
Effects of fluorine on the structure of fluorohydroxyapatite: a study by XRD, solid-state NMR and Raman spectroscopy†
Jinshuai
Chen‡
ab,
Zhiwu
Yu‡
c,
Peizhi
Zhu
*ab,
Junfeng
Wang
c,
Zhehong
Gan
d,
Jie
Wei
e,
Yinghui
Zhao
f and
Shicheng
Wei
*f
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Jiangsu, P.R. China 225009. E-mail: pzzhu@yzu.edu.cn
bJiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou, 225009, P.R. China
cHigh Magnetic Field Laboratory, Hefei Institutes of Physical Science, Chinese Academy of Sciences, 350 Shushanhu Road, Hefei 230031, Anhui Province, P.R. China
dCenter of Interdisciplinary Magnetic Resonance, National High Magnetic Field Laboratory, Tallahassee, FL 32310, USA
eKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai, P.R. China 200237
fDepartment of Oral and Maxillofacial Surgery, Laboratory of Interdisciplinary Studies, School and Hospital of Stomatology, Peking University, Beijing 100081, P.R. China. E-mail: sc-wei@pku.edu.cn
First published on 5th November 2014
Abstract
For the first time we observed well-resolved Ca(I) and Ca(II) signal changes in fluorohydroxyapatites with different fluorine contents by solid state NMR. The experiment results show that fluorine ions perturb the chemical environment of Ca(II) ions and OH− ions more than phosphorus tetrahedra and Ca(I) ions.
Hydroxyapatite (HA) is the main mineral in teeth and bones within the human body. Tooth enamel, the hardest and most highly mineralized substance in the human body, contains roughly 96 percent of hydroxyapatite. Fluorine-substituted hydroxyapatite (FHA) is chemically more stable than hydroxyapatite in acid environments.1 A higher concentration of FHA in tooth enamel decreases tooth dissolution and therefore decreases the incidence of tooth decay. It has also been reported that fluorine-substituted hydroxyapatites support cellular proliferation and colonization and promote bone growth.2
Over recent years, fluorohydroxyapatite has been used as bioactive ceramic coatings due to its enhanced biocompatibility as compared to other ceramic coatings.3,4 Fluoride-containing bioactive glasses are of particular interest in many fields of dentistry and orthopedics because they are osteoconductive and can combine the benefits of fluorapatite (FA) with the bone-regenerative properties of bioactive glasses.5–10 In the FHA composition, part of the OH− ions are substituted by F− ions in order to improve the material stability. However, most studies so far have focused on the preparation methods of FHA and its thermal and chemical stability.11 The effects of different fluorine contents on the structure have seldom been studied systematically. The resistance of fluorohydroxyapatite to acids depends largely on its chemical structure; it is of great interest to determine its structure that prevents the process of erosion.
The hydroxyapatite structure and substitution of hydroxyapatite by different anions and cations including Pb2+, Mg2+, Ti4+, CO32−, Cl− and F− ions12–18 have been extensively studied and characterized by X-ray diffraction (XRD),5–10,14–16,23 solid state NMR12–14,17–23 and Raman spectroscopy.18 Molecular dynamics simulations have also been used to investigate the transport of fluorine in fluorapatite and this study found that a sequence of F− ions move along the c-axis in a concerted mechanism, via lattice and interstitial sites.24 In this study, a series of FHAs with varying fluorine levels (Ca10(PO4)6(OH)2−xFx from x = 0 to 2) were synthesized by the wet precipitation method using Ca(NO3)2·4H2O and (NH4)2HPO4 as starting materials and NH4F as a source for fluorine incorporation (see ESI† for material synthesis and experimental details). The structures of the synthesized FHAs were characterized by 1H, 43Ca, 31P and 19F NMR MAS solid-state NMR, XRD and Raman spectroscopy to examine the effect of fluorine substitution on the hydroxyapatite structure.
XRD patterns of fluorohydroxyapatites containing different fluorine levels are shown in Fig. 1. The typical peaks of apatites are at 28.1°, 28.9°, 31.7°, 32.8° and 34.0°, which correspond to the (1 0 2), (2 1 0), (2 1 1), (3 0 0), (2 0 2), (3 1 0), (2 2 2), (2 1 3) and (0 0 4) Miller planes, respectively.23 With the increase of F− ion content in HA, some small peaks in the range of 45–50° become noisy and disappear, and peaks of (2 1 1), (3 0 0) and (2 0 0) gradually shift to the right-hand side with an increase in F− ions incorporated within the apatite lattice. The shifts are caused by the decrease in the a-axis length of the hexagonal crystal lattice induced by the lower ionic radius of F− ions.11
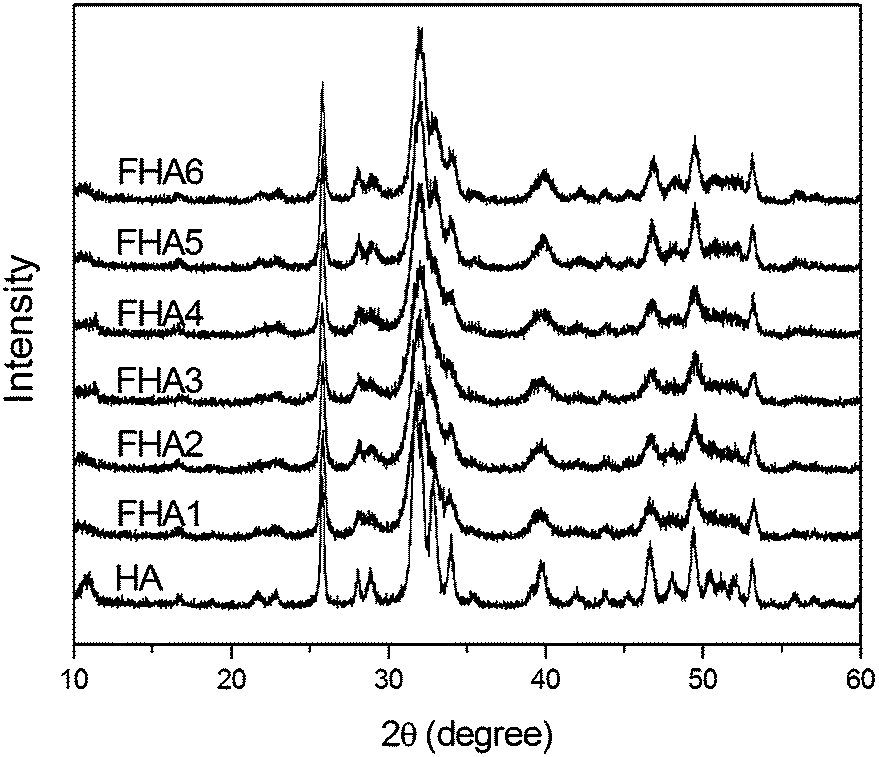 | ||
| Fig. 1 XRD patterns of fluorohydroxyapatites containing different fluorine levels (HA: 0 wt%, FHA1: 0.54 wt%, FHA2: 0.83 wt%, FHA3: 1.59 wt%, FHA4: 1.93 wt%, FHA5: 2.2 wt%, and FHA6: 2.94 wt%). | ||
The 1H MAS NMR spectra of hydroxyapatite (HA) is shown in Fig. 2a. Two main peaks are observed, one peak at 0.3 ppm corresponding to hydroxyl ions and the other at 4.95 ppm corresponding to water molecules on the surface of hydroxyapatite, which agree with previous reports.20,21 As the fluorine content increases, the chemical shift of OH− ions in the lattice of apatites gradually moves upfield and the height of the peak decreases due to substitution of F− ions. With the 0.54% fluorine added to the hydroxyapatite lattice shown in the spectrum of FHA1, the signal of hydroxyl ions significantly decreases and gets broader but a new peak at 1.8 ppm appears, which can be assigned to structure water molecules forming strong bonding with the surface vacancies of the crystal lattice of apatite. As the fluorine content increases to 0.83 wt% (FHA2), the signal of hydroxyl ions continues to decrease and the peak at 1.8 ppm can also be observed. When the fluorine content increases to 1.59 wt% (FHA3), the signal of hydroxyl ions almost disappears and two peaks at 1 ppm and 1.8 ppm are observed. These two peaks can be both assigned to structure water molecules. As the fluorine content increases to 1.93 wt% (FHA4), the surface water peak at 4.97 ppm and two structure water peaks at 0.8 ppm and 1.8 ppm can be observed. When the fluorine content increases to 2.2 wt% (FHA5) and 2.94 wt% (FHA6), only the surface water peak at 4.97 ppm and the structure water peak at 1.8 ppm are observed, indicating that OH− ions have been completely substituted by F− ions.
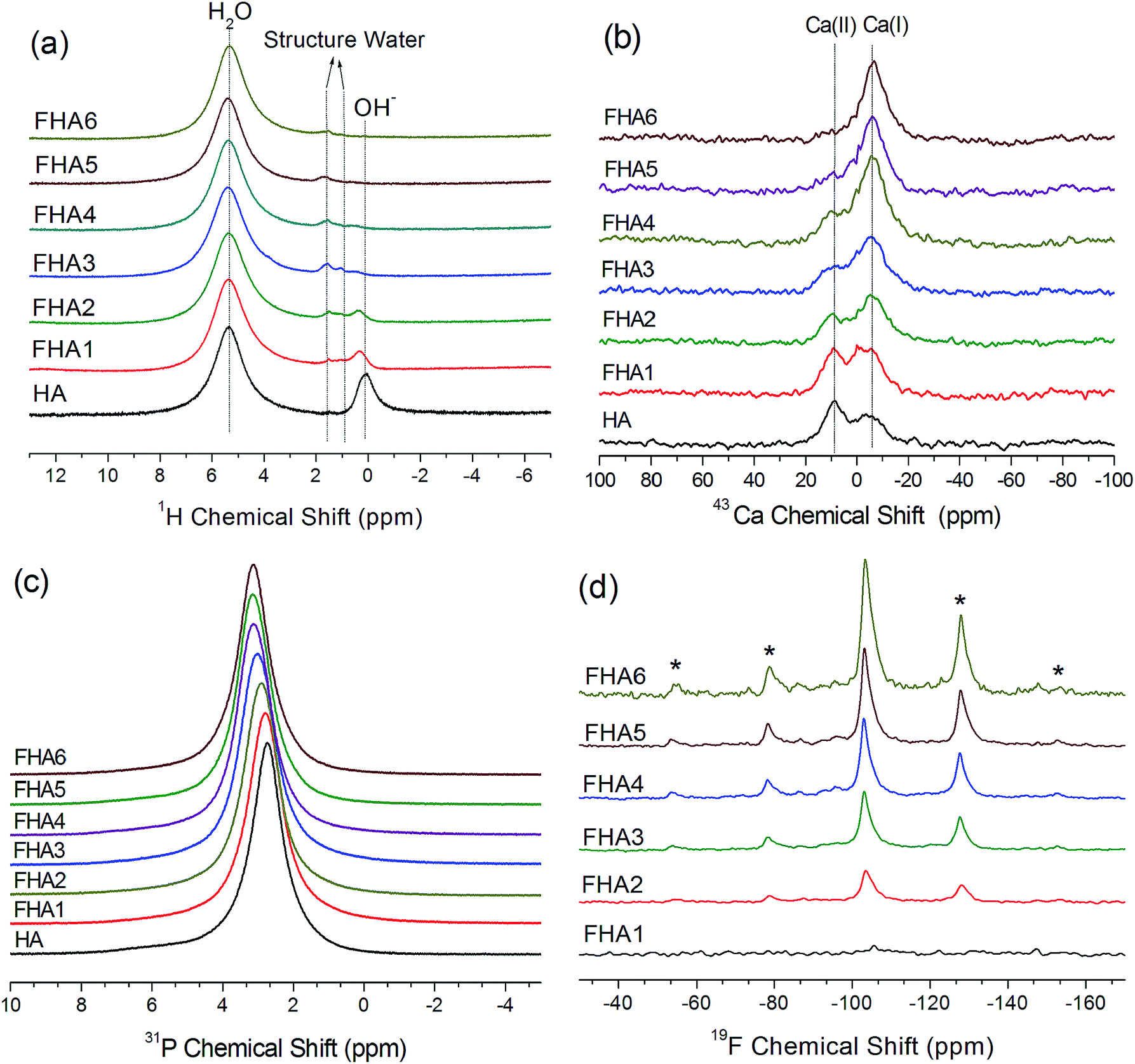 | ||
| Fig. 2 (a) 1H NMR spectra, (b) 43Ca NMR spectra, (c) 31P NMR spectra, and (d) 19F NMR spectra of fluorohydroxyapatites containing different fluorine levels (HA: 0 wt%, FHA1: 0.54 wt%, FHA2: 0.83 wt%, FHA3: 1.59 wt%, FHA4: 1.93 wt%, FHA5: 2.2 wt%, and FHA6: 2.94 wt%). The acquisition time for each 1H, 43Ca and 31P spectrum was 5 min, 24 hours and 1 hour, respectively. All 1H and 31P NMR spectra were obtained using a Varian VNMRS 400 MHz solid-state NMR spectrometer and a 6 mm double-resonance MAS probe with a spinning frequency of 8 kHz. All 43Ca spectra were obtained on a 830 MHz solid-state NMR spectrometer using a single-resonance 4 mm MAS probe with a spinning frequency of 10 kHz at room temperature (25 °C). Solid-state 19F NMR experiments were performed on a Bruker 600 MHz NMR spectrometer using a spin echo pulse excitation. A π/2 pulse length of 5.57 μs, a recycle delay of 2 s, a spinning rate of 14 kHz and the echo time set to a five rotor period were used for 19F MAS NMR experiments. Asterisks denote spinning sidebands. | ||
Fig. 2b shows natural abundance 43Ca NMR spectra of fluorohydroxylapatites containing different fluorine levels. The spectrum of HA shows Ca(I) and Ca(II) peaks of hydroxyapatite at 9.6 ppm and −5.6 ppm, respectively.12,17 As the fluorine content increases, the Ca(II) peak shifts downfield, while the Ca(I) position is unchanged, indicating that the increased fluorine content significantly changes the chemical environment of Ca(II) ions by reducing the Ca(II)–O bond length but the impact on Ca(I) is minimal. The NMR chemical shift is sensitive to the shielding of electrons which usually causes a downfield shift in low electron density environments. It follows that highly fluorine-level apatites have a lower chemical shift, as confirmed in this study. These results are consistent with XRD studies. The Ca(I) ion is coordinated to nine oxygen atoms in the arrangement of a tricapped trigonal prism and the Ca(I) polyhedron shows little response to the incorporation of different anion ions. However, in the Ca(II) polyhedron, the Ca(II) ion bonds to six oxygen atoms and one column anion. Therefore, the major structure substitution of OH− ions by F− ions will impact Ca(II) ions.
The 31P MAS NMR spectra of the fluorohydroxyapatites with various fluorine contents are shown in Fig. 2c. The 31P NMR spectrum of all samples exhibits a single well-resolved resonance at 2.8 ppm. This resonance is shifted upfield to higher ppm values as fluorine contents increase from 0 wt% to 1.59 wt%, then remain at the same 3.4 ppm from 1.93 wt% to 2.94 wt%. The phosphorus signal become progressively narrow as fluorine contents increase, indicating the increased crystallinity by the incorporation of fluorine into the HA lattice. Phosphorus atoms exist in apatite in tetrahedral coordination with four oxygen atoms. These rigid tetrahedra show a bit chemical environment change with fluorine substitution from 0 wt% to 1.59 wt%, but almost no chemical shift change from 1.93 wt% to 2.94 wt% due to the high structural similarity of the phosphorus atoms in these fluorine substituted hydroxyapatites.
The 19F MAS NMR spectra of FHAs containing different fluorine levels are shown in Fig. 2d. The 19F NMR spectra exhibit one peak at around −103 ppm and a few sidebands, which are similar to the previous results for fluorapatites.23 The height of the fluorine signal increases with fluorine levels, showing that OH− ions in the apatite lattice are gradually substituted by F− ions. With the 0.54% fluorine added to the hydroxyapatite lattice shown in the spectrum of FHA1, the signal of F− ions is very weak because of the low fluorine content. As the fluorine content increases, F− ions in the apatite lattice become less shielded and the fluorine chemical shift moves downfield.
Fig. 3a shows the Raman spectra of all seven apatite samples with different fluorine contents. All spectra have a strong PO43− band at ∼960 cm−1. As the fluorine content increases, the PO43− band associated with the P–O stretch shifts upfield, which suggests a progressive shortening of the P–O bond due to the increasing content of fluorine ions. The replacement of OH− ions by smaller F− ions increases the electrostatic attraction between the oxygen atoms in the phosphate tetrahedra, which then produces a shortening of the P–O bonds and an increase in vibrational frequency. The Full Width at the Half-Height (FWHH) of the phosphate symmetric stretch at 960 cm−1 as a measurement of crystallinity increases with fluorine levels.18,19Fig. 3b shows the decrease in the intensity of the O–H stretch at about 3580 cm−1 (normalized to the intensity of the 960 cm−1 peak) with increasing fluorine content. The peak remains at about the same position but shows broadening and development of a shoulder at about 3545 cm−1 side of the major O–H band at 3580 cm−1. As the fluorine content increases to 0.83 wt% (FHA3), two peaks at 3545 cm−1 and 3580 cm−1 can be observed. While the fluorine content increases to 1.59 wt% (FHA4), the band at 3580 cm−1 of O–H continues to decrease and its signal becomes weaker than the band at 3545 cm−1. As the fluorine content increases to 2.2 wt% (FHA5), the O–H band at 3580 cm−1 disappears and only one weak band at 3545 cm−1 can be observed. For the FHA6 sample, no peak can be observed, indicating that hydroxyl ions have been completely substituted by F− ions, which agrees well with 1H NMR results shown in Fig. 2a. Therefore, extensive substitution of O–H groups by F− ions may sufficiently alter the environment of the channel sites to cause a shift in the vibrational frequency of some O–H groups, leading to the development of a shoulder band at high fluorine concentrations. Alternatively, at high fluorine contents, fluorine may begin to substitute preferentially into the channel site.23,24
 | ||
| Fig. 3 Raman spectra of fluorohydroxyapatites containing different fluorine levels (HA: 0 wt%, FHA1: 0.54 wt%, FHA2: 0.83 wt%, FHA3: 1.59 wt%, FHA4: 1.93 wt%, FHA5: 2.2 wt%, and FHA6: 2.94 wt%) in the range of (a) 900–1050 cm−1 and (b) 3100–3800 cm−1. | ||
HA, FHAs and FA are widely used as biomaterials in bone tissue engineering and dental caries treatment. The chemical composition and surface characteristics of the biomaterials used as bone implants are important factors in affecting the cells at the interface between the material and the surrounding tissues.3,4 The substitution of OH− ions by F− ions and the content of F− ions in the HA lattice can be controlled in the synthesis process (see Material synthesis in the ESI†). When all OH− ions have been replaced by F− ions, fluorapatite is formed. The addition of F− ions to the HA lattice caused the increase of the thermal and chemical stability of HA.11 In the crystal structure of HA, the atoms of OH− ions sit in the atomic interstices neighbouring the oxygen atoms and O–H groups are oriented randomly, which brings a certain degree of disorder to the crystal structure of HA.25 When the OH− ions are partially substituted by the F− ions, the existing hydrogen atoms of the OH groups are bound to the nearby F− ions and become more ordered. In the crystal lattice of FHAs, OH− ions and F− ions are only surrounded by Ca(II) ions. When OH− ions are gradually substituted by F− ions, the higher affinity and smaller size of the fluorine atoms with respect to the oxygen atoms, and the improved bonding between F− ions and Ca(II) ions produce an increasingly compact and ordered apatite structure and also change the chemical environment of the HA surface, which are shown by solid-state NMR results in Fig. 2.
In summary, a series of fluorohydroxyapatites were synthesised and characterized to evaluate the effects of fluorine on the structure of hydroxyapatite. For the first time we observed the well-resolved Ca(I) and Ca(II) signal change in fluorohydroxylapatite with different fluorine contents at natural abundance. Compared with small variations of 31P NMR chemical shifts induced by the incorporation of fluorine, the significant 43Ca NMR signal change of Ca(II) ions and 1H NMR signal change of OH− ions indicate that the fluorine perturbs the chemical environment of Ca(II) ions and OH− ions more than phosphorus atoms. Fluorohydroxyapatite has extensive applications in teeth and bone materials. The combination of 1H, 43Ca, 31P and 19F solid-state MAS NMR, XRD and Raman spectroscopy can probe the structure of fluorohydoxyapatite at the atomic level. Especially the solid state 43Ca NMR can selectively and sensitively detect the chemical environments of calcium ions, which could be a powerful tool in the field of biomaterials, bioglass ceramics and geological materials.
Acknowledgements
This work was supported by Jiangsu Province for specially appointed professorship to Dr P. Z. Zhu, research funds from Yangzhou University and the support from Testing Center of Yangzhou University. Prof. S. C. Wei thanks the support from the State Key Development Program for Basic Research of China (Grant 2007CB936103) and Peking University's 985 Grant. This work was also supported by the National High Magnetic Field Laboratory through the National Science Foundation Cooperative Agreement (DMR-0084173) and by the State of Florida.Notes and references
- M. Okazaki, Y. Miake, H. Tohda, T. Yanagisawa, T. Matsumoto and J. Takahashi, Biomaterials, 1999, 20, 1421–1426 CrossRef CAS.
- J. Harrison, A. J. Melville, J. S. Forsythe, B. C. Muddle, A. O. Trounson and K. A. Gross, Biomaterials, 2004, 25, 4977–4986 CrossRef CAS PubMed.
- D. S. Brauer, N. Karpukhina, R. V. Law and R. G. Hill, J. Mater. Chem., 2009, 19, 5629–5636 RSC.
- D. Campocciaa, C. R. Arciolaa, M. Cervellatia, M. C. Maltarelloc and L. Montanaro, Biomaterials, 2003, 24, 587–596 CrossRef.
- L. Montanaro, C. R. Arciola, D. Campoccia and M. Cervellati, Biomaterials, 2002, 23, 3651–3659 CrossRef CAS.
- D. S. Brauer, M. N. Anjum, M. Mneimne, R. M. Wilson, H. Doweidar and R. G. Hill, J. Non-Cryst. Solids, 2012, 358, 1438–1442 CrossRef CAS PubMed.
- E. Lynch, D. S. Brauer, N. Karpukhina, D. G. Gillam and R. G. Hill, Dent. Mater., 2012, 28, 168–178 CrossRef CAS PubMed.
- A. Pedone, T. Charpentier and M. C. Menziani, J. Mater. Chem., 2012, 22, 12599–12608 RSC.
- I. Kansal, A. Goel, D. U. Tulyaganov, L. F. Santos and J. M. F. Ferreira, J. Mater. Chem., 2011, 21, 8074–8084 RSC.
- M. Mneimne, R. G. Hill, A. J. Bushby and D. S. Brauer, Acta Biomater., 2011, 7, 1827–1834 CrossRef CAS PubMed.
- Y. M. Chen and X. G. Miao, Biomaterials, 2005, 26, 1205–1210 CrossRef CAS PubMed.
- H. Pizzala, S. Caldarelli, J. Eon, A. M. Rossi, D. Laurencin and M. E. Smith, J. Am. Chem. Soc., 2009, 131, 5145–5152 CrossRef CAS PubMed.
- D. Laurencin, N. Almora-Barrios, N. H. de Leeuw, C. Gervais, C. Bonhomme, F. Mauri, W. Chrzanowski, J. C. Knowles, R. J. Newport, A. Wong, Z. Gan and M. E. Smith, Biomaterials, 2011, 32, 1826–1837 CrossRef CAS PubMed.
- A. A. Chaudhry, J. Goodall, M. Vickers, J. K. Cockcroft, I. Rehman, J. C. Knowles and J. A. Darr, J. Mater. Chem., 2008, 18, 5900–5908 RSC.
- P. N. Gunawidjaja, I. Izquierdo-Barba, R. Mathew, K. Jansson, A. García, J. Grins, D. Arcos, M. Vallet-Regí and M. Edén, J. Mater. Chem., 2012, 22, 7214–7223 RSC.
- C. J. L. Silwood, I. Abrahams, D. C. Apperley, N. P. Lockyer, E. Lynch, M. Motevalli, R. M. Nix and M. Grootveld, J. Mater. Chem., 2005, 15, 1626–1636 RSC.
- J. Xu, P. Zhu, Z. Gan, N. Sahar, M. M. J. Tecklenburg, M. D. Morris, D. H. Kohn and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 11504–11509 CrossRef CAS PubMed.
- M. D. O'Donnell, R. G. Hill, R. V. Law and S. Fong, J. Eur. Ceram. Soc., 2009, 29, 377–384 CrossRef PubMed.
- J.-D. P. McElderry, P. Zhu and K. H. Mroue, et al. , J. Solid State Chem., 2013, 206, 192–198 CrossRef CAS PubMed.
- E. E. Wilson, A. Awonusi, M. D. Morris, D. H. Kohn, M. M. J. Tecklenburg and L. W. Beck, Biophys. J., 2006, 90, 3722–3731 CrossRef CAS PubMed.
- G. Cho, Y. Wu and J. L. Ackerman, Science, 2003, 300, 1123–1127 CrossRef CAS PubMed.
- D. Laurencin, A. Wong, J. V. Hanna, R. Dupree and M. E. Smith, J. Am. Chem. Soc., 2008, 130, 2412–2413 CrossRef CAS PubMed.
- F. M. Mccubbin, H. E. Mason, H. Park, B. L. Phillips, J. B. Parise, H. Nekvasil and D. H. Lindsley, Am. Mineral., 2008, 93, 210–216 CrossRef CAS.
- E. E. Jay, M. J. D. Rushton and R. W. Grimes, J. Mater. Chem., 2012, 22, 6097–6103 RSC.
- H. Eslami, M. Solati-Hashjin and M. Tahriri, Adv. Appl. Ceram., 2010, 109, 200–212 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Material synthesis and experimental details including X-ray diffraction (XRD) measurements, X-ray photoelectron spectroscopy (XPS) measurements, solid state NMR spectroscopy, and Raman measurements. See DOI: 10.1039/c4tb01561d |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |