIPs unsuitable for repeated use, e.g. as stationary phases in chromatography separation.13 Thus, in order to achieve protein selective materials with high mechanical stability, new synthetic methods need to be developed.
Recently, MIPs carrying surface molecularly imprinted sites have been prepared as promising affinity materials for protein separation and analysis.18–23 In surface molecular imprinting, silica beads, glass slides and silica-modified magnetic nanoparticles are usually used as substrate to support a thin layer of MIP film, for which the thickness of the MIP film is controlled to allow protein binding sites to be accessible. In this way the protein template can be removed more easily from the MIP, and the kinetics of protein binding in intended applications can be accelerated. Although different surface imprinting methods have shown promising results,24–27 most of the reported methods use complicated and time-consuming steps to present protein template at the reactive interface in a heterogeneous imprinting system.
In our recent studies, we have demonstrated that nanoparticle-stabilized emulsions (Pickering emulsions) offer a versatile system for preparing MIP microspheres.28–31 In addition to MIP beads selective for small organic molecules, we have shown that hydrophilic MIP beads containing protein-imprinted sites can be synthesized by Pickering emulsion polymerization.32 In the previous protein-imprinting work, we used silica nanoparticles to stabilize the water-in-oil emulsion, where the water phase contained functional monomer and the protein template. The hydrophilic MIP beads, although showed high protein selectivity, had low mechanical strength due to the low crosslinking density in the interior of the polymer particles. In this work, we show that by presenting the protein template on the surface of the stabilizing nanoparticles, it is feasible to synthesize protein imprinted sites on MIP surfaces. This new method involves the use of protein coated silica as the stabilizing particles to establish an oil-in-water Pickering emulsion. The oil phase contains crosslinking monomer and initiator, and the functional monomer that interacts with the protein template is enriched at the oil–water interface due to the protein template (Fig. 1). Herein we used Hb as a model protein template, since it has important medical relevance and is a common model for studying protein–MIP interactions in the literature.21,23,32 This interfacial protein imprinting leads to the formation of protein recognition sites on the surface of cross-linked polymer beads. The advantage of this synthetic method resides in its general applicability and being able to be scaled up for preparation of large quantities of protein selective MIPs.
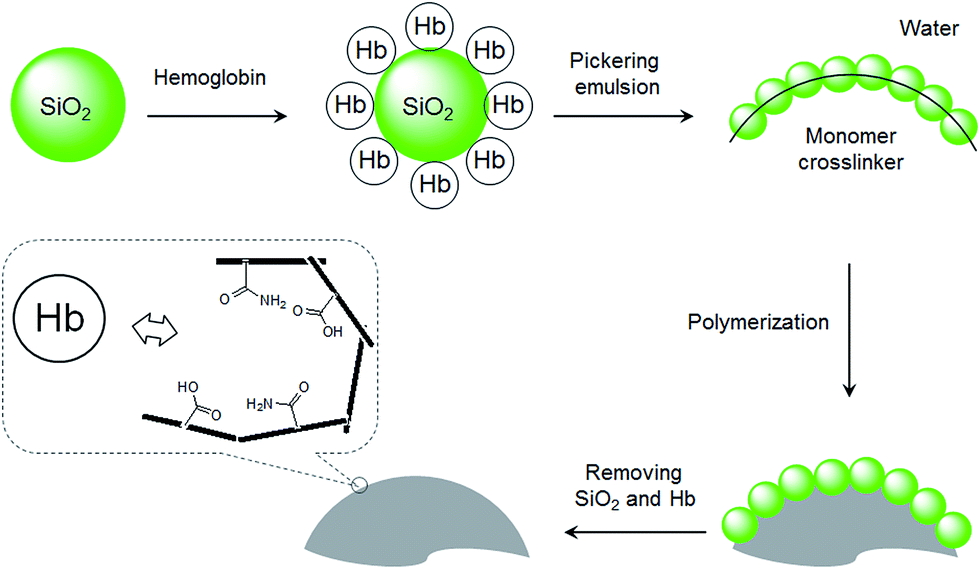 | ||
| Fig. 1 Schematic illustration of the interfacial protein imprinting process. | ||
Material and methods
Materials
Silica nanoparticles (diameter 10 nm), acrylamide (Am, ≥98%), ethylene glycol dimethacrylate (EGDMA, 98%), adult human hemoglobin (Hb, MW 65 kDa, pI 6.8–7.0), bovine serum albumin (BSA, MW 69 kDa, pI 4.9), myoglobin (Mb, MW 17.5 kDa, pI 6.8–7.2), lysozyme (Lyz, MW 14.3 kDa, pI 6.99) and ovalbumin (OVA, albumin from egg, MW 43.0 kDa, pI 4.5) were purchased from Sigma-Aldrich. Methacrylic acid (MAA, 98.5%), azobisisobutyronitrile (AIBN, 98%), sodium dodecyl sulphate (SDS, 98%) and Triton X-100 (99.5%) were obtained from Merck (Darmstadt, Germany). Other solvents and inorganic salts were of analytical reagent grade and were used without further purification. Polyacrylamide gels (12%, Mini-protean 3) used in SDS-PAGE were obtained from Bio-Rad.Preparation of MIP beads by Pickering emulsion polymerization
Protein imprinted polymer beads were prepared by Pickering emulsion polymerization. The Pickering emulsion was composed of an oil phase dispersed in an aqueous phase, which was stabilized by protein coated silica nanoparticles. The water phase was first prepared as the following: 10 mg of Hb was dissolved in 3 mL of water before 10 mg silica nanoparticles were added. The suspension was gently stirred on a rocking table for 1 h. After centrifugation, the supernatant was removed. The recovered silica nanoparticles coated with the Hb (∼3 mg, estimated from the Hb in the supernatant) were then mixed with 3 mL water, followed by addition of the functional monomer. When only Am was used as the functional monomer, 100 mg Am was added. When only MAA was used as the functional monomer, 136 μL MAA and 250 μL NaOH (3 M) were added. When both Am and MAA were used as the functional monomers, a mixture of Am (100 mg), MAA (136 μL) and NaOH (3 M, 250 μL) were added. The oil phase was prepared by mixing toluene (100 μL), EGDMA (400 μL, 2.1 mmol) and AIBN (10 mg). At last the two phases were mixed and shaken vigorously by hand for 1 min to give a stable Pickering emulsion. Polymerization was incurred by setting the Pickering emulsion in an oven at 70 °C for 16 h. After polymerization, the solid beads were collected and washed with methanol for 2 times. For removal of the silica nanoparticles, the composite beads were transferred into a plastic tube and stirred in a mixture of methanol (30 mL) and HF (30%, 1 mL) at room temperature for 1 h. Further template removal was ensured by washing the polymer beads with water containing 10% acetic acid and 5% SDS for 6 times. Finally the polymer beads were washed with water for 6 times before further drying in a depressurized desiccator. As a reference polymer, non-imprinted polymer (NIP) beads were prepared in the same way as the MIP beads, except that the Hb in the water phase was replaced by Triton X-100 (10 mg). Note: silica nanoparticles alone did not give a stable emulsion.Surface characterization of the MIP beads
A scanning electron microscope (Thermal Field Emission SEM LEO 1560, Zeiss, Oberkochen, Germany) was used to observe the surface morphology of the MIP beads.Protein binding analysis
Polymer particles (5 mg) were placed into 1 mL phosphate buffer (20 mM) containing different amount of proteins. The mixture was gently stirred at room temperature for 16 h. After centrifugation, about 700 μL of supernatant was collected. The concentrations of the proteins were measured with a DU 800 spectrophotometer (Beckman Coulter). Mb, OVA, BSA and Lyz were selected as reference proteins to investigate the selectivity of the MIP beads. The UV detection wavelengths were fixed at 405 nm and 408 nm for Hb and Mb, and at 278 nm for OVA, BSA and Lyz, respectively.In competitive protein binding experiments, polymer particles (5 mg) were stirred with 1 mL of protein mixture in phosphate buffer (containing 0.2 mg Hb and 2 mg of other competing proteins). After incubation at room temperature for 16 h and centrifugation, 10 μL of supernatant was collected and analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Protein binding kinetics
To investigate the kinetics of protein binding, polymer particles (5 mg) were mixed with 1 mL Hb buffer solution with an initial concentration of 0.2 mg mL−1. At different time intervals, the concentration of Hb in the solution was determined using a spectrophotometer. The tests were conducted in duplicate.Results and discussion
Synthesis of polymer beads by interfacial molecular imprinting
As shown in Fig. 1, the protein template Hb was presented at the oil–water interface during the imprinting reaction. The protein template was non-covalently adsorbed on the surface of the silica nanoparticles, which turned the silica nanoparticles to become more suitable to stabilize the oil-in-water emulsion. Compared to earlier protein imprinting using surface-immobilized template,18,20,33 the present method allows a larger number of protein molecules to act as macromolecular template due to the significantly larger area of the oil–water interface. In the literature, polyacrylamide is known to be biocompatible and acrylamide has been used as a main functional monomer for protein imprinting.34 Other functional monomers, such as methacrylic acid,12 dopamine21 and N-[3-(dimethylamino)propyl]-methacrylamide35 have also been used for protein imprinting. The choice of monomer is critical for successful protein imprinting, as it has a great impact on the adsorption capacity of the MIP. Moreover, it affects the non-specific protein binding under different solvent conditions. Since both MAA and Am previously have given successful Hb imprinted polymers,36 in this study we selected to use MAA, Am, and their binary mixture as functional monomers. The Pickering emulsion polymerization resulted in cross-linked polymer beads bearing different types and amounts of the functional monomers, depending on the strength of the monomer–protein interactions at the oil–water interface. During the protein imprinting reaction, the pH of the Pickering emulsion was kept at ∼6 in order to obtain a stable emulsion. Also, at this pH value we expected the Hb template (with an isoelectric point (pI) of 6.8–7.0) to attract the functional monomer MAA (pKa ≈ 4.66) through ionic interaction and Am via hydrogen bonding. After polymerization, the silica nanoparticles were removed by chemical dissolution in dilute HF, and any remaining protein template was removed by exhaustive washing. The morphologies of the MIP and the NIP are shown in Fig. 2. The average particle sizes of the MIP and the NIP beads were estimated to be 25 ± 8 μm and 122 ± 34 μm, respectively. The difference in particle size between the MIP and the NIP is due to the different surface-active molecules used during the Pickering emulsion polymerization. The surface bound Hb was more efficient than Triton X-100 in minimizing the surface energy (Fig. S1†), leading to smaller Hb-imprinted polymer beads. Fig. 2a and b shows the SEM images of Hb-imprinted poly(MAA-co-Am-co-EGDMA) beads and Fig. 2e and f are the SEM images for non-imprinted poly(MAA-co-Am-co-EGDMA) beads. | ||
| Fig. 2 Hb imprinted poly(MAA-co-Am-co-EGDMA) beads before (a and b) and after (c and d) protein binding experiment. Non-imprinted poly(MAA-co-Am-co-EGDMA) beads before (e and f) and after (g and h) protein binding experiment. The protein binding experiment was carried out at pH 5.4. | ||
Protein binding characteristics
The characteristic of protein binding was first studied by comparing Hb adsorption on the imprinted and the reference polymer beads. In this work, we prepared the non-imprinted polymer beads using a nonionic surfactant Triton X-100 to replace the Hb protein in the Pickering emulsion. The NIP beads therefore contain no protein imprinted sites on their surface but otherwise have morphology and functional groups similar to the imprinted beads. Despite the different sizes of the MIP and the NIP beads, we measured Hb adsorption on the polymer beads, and used the differential binding between the MIP and the NIP to evaluate the effect of the protein imprinting.As shown in Fig. 3, when MAA and Am were used separately as the functional monomer, the obtained MIP and the corresponding NIP beads showed very little Hb binding. Only when both MAA and Am were used as the functional monomers, the obtained MIP beads displayed significantly higher Hb binding than the NIP beads. This result is in agreement with previous findings that multiple functional monomers often lead to successful protein imprinted polymers.16,37 In this work, although MAA and Am have somewhat different solubility in the water and the oil phases, we suggest that the final population of the carboxyl and amide functional groups on the surface of the imprinted polymer beads is determined by the strength of the interaction between each monomer and the protein template.
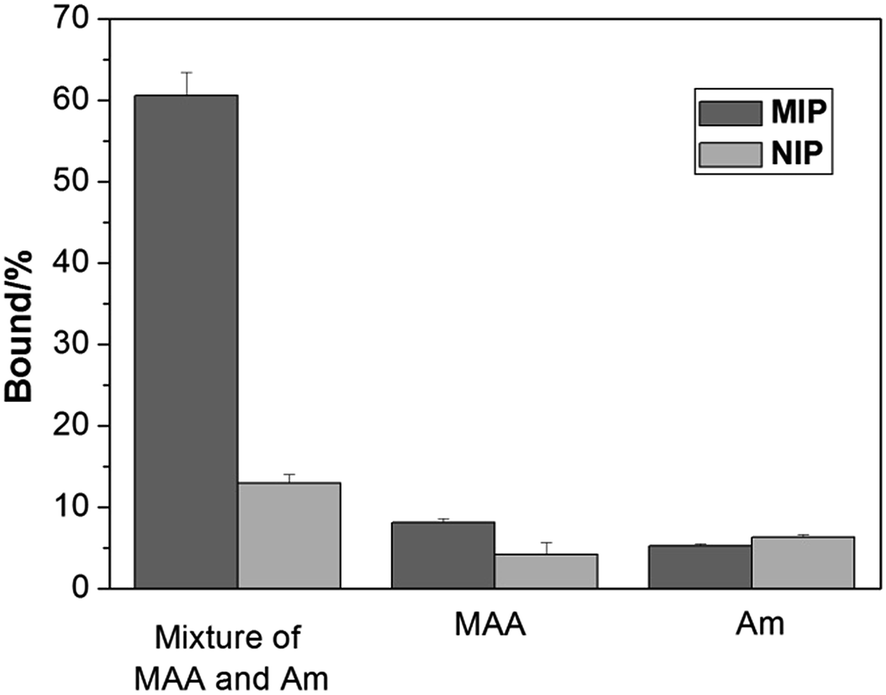 | ||
| Fig. 3 Hb binding to the polymer beads prepared using different functional monomers at pH 5.4. The concentrations of Hb and polymer beads were 0.2 mg mL−1 and 5 mg mL−1, respectively. | ||
From Fig. 2c and d, it is clear that after protein binding, the previously smooth surface of the MIP beads became rough, and the apparent protein aggregates were present on the MIP beads, which may be explained as a result of protein–protein interactions under the experimental condition, where the first layer of the bound Hb attracted further Hb molecules from the solution. In the case of the NIP beads (Fig. 2g and h), no obvious Hb aggregates were observed due to the lack of Hb binding sites in the first place. We should note however that the protein aggregates in Fig. 2c and d were observed under dehydrated condition, therefore it may not represent how the actual protein–protein complexes look like in solution.
Isoelectric point (pI) is an important parameter affecting protein binding. Protein will become charged at pH values lower or higher than its pI due to the acceptance or loss of protons (H+). The net charge of proteins will determine the electrostatic interaction of the protein with a charged surface, therefore it is important to investigate the impact of pH on protein binding with the imprinted polymers. Fig. 4 shows the effect of pH on Hb binding to the imprinted and the non-imprinted poly(MAA-co-Am-co-EGDMA) beads.
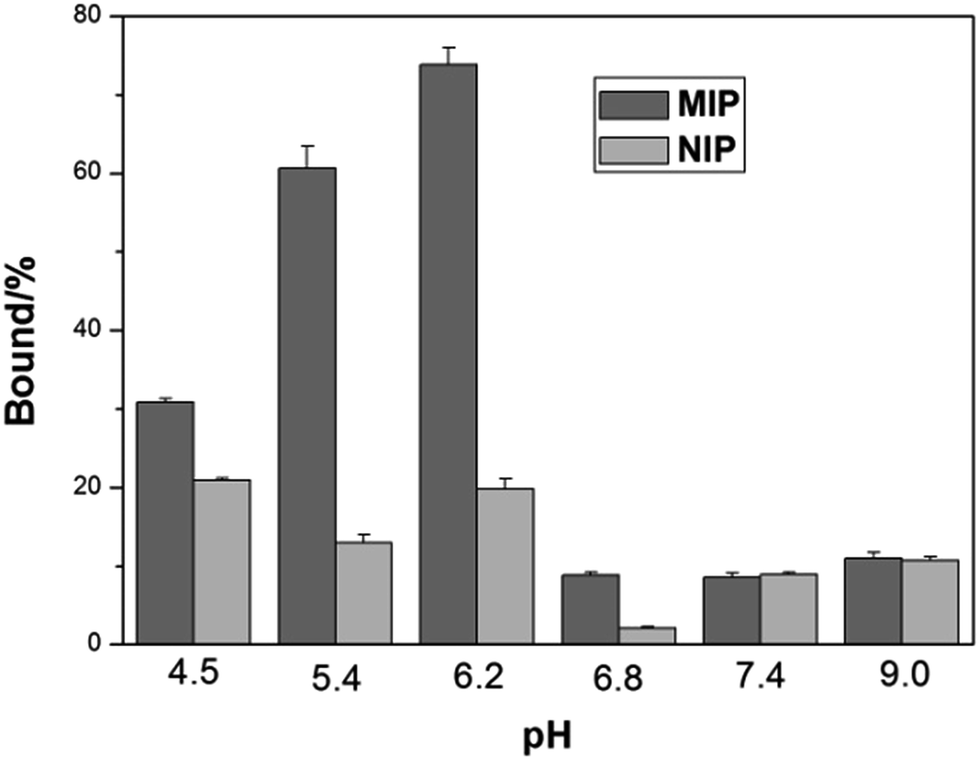 | ||
| Fig. 4 Effect of pH on Hb binding to poly(MAA-co-Am-co-EGDMA) beads. The concentrations of Hb and polymer beads were 0.2 mg mL−1 and 5 mg mL−1, respectively. | ||
It is interesting to see that the optimal Hb binding is not achieved at the pI of Hb (6.8–7.0), but at pH 5.4–6.2. This can be explained by that the MIP beads were prepared at pH ∼ 6, at which the functional monomers form the strongest interaction with the protein template, in agreement with previous observations.38 At pH higher than 6.8, the MIP beads become negatively charged leading to very low adsorption of the negatively charged Hb. On the other hand, when the pH changed to 4.5, the MIP beads start to become neutral, and the lack of ionic interactions lead to decreased protein binding, accompanied by an increased non-specific adsorption represented by the NIP beads. As pH 5.4 results in lower non-specific Hb binding than pH 6.2, we carried out the remaining protein binding experiments in pH 5.4 buffer.
Fig. 5 shows the binding isotherm of Hb on the imprinted and non-imprinted poly(MAA-co-Am-co-EGDMA) beads measured at pH 5.4. While the NIP beads show a characteristic non-specific adsorption, the MIP beads display saturated binding at protein concentration above 0.2 mg mL−1. The maximum capacity of the MIP is estimated to be 25 mg g−1, with an apparent dissociation constant of 3.8 × 10−8 M. The capacity of the present MIP is more than 5 times of a previously reported hemoglobin MIP grafted on silica-modified magnetic nanoparticles,21 and exceeds what can be expected from the template loading used in the imprinting reaction. This apparently high protein binding can be explained by possible Hb–Hb interaction, as we have discussed previously for Fig. 2c and d.
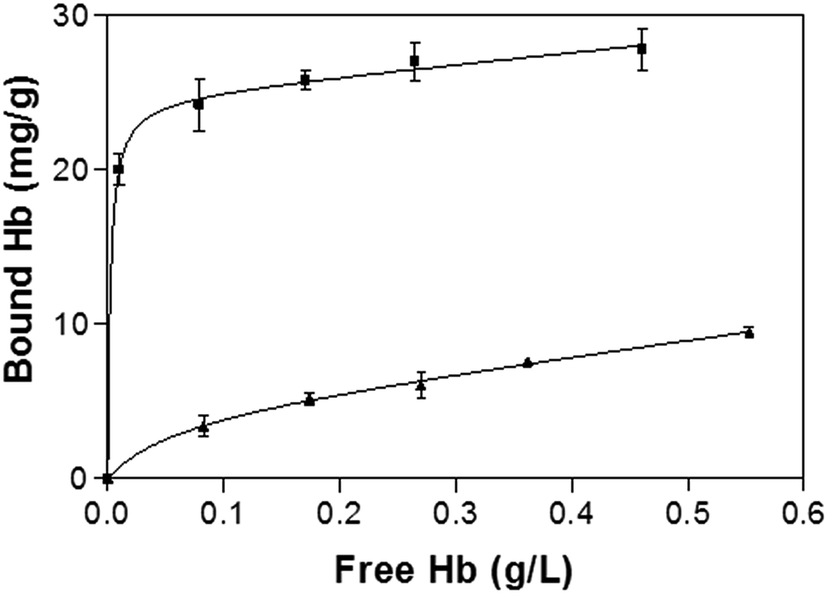 | ||
| Fig. 5 Hb binding isotherm measured at pH 5.4. The concentration of polymer beads was 5 mg mL−1. The amount of bound Hb was calculated as the difference between the total and the free Hb. | ||
To prove that the interfacial protein imprinting indeed leads to surface-exposed protein binding sites, we carried out kinetic Hb binding experiments. As seen in Fig. 6, Hb binding to the MIP beads reaches equilibrium within 1 h, and 80% of the binding takes place within 30 min, indicating that the new protein recognition sites are indeed located on the particle surface. In addition, since the core of the MIP beads have a high crosslinking density, it is unlikely that any protein binding will take place in the interior of the particles. The high cross-linking density in the interior also provide the MIP beads with a high mechanical strength, which is beneficial for material regeneration and for chromatography separation purposes. Compared to the MIP beads, the NIP beads show significantly lower but faster Hb binding, which can be explained as a result of non-selective adsorption. Obviously, large protein molecules require longer time to access the imprinted sites in the MIP than to simply adsorb on the NIP surface.
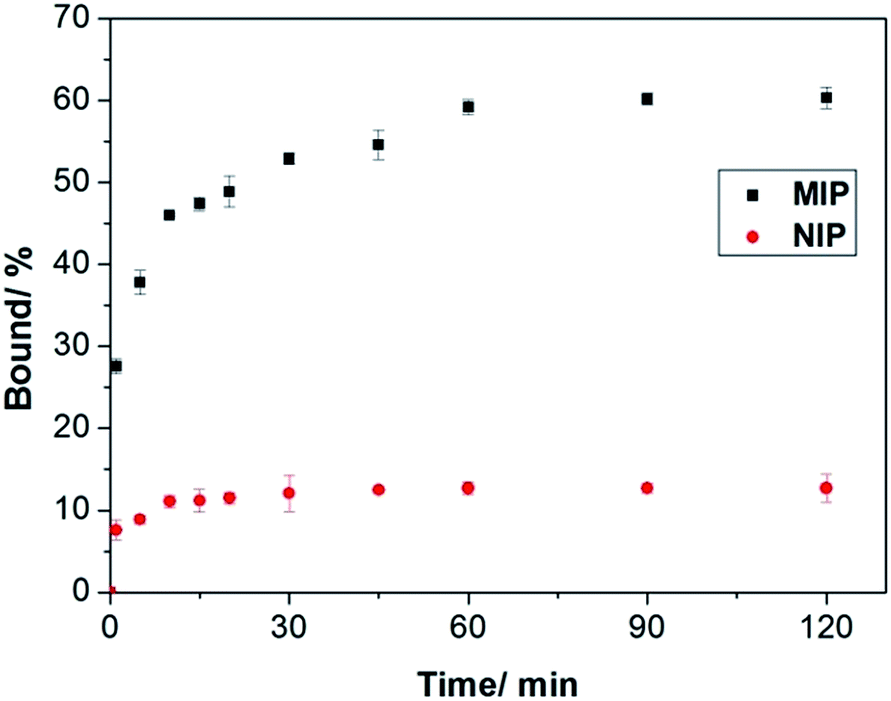 | ||
| Fig. 6 Kinetics of Hb binding to the poly(MAA-co-Am-co-EGDMA) beads. The concentrations of Hb and polymers were 0.2 mg mL−1 and 5 mg mL−1, respectively. | ||
Protein selectivity of Hb imprinted polymer
To investigate the protein selectivity of the Hb-imprinted poly(MAA-co-AM-co-EGDMA) beads, the adsorption of several proteins on the MIP and NIP beads was examined. These tested proteins are different from Hb either in molecular size or pI, and provide useful insight for understanding the main factors affecting the protein selectivity. As shown in Fig. 7, the MIP beads display significantly higher Hb binding (60.6%) than all the other proteins (10–30%). This result confirms that the MIP beads have an unambiguous selectivity for the template protein. Except for OVA, all proteins exhibit low binding to the NIP, which is desirable for minimizing non-specific protein adsorption. The relatively high OVA binding to the NIP may be caused by the polysaccharide chains on this glycoprotein, which can form multiple interactions with the polymer surface to increase protein adsorption. In the case of the MIP, the low pI (4.5) and the small size of OVA make it difficult to fit snugly in the Hb-imprinted sites, which lead to reduced OVA binding to the MIP. These results confirm that the selectivity of the MIP for Hb is contributed by the imprinted sites rather than by randomly distributed carboxyl or amide groups on the polymer surface.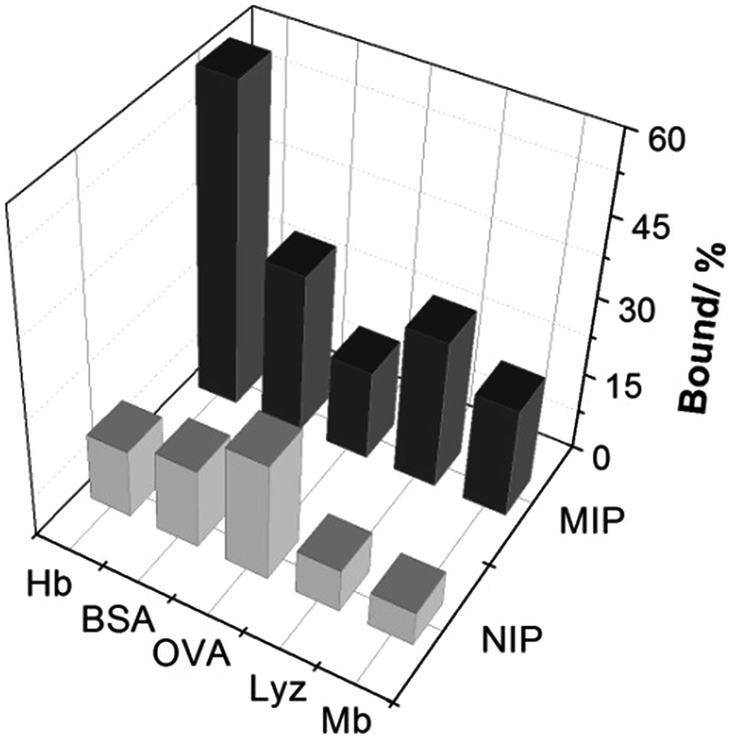 | ||
| Fig. 7 Uptake of Hb and other test proteins (0.2 mg mL−1) by 5 mg mL−1 poly(MAA-co-Am-co-EGDMA) beads. | ||
The high selectivity of the Hb-imprinted polymer is intriguing for practical protein separations. To demonstrate the potential of the MIP beads, a competitive protein depletion experiment has been carried out, where the MIP beads have been utilized to remove Hb in the presence of an excess of interfering proteins. Among the three interfering proteins used here, BSA (69 kDa) is similar in size to Hb (65 kDa). The difference is in their pI values, and that Hb is composed of four polypeptide chains with an overall biconcaval shape, whereas BSA consists of one polypeptide with an ellipsoidal form.34 Mb is a monomeric protein with a structure and pI similar to that of the individual subunit of Hb (alpha and beta subunits, ∼15 kDa). Therefore Mb is an ideal model to study the impact of molecular size on protein binding. After incubating the MIP beads in a solution of Hb and 10-fold excess of each of the interfering proteins (BSA, OVA and Mb), the proteins remaining in solution were analysed by SDS-PAGE. As shown in Fig. 8, in all cases the MIP beads successfully depleted Hb, as the protein band corresponding to Hb disappeared almost completely (Fig. 8a, Lanes 3 and 6). Interestingly, the MIP was able to deplete Hb even in the presence of excess Mb (Fig. 8b, Lanes 3 and 4), indicating the dependence of the selectivity on shape and size. These results indicate clearly that the protein imprinted polymer beads possess the desired molecular selectivity useful for practical protein separation and analysis.
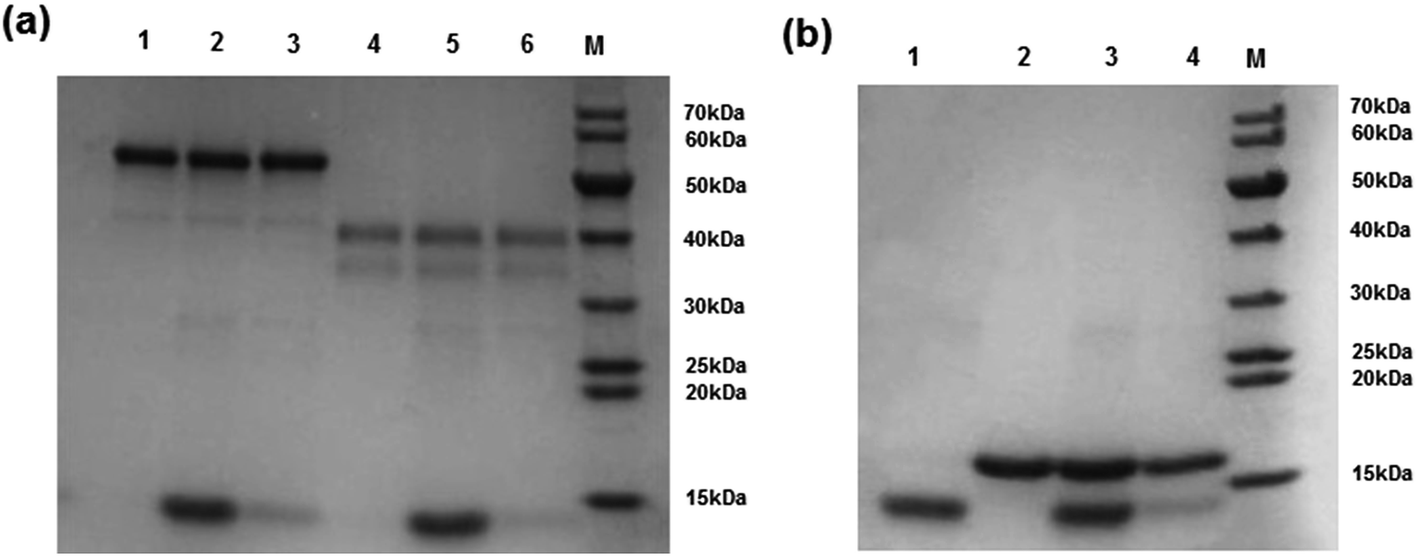 | ||
| Fig. 8 SDS-PAGE of protein solution after treatment with Hb-imprinted poly(MAA-co-Am-co-EGDMA) beads (5 mg mL−1). The protein solution contained Hb (0.2 mg mL−1) and one of the interfering proteins (2 mg mL−1). (a) Depletion of Hb in the presence of BSA or OVA. Lane 1: BSA standard; Lane 2: solution of Hb and BSA before depletion with the MIP; Lane 3: solution of Hb and BSA after depletion with the MIP; Lane 4: OVA standard; Lane 5: solution of Hb and OVA before depletion with the MIP; Lane 6: solution of Hb and OVA after depletion with the MIP; Lane M: molecular weight markers. (b) Depletion of Hb in the presence of Mb. Lane 1: Hb standard; Lane 2: Mb standard; Lane 3: solution of Hb and Mb before depletion with the MIP; Lane 4: solution of Hb and Mb after depletion with the MIP; Lane M: molecular weight markers. | ||
The protein imprinting conditions used in this work deserve further discussion. Although protein can be denatured at elevated temperature, it is possible that the surface-bound Hb remained intact at the beginning of the polymerization and acted as native protein template. From the literature, it is known that the stability of enzymes can be improved by immobilization.39,40 In addition, proteins have been shown to remain their native structure after short contact with a high temperature surface.41 We also found that the Hb eluted from the MIP beads after the binding experiments maintained its biological activity as determined by spectroscopic characterization (Fig. S2†), suggesting that the MIP does not cause protein unfolding. Therefore, the new protein imprinting method can be potentially developed to create macromolecular receptors for other types of proteins.
Conclusions
In this work, we developed a new interfacial protein imprinting technique to synthesize protein selective polymer beads using Pickering emulsion polymerization. The imprinted polymer beads have selective protein binding sites on surface and display fast and selective protein recognition. The synthetic approach involves coating silica nanoparticles with the protein template, using the obtained nanoparticles to stabilize monomer emulsion in water, polymerizing the monomer phase followed by removal of the stabilizing nanoparticles. Using Hb as a model, we demonstrated that the imprinted polymer beads prepared via the Pickering emulsion polymerization have protein selective binding sites on their surface. The high selectivity of the protein imprinted beads and their high stability are attractive for a number of applications involving bioseparation processes, for example for protein purification and selective depletion of abundance proteins in proteomics research.Acknowledgements
This work was supported by the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS, 212-2013-1305), the Danish Council for Strategic Research (project FENAMI, DSF-10-93456) and the Crafoord Foundation.References
- Z. Y. Chen, Z. D. Hua, L. Xu, Y. Huang, M. P. Zhao and Y. Z. Li, J. Mol. Recognit., 2008, 21, 71–77 CrossRef CAS PubMed.
- S. Ambrosini, M. Serra, S. Shinde, B. Sellergren and E. De Lorenzi, J. Chromatogr. A, 2011, 1218, 6961–6969 CrossRef CAS PubMed.
- G. Wulff, Chem. Rev., 2002, 102, 1–27 CrossRef CAS PubMed.
- R. Gupta and A. Kumar, Biotechnol. Adv., 2008, 26, 533–547 CrossRef CAS PubMed.
- B. Sellergren, Nat. Chem., 2010, 2, 7–8 CrossRef CAS PubMed.
- E. Saridakis, S. Khurshid, L. Govada, Q. Phan, D. Hawkins, G. V. Crichlow, E. Lolis, S. M. Reddy and N. E. Chayen, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 11081–11086 CrossRef CAS PubMed.
- M. J. Whitcombe, Nat. Chem., 2011, 3, 657–658 CrossRef CAS PubMed.
- H. Sambe, K. Hoshina and J. Haginaka, J. Chromatogr. A, 2007, 1152, 130–137 CrossRef CAS PubMed.
- L. J. Fang, S. J. Chen, Y. Zhang and H. Q. Zhang, J. Mater. Chem., 2011, 21, 2320–2329 RSC.
- Y. G. Zhao, X. H. Chen, S. D. Pan, H. Zhu, H. Y. Shen and M. C. Jin, J. Mater. Chem. A, 2013, 1, 11648–11658 CAS.
- S. Fireman-Shoresh, I. Turyan, D. Mandler, D. Avnir and S. Marx, Langmuir, 2005, 21, 7842–7847 CrossRef CAS PubMed.
- S. A. Yu, A. Q. Luo, D. Biswal, J. Z. Hilt and D. A. Puleo, Talanta, 2010, 83, 156–161 CrossRef CAS PubMed.
- X. S. Pang, G. X. Cheng, R. S. Li, S. L. Lu and Y. H. Zhang, Anal. Chim. Acta, 2005, 550, 13–17 CrossRef CAS PubMed.
- A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699–14702 CrossRef CAS PubMed.
- S. M. Reddy, Q. T. Phan, H. El-Sharif, L. Govada, D. Stevenson and N. E. Chayen, Biomacromolecules, 2012, 13, 3959–3965 CrossRef CAS PubMed.
- S. H. Ou, M. C. Wu, T. C. Chou and C. C. Liu, Anal. Chim. Acta, 2004, 504, 163–166 CrossRef CAS.
- D. M. Hawkins, A. Trache, E. A. Ellis, D. Stevenson, A. Holzenburg, G. A. Meininger and S. M. Reddy, Biomacromolecules, 2006, 7, 2560–2564 CrossRef CAS PubMed.
- H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS PubMed.
- C. J. Tan and Y. W. Tong, Anal. Chem., 2007, 79, 299–306 CrossRef CAS PubMed.
- D. R. Kryscio and N. A. Peppas, Anal. Chim. Acta, 2012, 718, 109–115 CrossRef CAS PubMed.
- X. P. Jia, M. L. Xu, Y. Z. Wang, D. Ran, S. Yang and M. Zhang, Analyst, 2013, 138, 651–658 RSC.
- H. Zhang, J. Jiang, H. Zhang, Y. Zhang and P. Sun, ACS Macro Lett., 2013, 2, 566–570 CrossRef CAS.
- W. Chen, W. Lei, M. Xue, F. Xue, Z. H. Meng, W. B. Zhang, F. Qu and K. J. Shea, J. Mater. Chem. A, 2014, 2, 7165–7169 CAS.
- T. Shiomi, M. Matsui, F. Mizukami and K. Sakaguchi, Biomaterials, 2005, 26, 5564–5571 CrossRef CAS PubMed.
- F. Bonini, S. Piletsky, A. P. F. Turner, A. Speghini and A. Bossi, Biosens. Bioelectron., 2007, 22, 2322–2328 CrossRef CAS PubMed.
- C. J. Tan, H. G. Chua, K. H. Ker and Y. W. Tong, Anal. Chem., 2008, 80, 683–692 CrossRef CAS PubMed.
- N. Sankarakumar and Y. W. Tong, RSC Adv., 2013, 3, 1519–1527 RSC.
- X. T. Shen and L. Ye, Chem. Commun., 2011, 47, 10359–10361 RSC.
- X. T. Shen and L. Ye, Macromolecules, 2011, 44, 5631–5637 CrossRef CAS PubMed.
- X. T. Shen, C. G. Xu and L. Ye, Soft Matter, 2012, 8, 7169–7176 RSC.
- T. C. Zhou, X. T. Shen, S. Chaudhary and L. Ye, J. Appl. Polym. Sci., 2014, 131, 39606 Search PubMed.
- X. T. Shen, T. C. Zhou and L. Ye, Chem. Commun., 2012, 48, 8198–8200 RSC.
- H. Q. Shi, W. B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593–597 CrossRef CAS PubMed.
- T. Y. Guo, Y. Q. Xia, G. J. Hao, M. D. Song and B. H. Zhang, Biomaterials, 2004, 25, 5905–5912 CrossRef CAS PubMed.
- Z. D. Hua, Z. Y. Chen, Y. Z. Li and M. P. Zhao, Langmuir, 2008, 24, 5773–5780 CrossRef CAS PubMed.
- L. Tan, C. C. Rang, S. Y. Xu and Y. W. Tang, Biosens. Bioelectron., 2013, 48, 216–223 CrossRef CAS PubMed.
- O. Y. Tov, S. Luvitch and H. Bianco-Peled, J. Sep. Sci., 2010, 33, 1673–1681 CrossRef CAS PubMed.
- A. Uysal, G. Demirel, E. Turan and T. Caykara, Anal. Chim. Acta, 2008, 625, 110–115 CrossRef CAS PubMed.
- A. M. Klibanov, Anal. Biochem., 1979, 93, 1–25 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- S. B. N. Biancardo, H. J. Pranov and N. B. Larsen, Adv. Mater., 2008, 20, 1825–1829 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01605j |
| This journal is © The Royal Society of Chemistry 2015 |