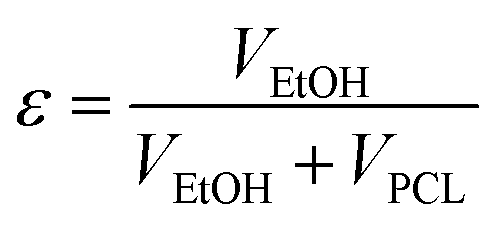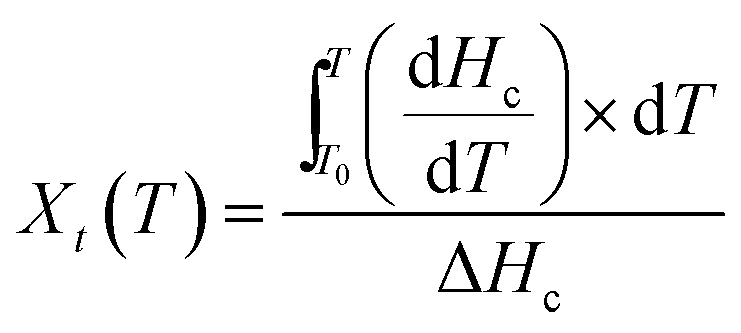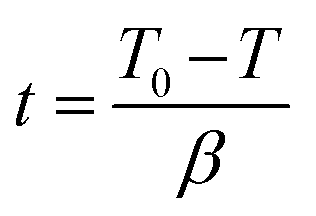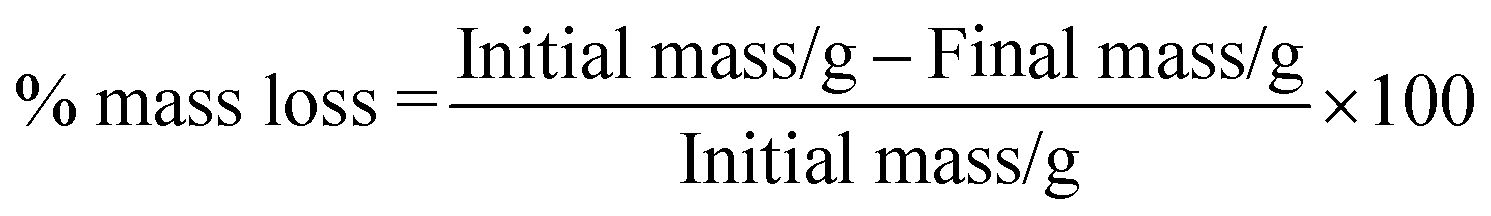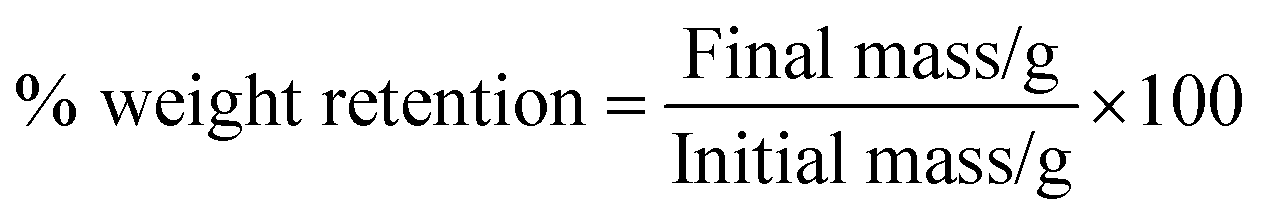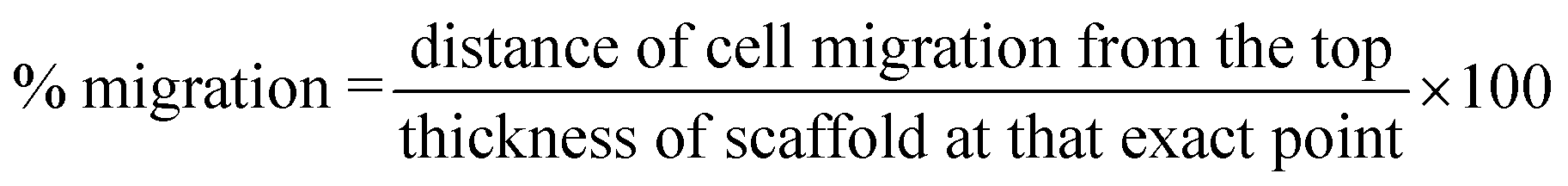Poly(ester-ether)s: III. assessment of cell behaviour on nanofibrous scaffolds of PCL, PLLA and PDX blended with amorphous PMeDX†
Received
14th August 2014
, Accepted 21st November 2014
First published on 26th November 2014
Abstract
The aim of this paper is to investigate the physico-chemical properties, degradation behaviour and cellular response of electrospun fibre-scaffolds of semi-crystalline PCL, PLLA and PDX blended with amorphous poly(methyl dioxanone) (PMeDX). Electrospun PCL/PMeDX and PLLA/PMeDX blend mats in varying weight ratios of the two components were fabricated and their overall performance was compared with similar composition PDX/PMeDX scaffolds. DSC analysis showed almost no change in crystallization temperature of PCL with increasing PMeDX content and TGA showed a different degradation profile as PMeDX content increased. The appearance of two crystallization peaks for PLLA/PMeDX blends suggested stereocomplex formation. As noted from AFM images, addition of PMeDX caused a change in the width of the lamellae from 14.8 ± 2.9 nm in 100/0 mat to 32.0 ± 11.5 nm in 85/15 mat. Moreover, PCL/PMeDX blend mats show a significant drop in Young's modulus for 93/7, 90/10 and 85/15 compositions compared to 100/0 and 98/2. On the other hand, no clear trend in mechanical properties was observed for espun PLLA/PMeDX mats with increasing PMeDX content. Based on these analyses, it was concluded that PCL and PMeDX were immiscible while miscible blends were obtained with PLLA and PMeDX. Initial degradation of electrospun mats over a period of 5 weeks appears to occur via a surface erosion mechanism. In vitro cell culture studies using HDFs showed that the scaffolds were bioactive and a greater density of viable cells was noted on electrospun PCL/PMeDX and PLLA/PMeDX scaffolds compared to PCL and PLLA mats respectively. HDFs infiltrated through the entire thickness of espun 85/15 PLLA/PMeDX scaffold due to a combination of factors including morphology, porosity, surface characteristics and mechanical properties.
Introduction
Scaffolds for tissue engineering applications serve as a biomimetic extracellular matrix (ECM) and play a critical role in supporting cells.1 They are designed to conform to a specific set of requirements which are often conflicting.2 They should be biocompatible, biodegradable, porous and possess appropriate mechanical properties.3 Electrospinning remains a preferred method due to its low cost, high throughput, ease of operation and system control. It allows the fabrication of non-woven mats containing fibres ranging from tens of microns to tens of nanometers in diameter, which can mimic both the form and function of the native ECM. Because of their mechanical properties and degradation rates that closely match those of proteins in soft and hard tissues, polymers are good candidates for the development of bone and vascular scaffolds.4 Several natural and synthetic polymers have been investigated to this end. The most commonly used synthetic polymers are the aliphatic polyesters, polycaprolactone and poly(L-lactide) due to their relatively good biocompatibility and mechanical performance. However, cell affinities towards synthetic polymers are often poor as a consequence of their hydrophobicity and lack of cell recognition sites.5,6
Several research groups have considered blending PCL and PLLA with other synthetic or natural polymers for optimized mechanical properties, degradation and bioactivity. Several studies have investigated the use of electrospun (espun) PCL/collagen7–12 and PCL/gelatin13–18 for biomedical applications. Lee et al.19 reported that compared to PCL, PCL/collagen scaffolds possessed enhanced biomechanical properties that could resist higher degrees of pressurized flow. Zhang et al.20 found that composite PCL and gelatin scaffold had higher elongation and better flexibility compared to PCL mat. Moreover, cells could not only grow and proliferate but also migrate inside the composite scaffold. Mehdinavaz Aghdam et al.21 showed that PGA increased the hydrophilicity, water uptake and mechanical properties of polycaprolactone/polyglycolic acid (PCL/PGA) nanofibrous mats. Kim et al.22 reported that the addition of PEI to PCL increased the hydrophilicity of the resulting espun mats and cell attachment on the blend mats was favoured due to the cationic nature of PEI. The miscibility of the two polymers in the blend is also an important parameter. For instance, Han et al.23 studied espun blends of semi-crystalline PCL and amorphous poly(tetramethylene carbonate) (PTMC) and showed that the polymers were phase-separated in the fibres. In a recent paper, Son et al.24 reported on in vitro and in vivo evaluation of espun polycaprolactone/poly(methyl methacrylate) (PCL/PMMA) fibrous scaffolds for bone regeneration. Addition of PMMA improved the wettability of the scaffolds, while decreasing PCL content caused a decrease in the tensile strength of the espun blend mats. Higher proliferation of MG-63 cells was observed on the 7/3 PCL/PMMA scaffolds.
PLLA has been blended with natural polymers such as gelatin,25,26 silk,27 chitosan28 as well as synthetic ones such as PLGA29 and PCL.30 Shalumon et al.28 reported that espun PLLA/chitosan blend nanofibres showed enhanced degradation and better human dermal fibroblasts (HDFs) cell growth compared to pure PLLA fibres. Liu et al.29 prepared PLGA/PLLA nanofibrous mats and analysed their thermal, morphological and mechanical properties. Elongation was found to decrease with increasing PLLA content. In addition, Young's modulus and tensile strength of espun blend mats of PCL/PLLA decreased with increasing PCL content.30 Scaffolds supported human adipose-derived stem cells (hASCs) well. However, the 1/1 wt ratio PLLA/PCL demonstrated better properties and cellular responses in all assessments.
Blending not only influences physico-chemical properties of the resulting espun mats but also impacts on their bioactivity and biological performance. It is well established that surface properties of scaffolds play a key role in cell–scaffold interactions, especially during the initial stage of cell-seeding. The topography created by the nanofibres has a strong influence on the biological response of cells seeded on its surface through contact guidance.31 In turn, cell morphology determines cell proliferation and infiltration within a scaffold.32 Previous studies have demonstrated that cells can recognize differences in substrate stiffness such that they can tune their internal stiffness to match that of the substrate, resulting in a change in their cytoskeletal structure.33,34 This causes fibroblasts to adopt a more spread phenotype on stiff substrates and the cell organizes the actin cytoskeleton into stress fibres.35 On the other hand, fibroblasts do not spread well on softer substrates and have a cortical actin cytoskeleton but no stress fibres. In fact, cell size and the spreading area increase with increasing stiffness of the substrate.36,37
Furthermore, Cui and Sinko38 showed that highly crystalline and rigid PCL/PGA surfaces were more efficient in supporting fibroblasts growth compared to amorphous and flexible ones. Yip et al.39 showed that fibroblast behaviour was governed by strain on substrates softer than 20 kPa while the latter was dependent on stress for stiffer substrates (>20 kPa). In another paper by Lo et al.,40 the authors concluded on fibroblasts preference for stiff substrates. They showed that fibroblasts generate more traction force and develop a broader and flatter morphology on stiff substrates than on soft ones.40 In addition to mechanical properties, cells are very sensitive to surface chemistry, surface energy and surface roughness.41 In fact, surface energy plays a key role in attracting specific proteins to the surface of scaffolds which, would in turn, affect cell affinity towards these materials. Surface roughness was also found to promote cell attachment and growth on PLLA scaffolds.41 Moreover, good hydrophobic–hydrophilic balance is crucial for optimized biocompatibility and cellular response.42 Indeed, recent studies have shown that cells adhere, spread and grow more easily on moderately hydrophilic substrates than on hydrophobic or very hydrophilic ones.43
In a recent study, we reported on blend films of semi-crystalline PDX and amorphous polyDL-3-methyl-1,4-dioxan-2-one (PMeDX) and showed that low amounts of PMeDX in the blends (of the order of 15 wt%) could act as plasticizer. Mechanical tests showed overall reduced tensile properties of the blend films. Interaction parameters from viscosity analysis and surface morphology images indicated immiscibility of the blend films over the range of compositions studied.44 In another paper,45 the thermal, mechanical and degradation characteristics of espun PDX/PMeDX mats were discussed. AFM images of the espun fibres showed an increasing degree of morphological heterogeneity with increasing PMeDX content. Hydrolytic degradation of espun mats was found to be mainly dependent on fibre diameter. Espun PDX/PMeDX nanofibrous scaffolds demonstrated excellent biocompatibility as demonstrated by HDF adhesion and proliferation.
In this paper, we analyse the properties of espun mats of semi-crystalline PCL and PLLA blended with amorphous PMeDX. Our objectives are: to better apprehend the physico-chemical characteristics of espun PCL/PMeDX and PLLA/PMeDX mats; to compare the response of these two nanofibrous scaffolds to HDFs behaviour with PDX/PMeDX scaffolds described previously;45 to attempt a correlation between physico-chemical properties and biological performance of scaffolds. The miscibility of the blends, the morphology of fibres, their thermal, mechanical properties and hydrolytic degradation as well as their efficacy to promote HDF cell growth and infiltration will be discussed. To the best of our knowledge, this is a first study where cell growth on PCL, PLLA and PDX scaffolds is compared under similar conditions.
Results and discussion
Physico-chemical and mechanical characterization of electrospun blend mats
Blends of semi-crystalline homopolymers (PCL and PLLA) and amorphous PMeDX in varying weight ratios (100/0, 98/2, 93/7, 90/10 and 85/15 wt%) were espun in HFIP at a concentration of 100 mg mL−1. Thermal behaviour, fibre morphology and mechanical performance of espun PCL/PMeDX and PLLA/PMeDX mats were analysed to get a better insight into blend miscibility and surface characteristics. In particular, the effect of increasing PMeDX content on fibre diameter, pore size and porosity were investigated and compared with PDX/PMeDX fibres.
Analysis of thermal properties
DSC analysis: crystallinity and crystallization kinetics of electrospun mats.
The melting temperature (Tm), enthalpy of melting (ΔHm), crystallization temperature (Tc), enthalpy of crystallization (ΔHc) of espun PCL/PMeDX and PLLA/PMeDX mats are summarized in Tables 1 and 2. The degree of crystallinity of the blends (χblend) and that of the PCL phase in the blends (χPCL) were calculated as reported previously.45 The enthalpy of melting for 100% crystalline PCL and PLLA were taken from the literature as 139.5 J g−1 (ref. 46) and 93.7 J g−1 (ref. 47) respectively.
Table 1 DSC analysis of PCL/PMeDX espun mats
| Blend composition (wt%) |
Espun PCL/PMeDX non-woven mats |
|
T
c/°C |
ΔHc/(J g−1) |
T
m/°C |
ΔHm/(J g−1) |
χ
blend/(%) |
χ
PCL/(%) |
| 100/0 |
31.2 |
52.9 |
57.9 |
44.9 |
32.2 |
32.2 |
| 98/2 |
32.1 |
52.8 |
57.3 |
43.1 |
30.9 |
31.5 |
| 93/7 |
33.0 |
50.9 |
56.7 |
43.3 |
31.0 |
33.3 |
| 90/10 |
33.3 |
51.0 |
56.3 |
40.8 |
29.2 |
32.5 |
| 85/15 |
33.9 |
42.4 |
55.9 |
35.0 |
25.1 |
29.5 |
Table 2 DSC analysis of PLLA/PMeDX espun mats
| Espun PLLA/PMeDX non-woven mats |
| Blend composition (wt%) |
T
c1/°C |
T
c2/°C |
ΔHc/(J g−1) |
T
m1/°C |
T
m2/°C |
ΔHm1/(J g−1) |
ΔHm2/(J g−1) |
χ
blend/(%) |
| 100/0 |
149.6 |
— |
56.9 |
178.0 |
— |
57.6 |
NA |
61.4 |
| 98/2 |
115.9 |
122.2 |
43.3 |
175.0 |
165.8 |
34.5 |
12.5 |
50.2 |
| 93/7 |
114.7 |
106.9 |
43.8 |
175.6 |
167.2 |
37.8 |
7.47 |
48.4 |
| 90/10 |
124.4 |
109.7 |
37.4 |
177.0 |
— |
43.9 |
— |
46.8 |
| 85/15 |
115.8 |
108.5 |
31.2 |
177.3 |
— |
42.5 |
— |
45.3 |
As can be noted from Table 1, espun PCL/PMeDX blend mats crystallize in the temperature range 31.2 to 33.9 °C depending on PMeDX content. The small difference in crystallization temperature suggests immiscibility of PCL and PMeDX homopolymers. It is known from the literature that when the glass transition temperature, Tg of the amorphous polymer in an immiscible blend is well below the Tc of the semi-crystalline polymer as in our case, the amorphous polymer doesn't affect Tc due to chain mobility at that temperature.48 In contrast, the crystallization temperatures of espun PDX/PMeDX fibres were found to increase from 51.0 to 80.4 °C as the PMeDX content increased.
The plots of relative crystallinity against crystallization time (Fig. 1A and B) show a sigmoid shape, indicative of a fast primary crystallization during the early stage and slow secondary crystallization in a later stage. Avrami constants, K and n which are related to crystallization kinetics and mechanism respectively were determined and the results summarized in Table 3. Variation in n values indicated that PMeDX interferes with PCL nucleation and crystallite formation with an overall increase in K value, as the PMeDX content increases. The increasing value of n in espun PCL/PMeDX blend mats compared to 100/0 denotes morphological change of crystallites, as will be discussed in the next section.
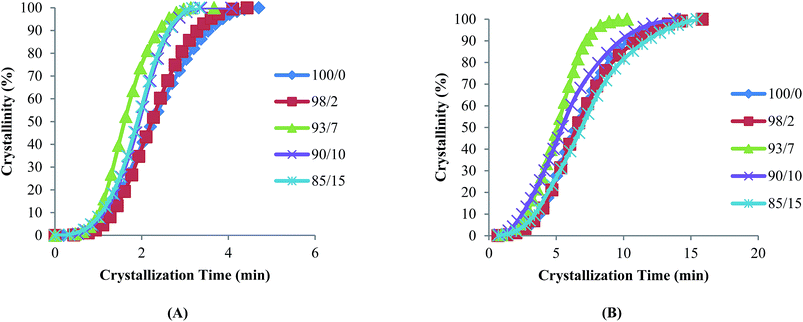 |
| | Fig. 1 Plots of relative crystallinity versus crystallization time for espun (A) PCL/PMeDX and (B) PLLA/PMeDX mats. | |
Table 3 Summary of Avrami parameters for espun PCL/PMeDX and PLLA/PMeDX
|
|
100/0 |
98/2 |
93/7 |
90/10 |
85/15 |
|
Espun PCL/PMeDX mats
|
|
K
|
0.29 |
0.24 |
0.37 |
0.31 |
0.36 |
|
n
|
2.95 |
3.53 |
3.64 |
3.59 |
3.29 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Espun PLLA/PMeDX mats
|
|
K
|
0.07 |
0.07 |
0.10 |
0.14 |
0.09 |
|
n
|
2.93 |
2.96 |
3.12 |
2.41 |
2.58 |
A shouldering of Tc is noted for all espun PLLA/PMeDX compositions in contrast to pure espun PLLA mat. This is attributed to stereocomplex formation. Indeed, the formation of stereocomplex has been reported for mixtures of PLLA and PDLLA where crystallization peaks at 110 and 130 °C were attributed to PLLA homocrystallites and PLLA/PDLLA stereocrystallites.49,50 The likely formation of a stereocomplex between PLLA and PMeDX translates miscibility or partial miscibility of the two homopolymers. The Avrami K values show an overall increase in crystallization rate with increasing PMeDX content.
As can be noted in Table 2, the Tm of PLLA/PMeDX blend mats are very close to that of PLLA. A slight shouldering of Tm is observed for the 98/2 and 93/7 composition which could originate from the thermal history of the polymers. The presence of two distinct melting transitions at 175 °C and 166 °C have previously been reported for α- and β-forms of PLLA.51,52 From Tables 1 and 2, it can also be noted that the enthalpy of fusion, ΔHm of espun PCL/PMeDX and PLLA/PMeDX mats decrease with increasing content of amorphous PMeDX.
Thermogravimetric analysis (TGA)
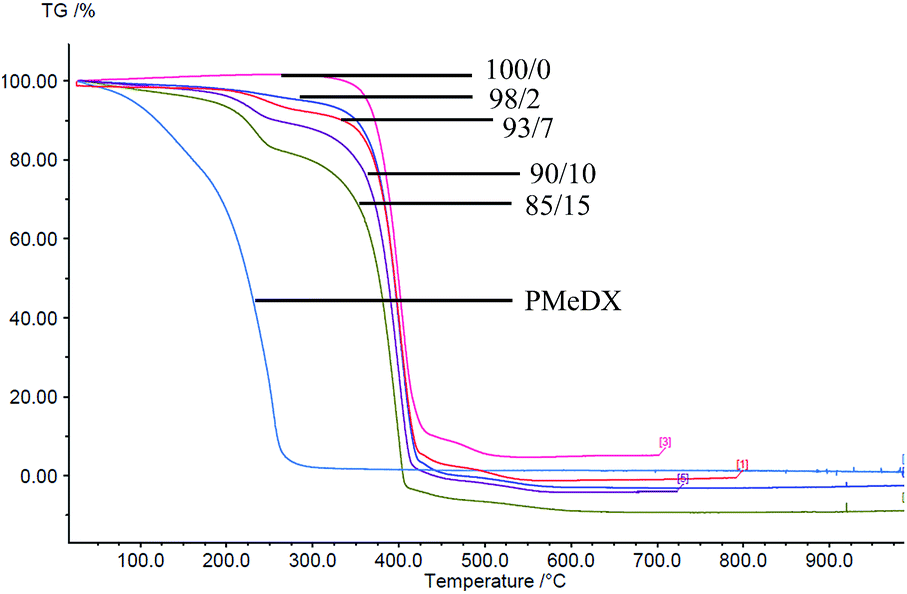
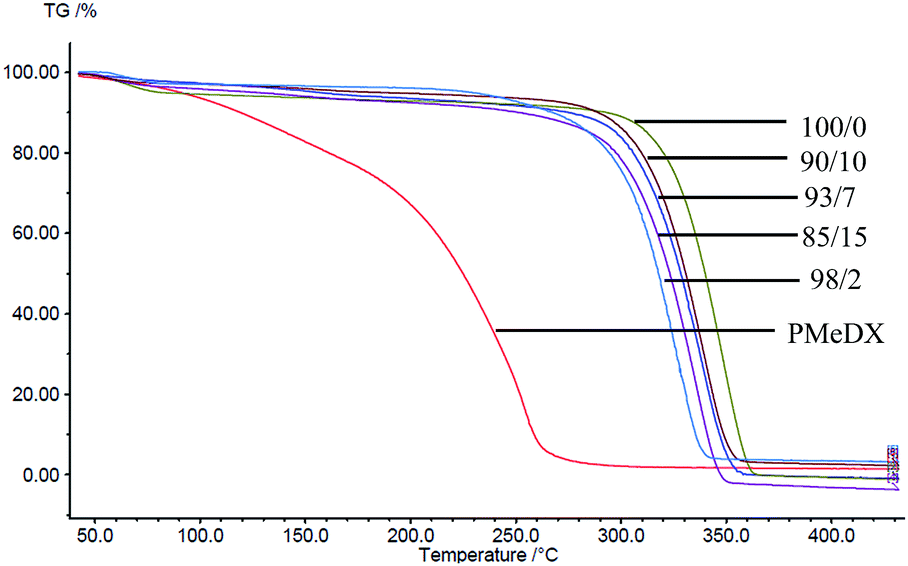
Thermal degradation profiles of espun PCL/PMeDX and PLLA/PMeDX mats are shown in Fig. 2 and 3 respectively. Espun PCL mat shows a two stage degradation profile with an onset degradation temperature, Tonset at 374.6 °C. PMeDX, on the other hand, degrades in a single step with a much lower Tonset of 78 °C. Espun 98/2 PCL/PMeDX follows a similar degradation profile as the 100/0 mat with no change in Tonset (Table 4). This suggests that PMeDX may be partially miscible at that composition. This is also confirmed by mass loss calculations whereby PCL and PCL/PMeDX 98/2 have comparable mass loss. However, with increasing PMeDX contents (7, 10 & 15 wt%), the degradation profile changes with a first-step degradation occurring at a lower temperature. Mass loss calculations ΔW1 is equivalent to the initial wt% of PMeDX for these blend compositions (Table 5), which denotes degradation of PMeDX in the blend. This supports immiscibility of PCL and PMeDX homopolymers, in line with DSC data.
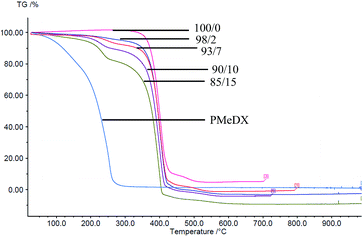 |
| | Fig. 2 TGA profiles of espun PCL/PMeDX mats. | |
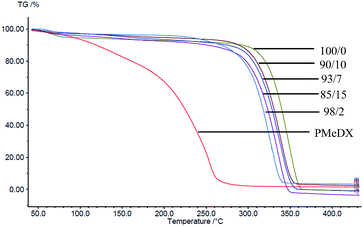 |
| | Fig. 3 TGA profiles of espun PLLA/PMeDX mats. | |
Table 4 Variation of onset degradation temperatures of espun fibresa
| Blend composition (wt%) |
Espun PCL/PMeDX fibres |
Espun PLLA/PMeDX fibres |
|
T
onset/°C |
T
′onset/°C |
T
max/°C |
T
onset/°C |
T
max/°C |
|
T
onset: onset degradation temperature for 100/0 and 98/2 (one stage). T′onset: onset degradation temperature for first stage degradation of 93/7, 90/10 and 85/15.
|
| 100/0 |
374.6 |
432.0 |
405.3 |
324.2 |
348.9 |
| 98/2 |
374.3 |
424.0 |
400.0 |
321.1 |
348.8 |
| 93/7 |
214.3 |
375.7 |
402.2 |
327.2 |
338.8 |
| 90/10 |
201.4 |
370.2 |
400.8 |
312.4 |
340.2 |
| 85/15 |
206.6 |
368.8 |
399.3 |
306.5 |
335.8 |
Table 5 Mass loss derived from TG degradation profilesa
| Blend composition (wt%) |
Espun PCL/PMeDX fibres |
Espun PLLA/PMeDX fibres |
| ΔW1/(wt%) |
ΔW2/(wt%) |
ΔW3/(wt%) |
ΔW1/(wt%) |
|
ΔW1: mass loss corresponding to first decomposition stage in 93/7, 90/10 and 85/15. ΔW2: mass loss corresponding to major decomposition stage in all blends. ΔW3: mass loss corresponding to second decomposition stage in 100/0 and 98/2 blend.
|
| 100/0 |
— |
91.7 |
4.09 |
97.9 |
| 98/2 |
— |
96.2 |
5.96 |
98.7 |
| 93/7 |
6.68 |
92.6 |
— |
99.1 |
| 90/10 |
10.3 |
90.8 |
— |
97.6 |
| 85/15 |
15.2 |
85.9 |
— |
99.9 |
Espun PLLA mat show a Tonset of 324.2 °C and increasing PMeDX content led to decreased thermal stability of PLLA (Table 4) with Tonset dropping from 324.2 to 306.5 °C. In contrast to PCL/PMeDX blend mats, only one degradation stage was noted for all espun PLLA/PMeDX mats. This further supports the formation of PLLA/PMeDX stereocomplex as proposed in the previous section.
On the basis of these results, it can be suggested that immiscibility increases with increasing PMeDX content for PCL/PMeDX, with the 98/2 blend being partially miscible. Moreover, the formation of stereocomplex between PLLA and PMeDX is confirmed by TGA.
FTIR analysis
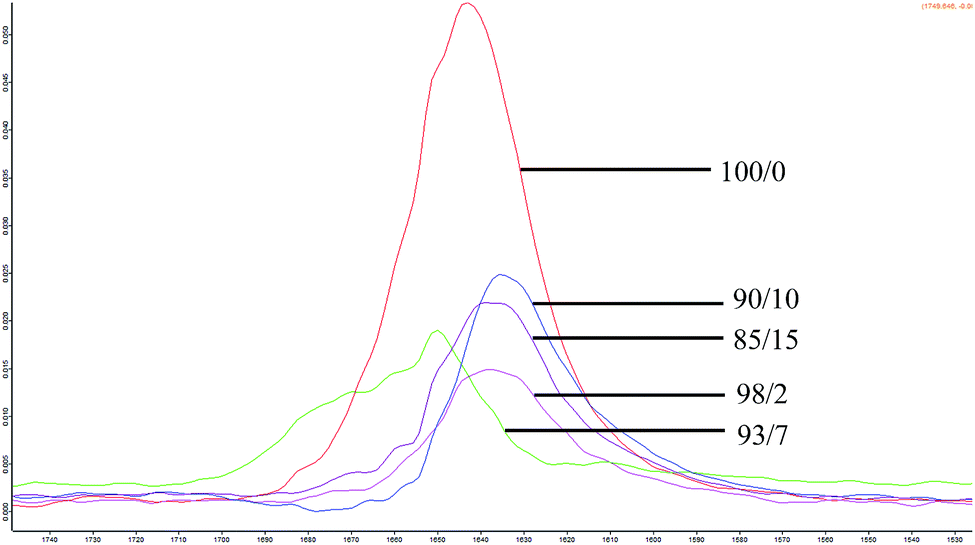
FTIR spectrum of espun PCL and PMeDX homopolymer showed characteristic C–O stretching bands at 1643 cm−1 and 1732 cm−1 respectively. Espun PCL/PMeDX blends showed bands between 1743–1696 cm−1 and between 1631–1650 cm−1 depending on PMeDX content (Fig. 4 and Table 6). Significant shift in PMeDX carbonyl for 98/2 blend composition (1696 cm−1) suggests some interaction with PCL.
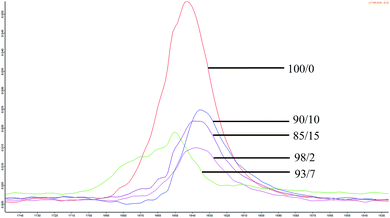 |
| | Fig. 4 FTIR spectra of espun PCL/PMeDX mats. | |
Table 6 Variation of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in espun PCL/PMeDX and PLLA/PMeDX
O) in espun PCL/PMeDX and PLLA/PMeDX
| Blend composition/(w/w)% |
ν
(CO)/cm−1 |
Shoulder at ν(CO)/cm−1 |
| 100/0 |
1643 |
— |
| 98/2 |
1638 |
1696 |
| 93/7 |
1650 |
1743 |
| 90/10 |
1635 |
1713 |
| 85/15 |
1639 |
1714 |
The stretching vibration of PLLA was found at 1631 cm−1. For espun PLLA/PMeDX mats, only one band was observed contrary to PCL/PMeDX mats. A slight shift in carbonyl stretching (4 cm−1) was noted with increasing PMeDX content.
In summary, FTIR analysis suggests immiscibility of espun PCL/PMeDX mats and plasticization at low PMeDX content (2 wt%) while PLLA and PMeDX appear to be miscible based on the appearance of a single band.
AFM analysis
Surface morphology.
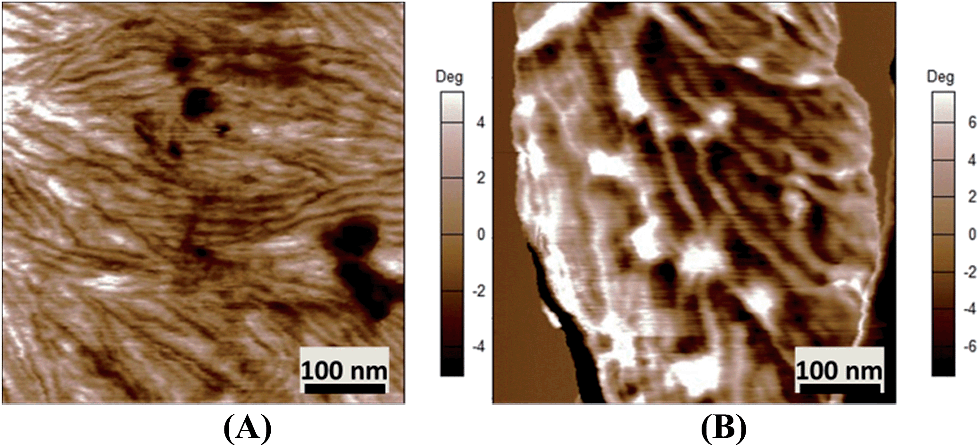
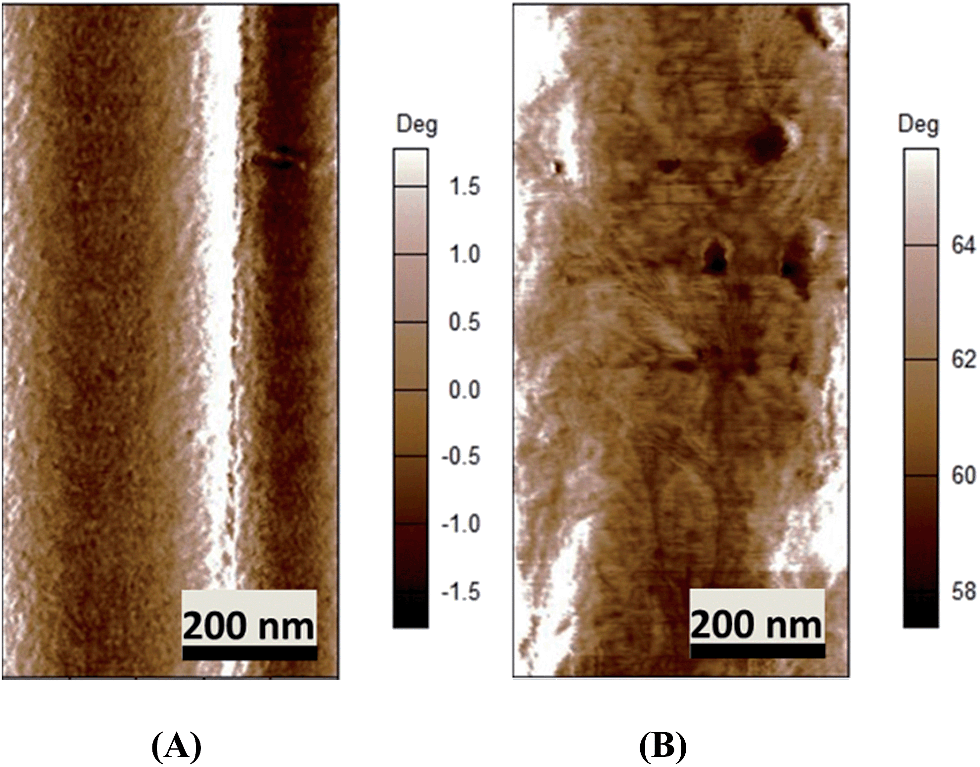
AFM images (Fig. 5A and B) show that both espun PCL and PCL/PMeDX nanofibres exhibit cylindrical morphology. Espun PCL mats had a smooth surface in contrast to blend nanofibres (85/15) which showed a rougher surface, possibly due to dispersion of PMeDX within the PCL matrix. Indeed, as reported by Leclair et al.,53 deformations of the rubbery amorphous polymer occur in an immiscible semi-crystalline/amorphous polymer blend if the latter is above its Tg during crystallization of the semi-crystalline polymer (Tg PMeDX = 20 °C, Tc PCL = 33 °C).
 |
| | Fig. 5 AFM phase images of espun PCL/PMeDX (A) 100/0 (B) 85/15 fibres. | |
More in-depth analysis of AFM images were carried out to investigate the nanostructure of the espun mats. Fig. 6A shows the lamellar crystal morphology within PCL spherulites crystallized at room temperature. A clear morphological change is observed with the addition of PMeDX as shown in Fig. 6B. Indeed, the presence bright domains within the lamellae can be seen in Fig. 6B. The bright regions possibly correspond to amorphous phase as reported by Gomez-Pachon et al.54
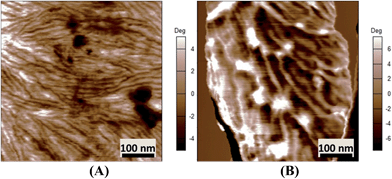 |
| | Fig. 6 AFM image of espun PCL (A) and espun 85/15 PCL/PMeDX mat (B) showing morphological change of crystallites. | |
Moreover, addition of PMeDX caused a change in the width of the lamellae from 14.8 ± 2.9 nm in 100/0 mat to 32.0 ± 11.5 nm in 85/15 mat, suggesting that PMeDX influences PCL crystallization as already noted from Avrami constants, K and n in the discussion on DSC analysis. This confirms that PMeDX interferes with PCL nucleation and crystallite formation.
PLLA/PMeDX fibres do not show clear structures as for PCL/PMeDX but exhibit corrugation-like patterns (Fig. 7) whereas some PLLA fibres show a regular wavy pattern perpendicular to the direction of the fibres. The lamellae appeared very thin and were arranged in a parallel manner but with changing orientation relative to the fibre, unlike in the case of espun PDX fibre (Fig. 8).
 |
| | Fig. 7 AFM phase images of espun PLLA/PMeDX (A) 100/0 and (B) 85/15. | |
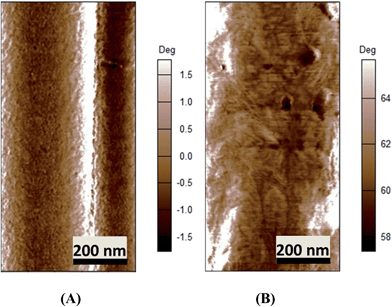 |
| | Fig. 8 AFM phase image of espun (A) PLLA/PMeDX (85/15) and (B) PDX fibre. | |
Fibre diameter and pore size.
Table 7 summarizes fibre diameters and pore sizes as determined by SEM as well as porosity values based on calculations described in the experimental section. Overall, fibre diameters decrease with increasing PMeDX wt% for both espun PCL/PMeDX and PLLA/PMeDX fibres while no clear trend was observed for PDX/PMeDX fibres, data was more erratic due to protrusions on the fibre surface and no clear trend was observed.45 The decrease is explained by the fact that fibre diameter of espun binary blends of incompatible polymers is dependent on the internal phase morphology and solution viscosity. Indeed, if the dispersed phase in the blend has a lower viscosity, as that of PMeDX, the solution jet will be stretched and elongated more by the electric forces during the electrospinning process.55 However, the decrease in fibre diameter was more pronounced for PCL/PMeDX fibres compared to PLLA/PMeDX due to its higher elasticity and stretchability.
Table 7 Effect of PMeDX wt% on fibre diameters
| Blend composition (w/w)% |
Fibre diameters/(μm) |
Inside pore size/(μm) |
Outside pore size/(μm) |
Porosity |
|
Espun PCL/PMeDX mats
|
| 100/0 |
0.35 ± 0.26 |
1.2 ± 0.79 |
0.83 ± 0.27 |
12.3 ± 0.9 |
| 98/2 |
0.27 ± 0.15 |
0.78 ± 0.34 |
0.68 ± 0.31 |
14.5 ± 1.3 |
| 93/7 |
0.28 ± 0.21 |
0.63 ± 0.24 |
0.56 ± 0.19 |
20.1 ± 1.1 |
| 90/10 |
0.25 ± 0.13 |
0.58 ± 0.22 |
0.60 ± 0.25 |
23.2 ± 1.5 |
| 85/15 |
0.23 ± 0.10 |
0.62 ± 0.23 |
0.72 ± 0.24 |
27.4 ± 1.7 |
|
|
Espun PLLA/PMeDX mats
|
| 100/0 |
0.33 ± 0.11 |
0.68 ± 0.34 |
0.73 ± 0.37 |
13.7 ± 0.6 |
| 98/2 |
0.34 ± 0.11 |
0.79 ± 0.43 |
0.89 ± 0.38 |
14.4 ± 1.6 |
| 93/7 |
0.34 ± 0.10 |
0.82 ± 0.33 |
0.57 ± 0.21 |
18.8 ± 1.2 |
| 90/10 |
0.30 ± 0.09 |
0.67 ± 0.24 |
0.81 ± 0.33 |
20.1 ± 1.8 |
| 85/15 |
0.30 ± 0.08 |
0.64 ± 0.26 |
0.71 ± 0.27 |
25.7 ± 2.1 |
However, as for espun PDX/PMeDX fibres, there was no clear cut trend in pore size for both espun PCL/PMeDX and PLLA/PMeDX fibres. Porosity values were found to increase slightly with increasing PMeDX content in both cases which may suggest a decrease in fibre packing density.
In general, the extent of whipping motion determines fibre diameter. When the electrospinning jet experiences more whipping motion with crystallization most likely occurring before the jet reaches the collector, small diameter fibres result with fibrillar structures. Large diameter fibres experience less whipping and crystallization is most likely to occur after the jet reaches the collector.56
Mechanical performance.
Mechanical properties of espun fibres depend on a combination of several factors such as fibre alignment, fibre diameter, fibre lay-ups and interface properties of fibre–fibre contact (fibre fusion).57,58 Tensile properties of fibres are affected by their structural morphology. In fact, the strength and elastic modulus of fibres is influenced by the lamellar and amorphous fractions of chains present within fibres. Elastomeric property of fibres is due to the amorphous phase of the fibres while dimensional stability is attributed to the crystalline phase.59 Thus, the mechanical deformation characteristic of the fibre is influenced by both the random amorphous and ordered crystalline phases in the fibre.59 The internal micro/nanostructural morphology of nanofibres determines their physical and mechanical properties.60 As reported in previous studies, changes in lamellae alignment and thickness result in variations of mechanical properties.61 Saffar et al.62 demonstrated that elongation at break decrease with crystalline orientation. Also, Bozic et al.63 showed that substrates with coarse lamellar structures possessed higher elongation compared to those with fine lamellar microstructures.
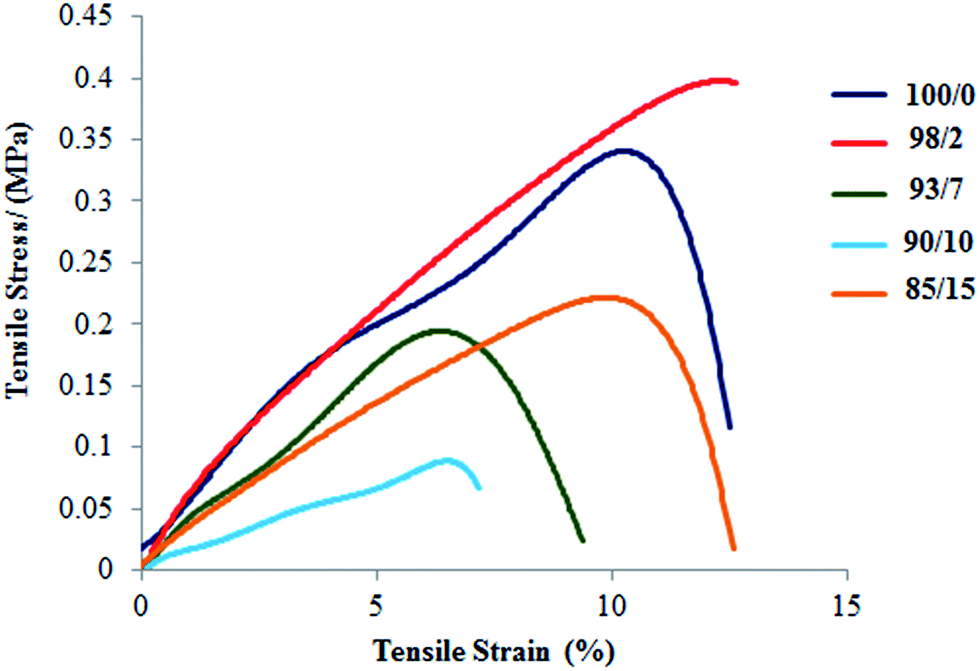
As reported in our previous paper,45 the Young's modulus of espun PDX/PMeDX mats decrease with increasing PMeDX wt%. Also, in general, both strain at break and peak stress increase with decreasing fibre diameters. PCL/PMeDX blend mats show a significant drop in Young's modulus for 93/7, 90/10 and 85/15 compositions compared to 100/0 and 98/2 (Table 8 and Fig. 9). It is likely that the formation of phase boundaries impact on mechanical performance more than crystallinity changes.64
Table 8 Mechanical properties of espun PCL/PMeDX mats
| PCL/PMeDX composition (wt%) |
Tensile strain (mm/mm) |
Modulus (MPa) |
Extension at break (mm) |
| 100/0 |
0.14 ± 0.03 |
5.33 ± 0.025 |
10.33 |
| 98/2 |
0.13 ± 0.007 |
5.19 ± 0.026 |
9.58 |
| 93/7 |
0.11 ± 0.002 |
3.54 ± 0.004 |
8.12 |
| 90/10 |
0.08 ± 0.002 |
1.41 ± 0.003 |
6.44 |
| 85/15 |
0.13 ± 0.005 |
2.86 ± 0.011 |
9.56 |
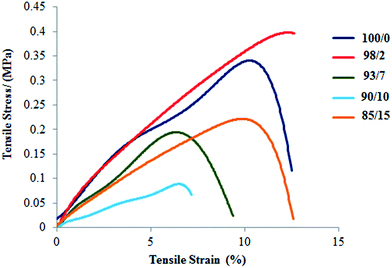 |
| | Fig. 9 Stress–strain curves of espun PCL/PMeDX mats. | |
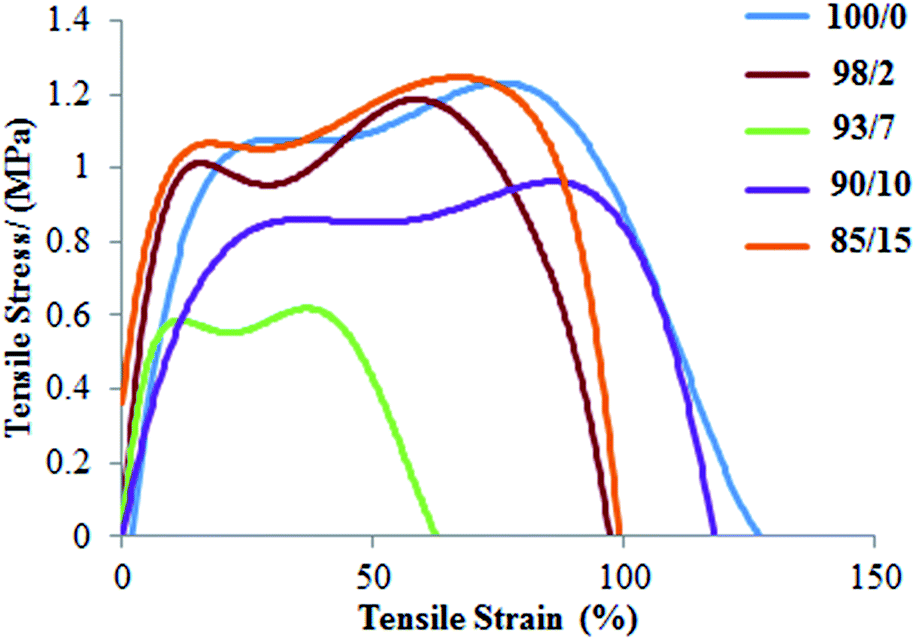
On the other hand, the interpretation of mechanical properties for PLLA/PMeDX blends is more difficult as no clear trend is observed with increasing PMeDX content (Table 9 and Fig. 10). This could be due to antagonist effects such as drop in crystallinity which impacts negatively on mechanical properties (98/2 composition) and formation of stereocomplex which could enhance (93/7 and 90/10) but in other cases (85/15) limit mechanical performance. This can be explained by the stereocomplex formation based on thermal analysis data where the percentages of homoPLLA crystallites and PLLA/PMeDX stereocomplex crystallites may affect mechanical performance.
Table 9 Mechanical properties of espun PLLA/PMeDX mats
| PLLA/PMeDX composition (wt%) |
Tensile strain (mm/mm) |
Modulus (MPa) |
Extension at break (mm) |
| 100/0 |
1.40 ± 0.022 |
9.65 ± 0.100 |
14.05 |
| 98/2 |
1.00 ± 0.007 |
4.57 ± 0.031 |
10.01 |
| 93/7 |
0.89 ± 0.001 |
17.24 ± 0.018 |
8.90 |
| 90/10 |
1.25 ± 0.0007 |
30.11 ± 0.018 |
12.55 |
| 85/15 |
1.00 ± 0.003 |
4.78 ± 0.019 |
10.01 |
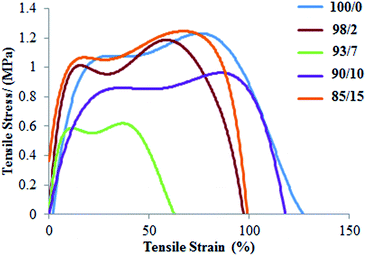 |
| | Fig. 10 Stress–strain curves of espun PLLA/PMeDX mats. | |
Hydrolytic degradation studies
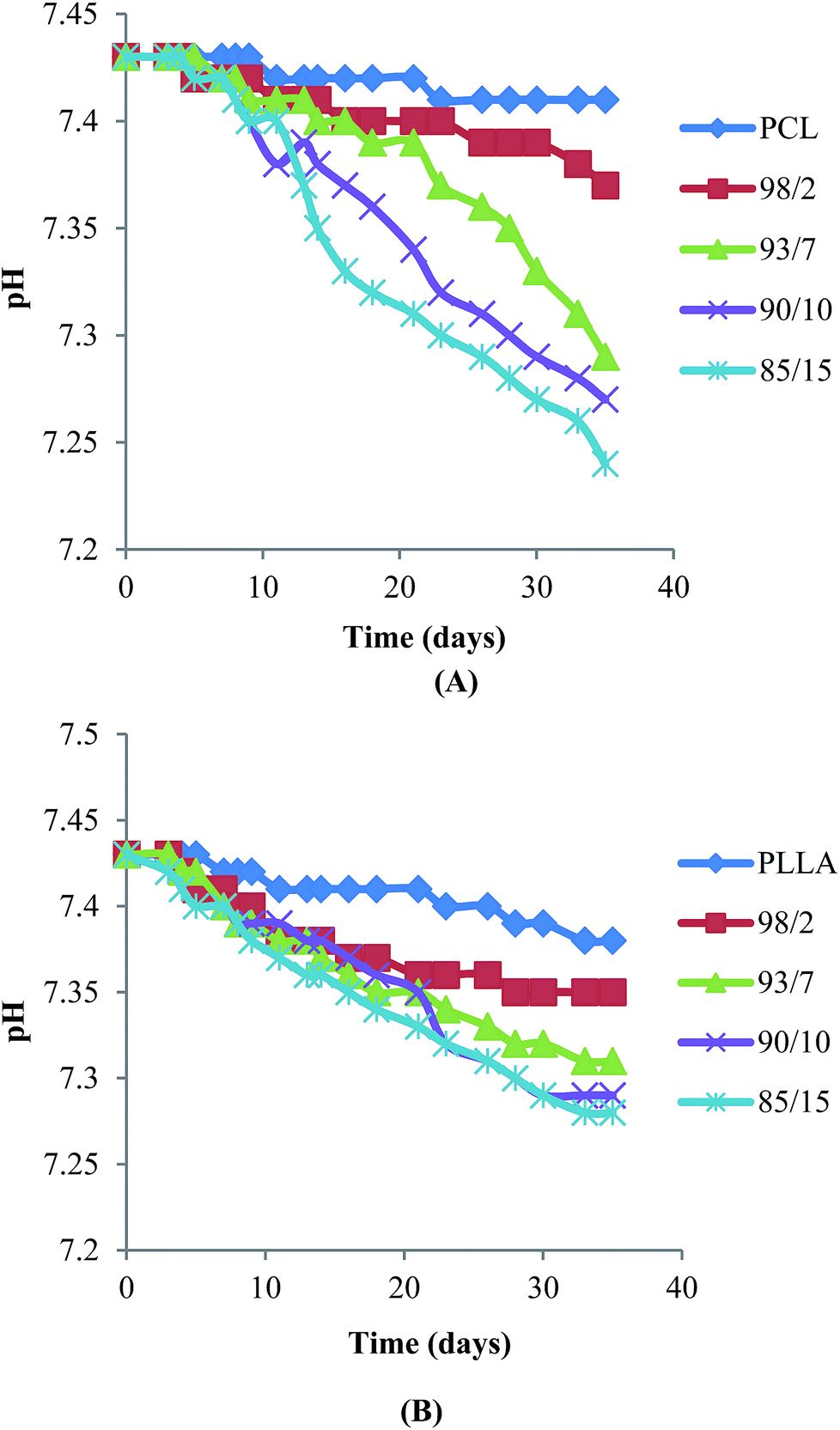
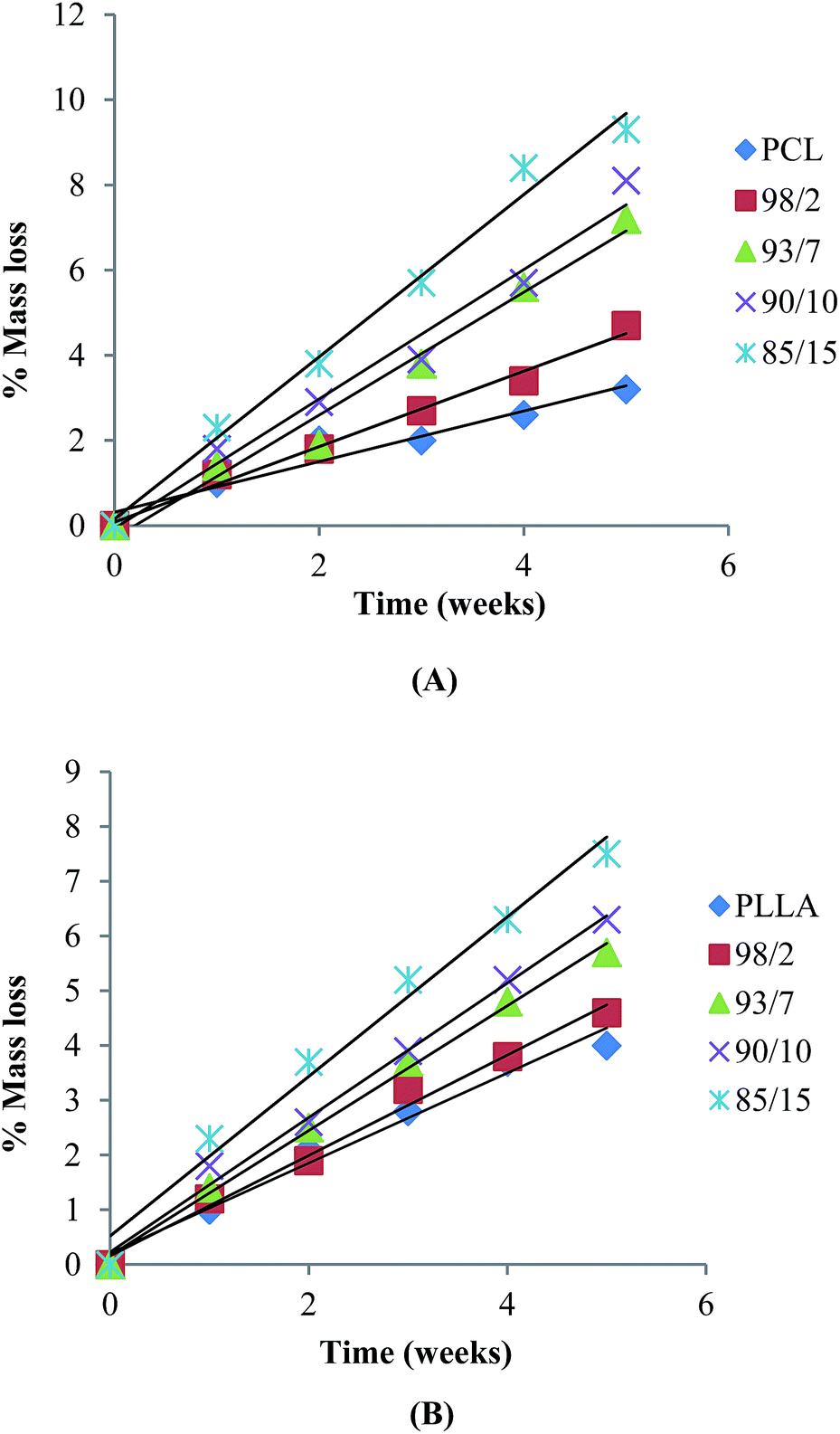
Hydrolytic degradation of espun mats was carried out in PBS at 37 °C for 5 weeks. We have shown recently that degradation of espun PDX/PMeDX mats occurred via surface erosion and was found to be dependent on fibre diameter of the fibres.45 Larger diameter espun 98/2 PDX/PMeDX mat degraded at a faster rate than smaller diameter 85/15 mat. Similarly, espun PCL/PMeDX and PLLA/PMeDX mats appear to degrade via a surface erosion mechanism during the time period investigated as noted by the slight drop in pH (Fig. 11) and linear mass loss profiles (Fig. 12). Mass loss profiles showed enhanced degradation with increasing PMeDX content which can possibly be explained by a combination of factors: decreased crystallinity in the blends, reduced fibre diameters and higher porosities. Mass loss profiles show that espun PCL/PMeDX and PLLA/PMeDX exhibit almost similar degradation rates. For instance, 93/7 PCL/PMeDX and PLLA/PMeDX mat had a mass loss of about 7.2 and 5.7% at week 5 and 85/15 PCL/PMeDX and PLLA/PMeDX mat had a mass loss of approximately 9.3 and 7.5% respectively at week 5. It has to be pointed out that mass loss values correspond to a relatively short degradation time. Zhao et al.65 also observed linear mass loss profiles for the hydrolytic degradation of espun PLLA and PLLA/hydroxyapatite mats.
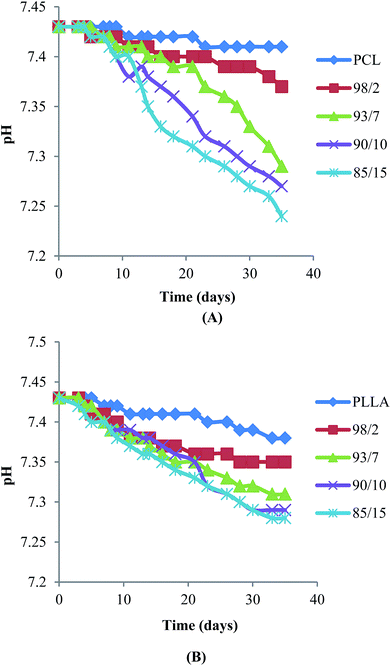 |
| | Fig. 11 pH evolution of (A) PCL/PMeDX and (B) PLLA/PMeDX mats with varying PMeDX wt%. | |
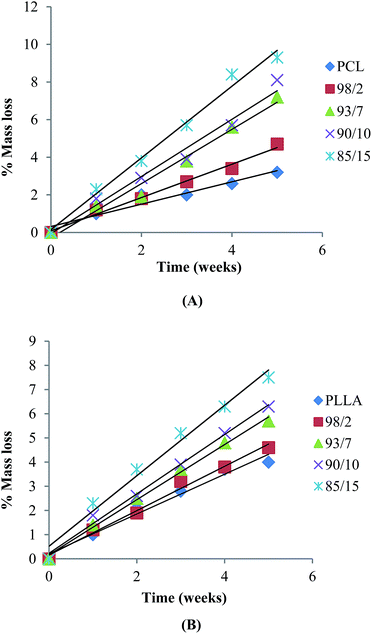 |
| | Fig. 12 Mass loss of (A) PCL/PMeDX and (B) PLLA/PMeDX mats as a function of hydrolysis time in PBS at 37 °C. | |
Degraded samples were analysed by SEM. Fig. 13 depicts SEM images of espun 98/2 and 85/15 PCL/PMeDX and PLLA/PMeDX mats after 5 weeks of degradation. Both 98/2 PCL/PMeDX and PLLA/PMeDX mats show no change in morphology at week 5. However, the 85/15 PCL/PMeDX mat shows fibre melting which is not observed in the corresponding PLLA/PMeDX mat. This can possibly be explained by the smaller fibre diameter of the 85/15 espun PCL/PMeDX mat.
 |
| | Fig. 13 SEM (3000× magnification, scale bar = 10 μm) of espun 98/2 and 85/15 PCL/PMeDX (top) and PLLA/PMeDX (bottom) mats at week 5. | |
In vitro biocompatibility studies
We have previously shown that addition of PMeDX to espun PDX fibres resulted in a greater density of viable human dermal fibroblasts compared to espun PDX mat with cells migrating up to a maximum of 45.1 ± 11.8% throughout the scaffold after 7 days.45 This was attributed to smaller fibre packing density and higher porosity. Espun PCL/PMeDX and PLLA/PMeDX mats were subjected to cell viability studies to investigate the effect of PMeDX incorporation in PCL and PLLA. Human dermal fibroblasts (HDFs) were cultured and seeded on the scaffolds for a period of up to 7 days.
Cell attachment and proliferation.
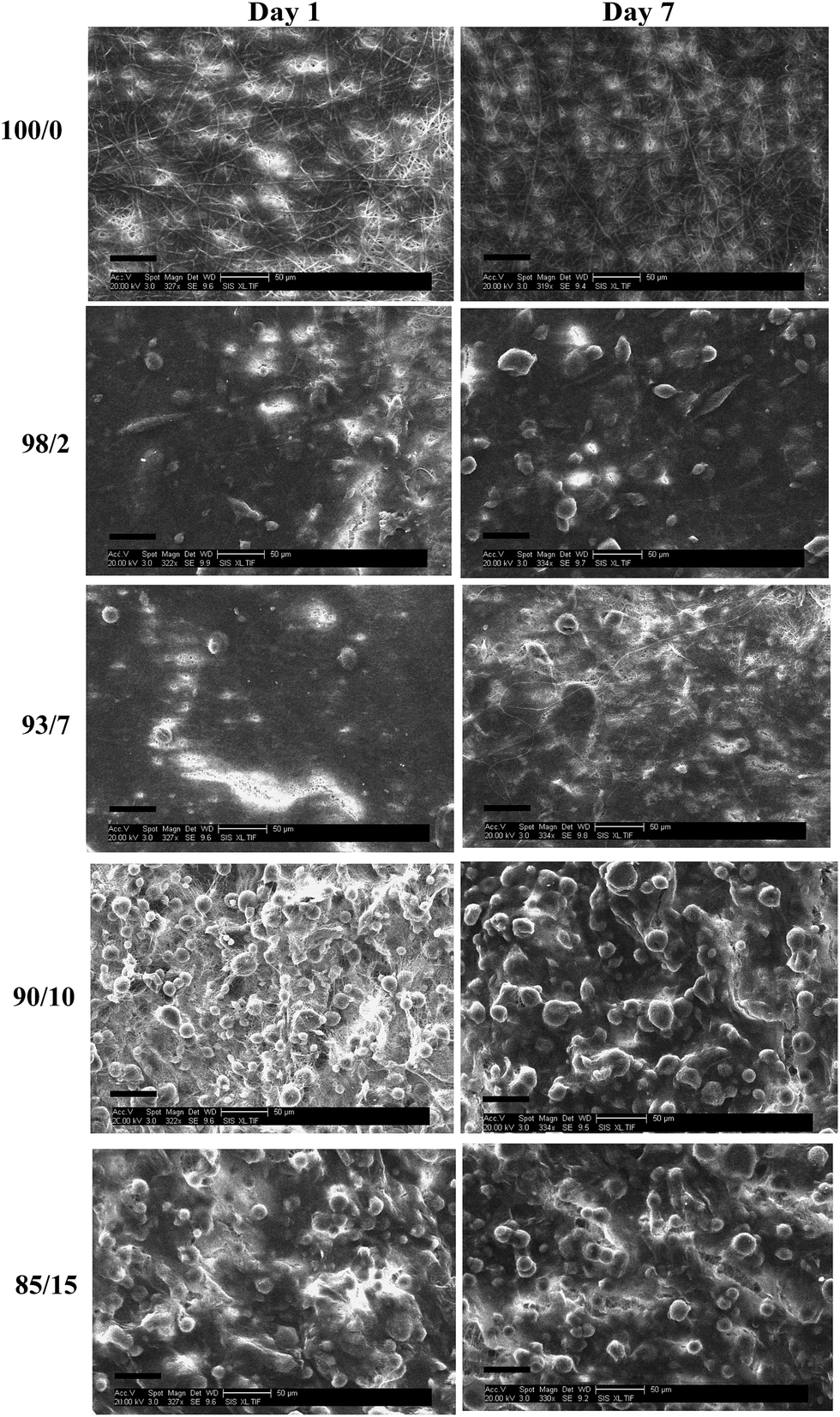
Fig. 14 and 15 show the SEM images of HDFs cultured on espun PCL/PMeDX and PLLA/PMeDX scaffolds after 1 and 7 days respectively. In general, it was observed that cells spread over the mat surface. Compared to espun PCL or PLLA mats, a higher density of cells was found on PCL/PMeDX and PLLA/PMeDX scaffolds as from day 1.
 |
| | Fig. 14 SEM images (scale bar = 50 μm) of cell seeded espun PCL/PMeDX scaffolds after days 1 and 7 respectively. | |
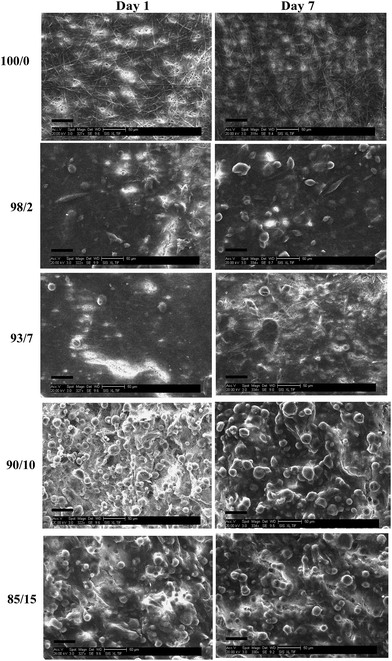 |
| | Fig. 15 SEM of (scale bar = 50 μm) cell seeded espun PLLA/PMeDX scaffolds after days 1 and 7 respectively. | |
A change in cell morphology was noted with increasing PMeDX content from bipolar spindle (as red marked in Fig. 14) to a cobble-stone morphology with poorly organized actin filaments (Table 10) which was more pronounced with PLLA/PMeDX than PCL/PMeDX. This suggests that higher PMeDX content results in poor cell adhesion, causing the cells to adopt a cobble-stone morphology with few filopodia as can be seen in espun 85/15 PCL/PMeDX mat (Fig. 16). The cobble-stone shaped cells seem to be covered with deposited material, which is most probably ECM. Despite poor adhesion to the substrate, the balled up cells proliferated and produced matrix to create a conducive environment. As discussed in previous sections, addition of PMeDX to PLLA alters mechanical properties and crystallinity more significantly compared to PCL and as highlighted in the introduction, cell growth is influenced by changes in crystallinity and mechanical properties of the substrate. The presence of cobble-stone HDF morphology was detected as from 15 wt% PMeDX for PCL/PMeDX mat and as early as 2 wt% PMeDX for PLLA/PMeDX mat. A change in fibroblast morphology from elongated, oriented to cobble-stone was observed with the application of shear stress as reported by Braddon et al.66 The change in HDF morphology can be explained by the change in microstructures of espun fibres. Ajami-Henriquez et al.67 reported that the presence of lamellae promotes “cell contact-guidance”. In fact, they showed that other factors such as chemical composition, degree of crystallinity and surface roughness did not play a major role in determining cell preference towards a specific material. Based on the above, we deduce that the presence of lamellae in espun PCL/PMeDX fibres may account for the fact that cells grown on these surfaces were mostly spindle-shaped. Furthermore, cobble-stone morphology observed on espun PLLA/PMeDX mats can possibly be related to the absence of lamellae on the mats.
Table 10 Influence of crystallinity and tensile strain on cell morphology
| Blend composition (w/w)% |
Cell morphology |
Crystallinity χblend/(%) |
Tensile strain/(mm/mm) |
|
Espun PCL/PMeDX
|
| 100/0 |
Bipolar spindle |
32.2 |
0.14 |
| 98/2 |
Bipolar spindle |
30.9 |
0.13 |
| 93/7 |
Bipolar spindle |
31.0 |
0.11 |
| 90/10 |
Bipolar spindle |
29.2 |
0.08 |
| 85/15 |
Cobble-stone |
25.1 |
0.13 |
|
|
Espun PLLA/PMeDX
|
| 100/0 |
Bipolar spindle |
61.4 |
1.40 |
| 98/2 |
Cobble-stone |
50.2 |
1.00 |
| 93/7 |
Cobble-stone |
48.4 |
0.89 |
| 90/10 |
Cobble-stone |
46.8 |
1.25 |
| 85/15 |
Cobble-stone |
45.3 |
1.00 |
 |
| | Fig. 16 SEM of (1327× magnification, scale bar = 20 μm) cell seeded espun PCL/PMeDX 85/15 scaffolds after day 7. | |
In contrast, no change in morphology was observed for PDX/PMeDX mat with 0–15 wt% PMeDX when subjected to cell growth. An increasing degree of heterogeneity and surface roughness noted with increasing amorphous PMeDX in espun PDX/PMeDX mats45 could explain enhanced cell adhesion. It was therefore hypothesized that surface roughness in espun PDX/PMeDX was the dominant factor on fibroblasts proliferation compared to the decrease in crystallinity.
Cell behaviour has been reported to be dependent on three factors namely, percentage cell coverage of the surface, pore size and cell type. Indeed, as demonstrated by Salem et al.,68 fibroblasts displayed a co-operative pattern of cell spreading whereby pores greater than cell dimensions were bridged by group of cells using their neighbours as supports.
As depicted in Table 10, the decreasing crystallinity of the blends with increasing PMeDX wt% here implies an enhanced flexibility of the corresponding mats and in accordance with the introductory paragraph explains poorer cell adhesion especially in the case of PLLA/PMeDX mats where only 2 wt% PMeDX causes a drop of 10% in crystallinity.
As discussed in the Introduction, previous reports have shown that fibre diameter, porosity and surface roughness influence surface hydrophobicity which also affects cell behaviour. To have a better insight into surface hydrophobicity, contact angles (CA) were measured for a few samples and are listed in Table 11. Compared to PCL, the contact angle of a 93/7 PCL/PMeDX mat is nearly halved which translates a significant increase in surface hydrophilicity. The same trend was previously noted for espun PDX/PMeDX where the contact angles decreased upon increasing content of PMeDX.45 Interestingly, cell adhesion appeared optimal for this 93/7 PCL/PMeDX mat, with extensive ECM secretion by day 7. This supports the fact that higher hydrophilic surfaces promote cell adhesion. On the other hand, increasing PMeDX in PLLA/PMeDX mats did not cause significant change in surface contact angles. This could partly explain the poor adhesion of cells onto PLLA/PMeDX mats, as we highlighted in the previous paragraphs.
Table 11 Summary of static contact angle measurements on espun PCL/PMeDX and PLLA/PMeDX mats
| Blend composition (w/w)% |
CA (mean ± SD) |
|
Espun PCL/PMeDX
|
| 100/0 |
131.6 ± 2.5 |
| 93/7 |
63.1 ± 2.1 |
|
|
Espun PLLA/PMeDX
|
| 100/0 |
127.2 ± 4.2 |
| 98/2 |
132.3 ± 5.0 |
| 85/15 |
130.0 ± 2.0 |
Cell infiltration.
Engineered scaffolds should not only support cell attachment and proliferation but should also allow cell infiltration throughout the scaffolds so as to promote uniform tissue regeneration.69 A major challenge therefore concerns the promotion of cellular ingrowth into espun scaffolds.
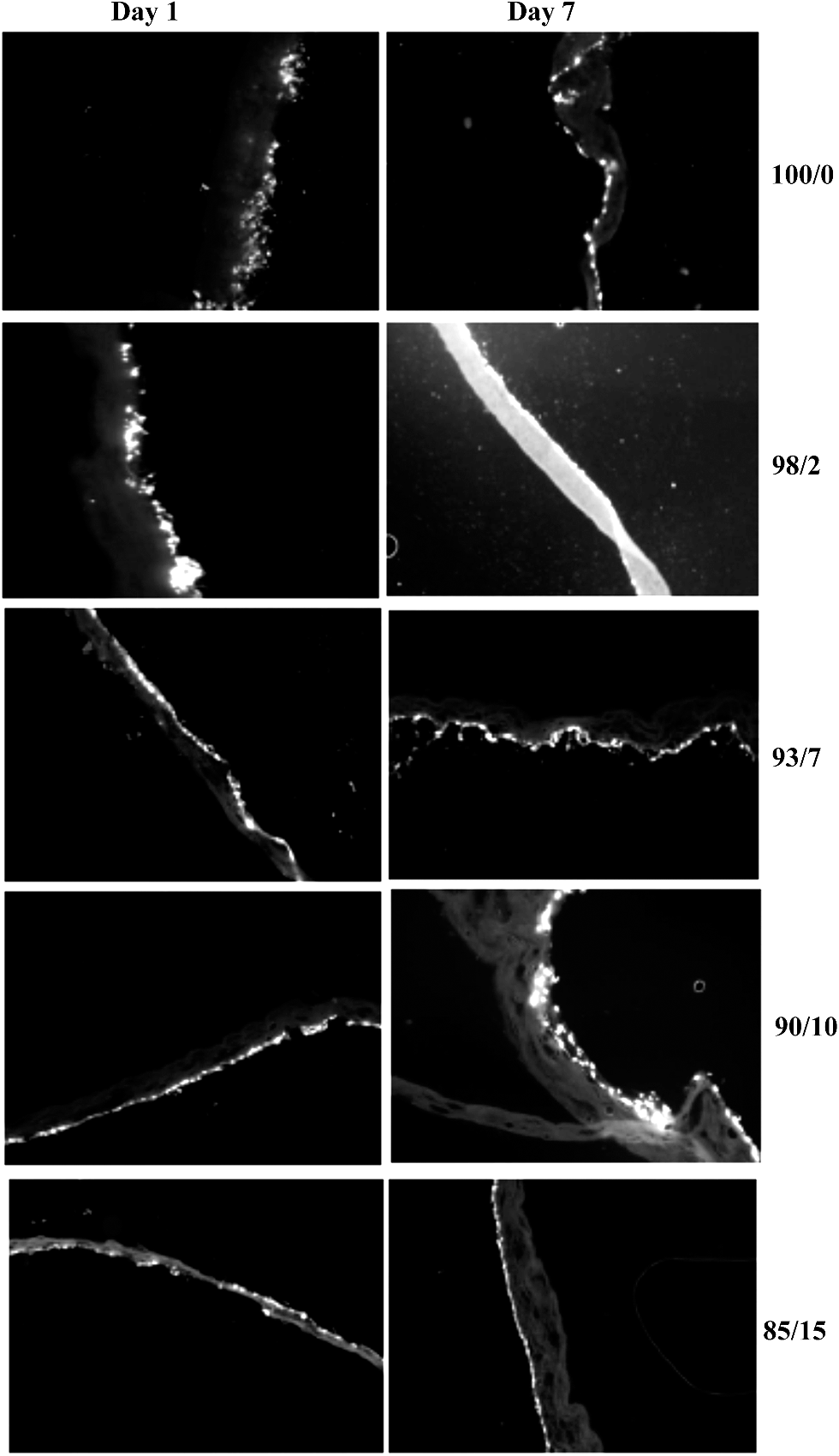
Cell-seeded espun PCL/PMeDX and PLLA/PMeDX mats were cryo-sectioned and stained with 4′-6-diamidino-2-phenylindole (DAPI) to image cell nuclei and determine cell migration. Fig. 17 and 18 depict the fluorescence microscopy images of HDFs cultured on the scaffolds. HDFs appear as bright dots as can be noted from Fig. 17. The depth of cellular infiltration was quantified and results summarised in Table 12. As detailed in the Experimental section, we have privileged % migration rather than distance migration. As observed previously, espun PDX/PMeDX mats show an infiltration up to 45.1%.45 No infiltration is noted for PCL/PMeDX mats independently of PMeDX composition. Infiltration is observed in varying percentages for PLLA/PMeDX depending on composition. Noteworthy is the fact that a 85/15 blend shows a 100% infiltration. Comparison of the different blend mats shows that cell morphology appears to be a dominating factor influencing cell infiltration. Indeed, in the case of PDX/PMeDX and PCL/PMeDX mats, HDFs adopt more of a spindle-shape whereas cells are more cobble-stone shaped in the case of espun PLLA/PMeDX mats. The pore size of the scaffold mat is also a determining factor. Indeed, literature reveals that HDFs start secreting ECM almost immediately in scaffolds with pore size less than 15 μm.
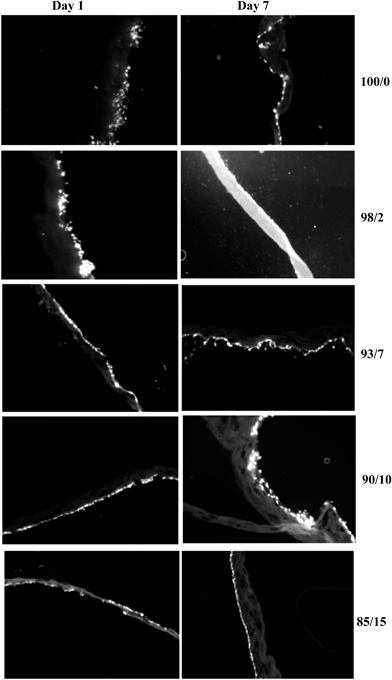 |
| | Fig. 17 Fluorescence microscopy images (magnification 10×) of HDFs on espun PCL/PMeDX mats after days 1 and 7 respectively. | |
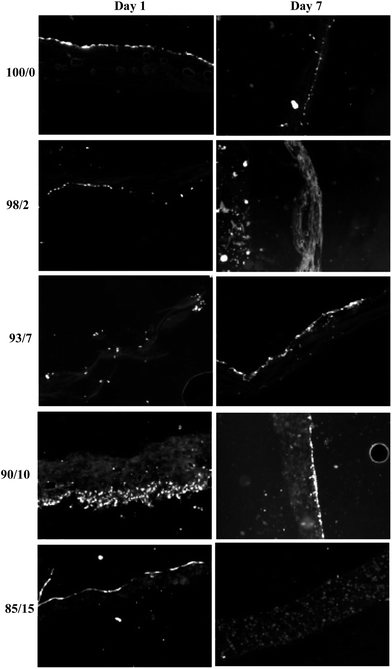 |
| | Fig. 18 Fluorescence microscopy images (magnification 10×) of HDFs on espun PLLA/PMeDX mats after days 1 and 7 respectively. | |
Table 12 Summary of HDF migration in espun PCL/PMeDX and PLLA/PMeDX mats
| Blend composition (w/w)% |
Depth of cellular migration (%) |
| Espun PDX/PMeDX |
Espun PCL/PMeDX |
Espun PLLA/PMeDX |
| 100/0 |
0 |
0 |
0 |
| 98/2 |
45.1 ± 11.8 |
0 |
| 93/7 |
0 |
15.1 ± 5.68 |
| 90/10 |
0 |
4.65 ± 10.6 |
| 85/15 |
0 |
100 |
It is possible that the presence of corrugation-like structures in 85/15 PLLA/PMeDX scaffold enhanced cell infiltration. Indeed, as reported previously,70 microstructure and morphological features such as lamellar thickness or interlamellar distance were found to guide human mesenchymal stem cells (hMSCs) towards the interior of silk scaffold, producing a multilamellar hybrid construct.70
HDFs penetrated the full thickness of the 85/15 PLLA/PMeDX scaffold and were homogeneously distributed at day 7. All cells resided on the surface of the 85/15 PLLA/PMeDX scaffold at day 1. This suggests that cells observed within the scaffold at day 7 is a result of cell migration and not initial cell seeding. We can therefore conclude that espun 85/15 PLLA/PMeDX mat had the right combination of fibre diameter, pore size, porosity, mechanical and degradation properties. Such extensive cell migration is quite rare for espun materials without modification in porosity. Indeed, a number of methodologies have been proposed for improving cell infiltration. For instance, electrospinning has been combined with salt leaching to produce a PCL scaffold with engineered delaminations.71 Up to 4 mm of cellular infiltration was observed after 3 weeks in culture. Another common method of increasing porosity of espun mats is via the selective removal of sacrificial fibres as reported in a study by Baker et al.72 Overall, cellular infiltration improved with increasing PEO content. At higher PEO contents, nearly complete infiltration was observed. However, some regions remained devoid of cells and cell distribution within the scaffold was non-homogeneous.
Overall cytocompatibility results indicate that the inclusion of PMeDX into espun PCL and PLLA scaffolds improves the in vitro bioactivity of the matrix through either chemical or mechanical signalling, or a combination of the two.
In summary, in vitro cell culture studies showed that compared to espun PCL and PLLA mats, a greater density of viable cells were observed on espun PCL/PMeDX and PLLA/PMeDX scaffolds respectively. Moreover, extensive HDF infiltration was noted in espun 85/15 PLLA/PMeDX mats.
Experimental section
Materials
Tin(II) octanoate (Alfa Aeser, 97%) was used as received. Polycaprolactone (CAPA™ 6500-inherent viscosity 2.45 at a concentration of 0.1 g dL−1 at 30 °C in 1,1,1,3,3,3-hexafluoroisopropanol) was obtained from Solvay Chemicals. Polylactic acid (Purasorb PL18, inherent viscosity of 1.8 at a concentration of 0.1 g dL−1 at 25 °C in chloroform) was purchased from Purac. D,L-3-methyl-1,4-dioxan-2-one was synthesized according to procedures previously described by us.73 1,1,1,3,3,3-hexafluoroisopropanol from Apollo Scientific Limited was used as received. Phosphate buffer solution (PBS) was purchased from Sigma-Aldrich and used as received.
Methods
Polymerization of 3-MeDX.
A typical polymerization is hereby described. A solution of Sn(Oct)2 was prepared by dissolving 0.1 g of Sn(Oct)2 in toluene (5 mL) in a glove box. 29 μL of this solution was transferred to a quick fit tube containing 3-MeDX (1.16 g). The tube was then placed in a preheated oil bath at 80 °C. After the desired polymerization time, the reaction was quenched in liquid nitrogen and the crude sample purified by dissolving in chloroform and precipitating in petroleum ether. The product was then dried under vacuum before characterization by 1H-NMR and 13C-NMR.
Electrospinning of PCL/PMeDX and PLLA/PMeDX blends.
PCL/PMeDX and PLLA/PMeDX were blended in the following ratios: 100/0, 98/2, 93/7, 90/10, and 85/15 w/w%. Each blend was immersed in HFIP in separate vials at a concentration of 100 mg mL−1 and left on a shaker plate overnight. The polymer solutions were then loaded into a 3 mL syringe and dispensed at a constant rate using a KD Scientific syringe pump. Electrospinning parameters were optimized (rate: 3.5 mL h−1, air-gap distance: 20 cm, voltage: +26 kV, grounded back target) to produce continuous, non-woven fibres which were collected on a rotating grounded rectangular mandrel (7.5 × 2.5 × 0.5 cm). After electrospinning, scaffolds were removed from the mandrel and stored in a desiccation chamber until further analysis.
Determination of porosity.
The porosity of the scaffolds was measured using a method described by Soliman et al.74 Briefly, 10 × 10 mm2 disks of the espun scaffolds were weighed and subsequently immersed in 70% ethanol overnight with slight mechanical agitation. This was done to allow the ethanol to penetrate into the scaffold pores. The surface of the samples was then blotted dry on a filter paper and weighed once more to determine the mass of the ethanol present within the scaffold. Measurements were made on three samples of each scaffold type. The density of ethanol, PCL and PLLA are 0.788, 1.123 and 1.23 g mL−1 respectively. The porosity (ε) was calculated as:| | 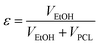 | (1) |
where VEtOH is the volume of the intruded ethanol and was calculated as the ratio of the observed mass change after intrusion and ρEtOH. VPCL is the volume of PCL fibres and was calculated as the ratio between the dry scaffold mass before intrusion and the density of PCL (ρPCL).
Non-isothermal crystallization kinetics.
Non-isothermal crystallization kinetics was investigated using eqn (2) and (3) as detailed in our recent paper.45| | 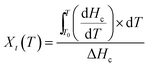 | (2) |
T0: onset temperature of crystallization, T: an arbitrary temperature, dHc/dT: variation of the enthalpy of crystallization as a function of temperature variation, ΔHc: total enthalpy of crystallization at a specific cooling rate.| | 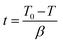 | (3) |
t: time, T: temperature, β: constant cooling rate.
Hydrolytic degradation of electrospun mats.
Espun mats were placed in PBS at 37 °C for five weeks. After each degradation period, the samples were washed and dried under vacuum. % mass loss and % weight retention were calculated according to eqn (4) and (5) respectively.| | 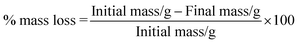 | (4) |
| | 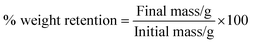 | (5) |
In vitro biocompatibility studies.
Cell culture and migration studies were carried out using human dermal fibroblasts. The experimental protocol was similar to the one used in our previous paper.45 To standardize the variation in scaffold thickness, cell migration was analysed as a percentage of distance travelled with respect to the thickness at that spot instead of solely distance travelled. % migration (n = 30) was calculated according to eqn (6).| |  | (6) |
Measurements
Differential Scanning Calorimetry (DSC) analysis was carried out using a Netzsch DSC 200 F3 Maia® thermal analyzer (Chennai, India). All PCL/PMeDX blend samples were heated from 30 to 80 °C, cooled to −30 °C and reheated to 80 °C at 3 °C min−1. All PLLA/PMeDX blend samples were heated from 30 to 180 °C, cooled to −30 °C and reheated to 180 °C at 3 °C min−1. Netzsch TG 209 F3 Tarsus® analyzer (Chennai, India) was used to measure and record the sample mass change with temperature over the course of the pyrolysis reaction. Thermogravimetric curves were obtained at a heating rate of 10 °C min−1 between 25 °C and 700 °C. Nitrogen was used as an inert purge gas to displace air in the pyrolysis zone, thus avoiding unwanted oxidation of the sample. The sample mass used in this study was approximately 10 mg. SEM data were acquired on a Philips XL 30 scanning electron microscope operated at an acceleration voltage of 20 kV using the secondary electron detector. The polymer samples were sputter-coated with gold (5 nm thick) before imaging (Edwards, UK). Inner (in contact with mandrel) surface of the espun mats were imaged. To determine fibre and pore size, the ImageTool 3.0 image analysis software package was used (Shareware provided by University of Texas Health Science Center at San Antonio). The software was calibrated using the micron scale bar of each picture. An average fibre diameter was determined by measuring the diameter of 60 different fibres, while an average pore size was determined by measuring the diameter of 60 different pores. Pores were identified as areas of void space bounded by fibres on all sides at or near the same depth of field, while their long and short diagonal axes were measured and averaged together to serve as their diameter.75 Intermittent contact (tapping) mode AFM imaging was done on as prepared samples on an Asylum MFP-3D atomic force microscope (Asylum Research, USA) using Olympus AC160TS cantilevers (with a resonance frequency of 300 kHz and a nominal spring constant of 40 N m−1) under ambient conditions. The rms amplitude of the cantilever was adjusted to 85 nm and a setpoint ratio of 0.8 was chosen. Constant amplitude images were acquired, depending on the scan size, with 512 pixels × 512 pixels (up to 2024 pixels × 2024 pixels); the phase shift was recorded simultaneously. The data was processed off line using the MFP-3D software. Tensile properties of the espun mats (40 × 10 × 4 mm3) were studied utilizing an Instron Tensile Tester 3343 (Instron, USA) at 27 °C and 60% relative humidity using a crosshead speed of 10 mm min−1, gauge length of 1 cm and 500 N load cell. Errors in Young's modulus and strain were calculated as reported in our previous paper.45 The contact angles of the modified surfaces were measured using water as a probe liquid (Milli-Q water from a Millipore Direct-Q 8 system with resistivity of 18.0MΩ cm−1) with an OCA 15plus instrument (Data Physics Instruments GmbH, Germany). Static contact angle data based on the sessile drop method were acquired immediately after deposition of a 1 μL drop on at least three positions for each sample and are stated as arithmetic mean. A short film sequence covering several seconds before and after deposition of the droplet was taken.
Conclusions
Espun PCL/PMeDX and PLLA/PMeDX mats were successfully fabricated from HFIP solutions. FTIR and TGA analysis showed that immiscibility increases with increasing PMeDX content for espun PCL/PMeDX fibres, with the 98/2 blend being partially miscible. AFM images revealed that espun PCL/PMeDX mats showed a rougher surface compared to espun PCL mat, possibly due to dispersion of PMeDX within the PCL matrix. Addition of PMeDX caused a change in the width of the lamellae as was noted from AFM images. Espun PLLA/PMeDX fibres exhibit corrugation-like patterns. The drop in mechanical properties of espun PCL/PMeDX was explained by the likely formation of phase boundaries which impact on mechanical performance more than crystallinity changes. No clear trend was observed with increasing PMeDX content for espun PLLA/PMeDX due possibly to antagonist effects such as drop in crystallinity which impacts negatively on mechanical properties (98/2 composition) and formation of stereocomplex. Degradation of espun mats occurred via a surface erosion mechanism. In addition, biocompatibility studies conducted using HDFs showed that in general, espun PCL/PMeDX and PLLA/PMeDX scaffolds supported cell growth better than the corresponding espun homopolymer mats. Correlation of physico-chemical properties with biological performance is very complex and the nature of the polymer plays a key role. Adjustment of physico-chemical and mechanical properties to match bio-performance requires careful investigation almost on a case by case basis.
Acknowledgements
We thank the Tertiary Education Commission (Mauritius) for awarding a PhD scholarship to N. Goonoo. We are grateful to the Mauritius Research Council (Mauritius) for supporting biomaterials and drug delivery research at the ANDI Centre of Excellence for Biomedical and Biomaterials Research (CBBR). We acknowledge the input from Gary Bowlin's group at University of Memphis for assistance with electrospinning and cell culture studies. Daniel Wesner and Holger Schönherr acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG instrument grant no. INST 221/87-1FUGG) and the European Research Council (ERC starting grant to HS, ERC grant agreement no. 279202).
Notes and references
- J. M. Holzwarth and P. X. Ma, J. Mater. Chem., 2011, 21, 10243–10251 RSC.
- M. N. Collins and C. Birkinshaw, Carbohydr. Polym., 2013, 92, 1262–1279 CrossRef CAS PubMed.
- N. Goonoo, A. Bhaw-Luximon, G. L. Bowlin and D. Jhurry, Polym. Int., 2013, 62(4), 523–533 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Adv. Biochem. Eng./Biotechnol., 2006, 102, 47–90 CrossRef CAS.
- Q. Cai, Y. Wan, J. Bei and S. Wang, Biomaterials, 2003, 24, 3555–3562 CrossRef CAS.
- Q. Cai, J. Yang and S. Wang, Biomaterials, 2002, 23, 4483–4492 CrossRef CAS.
- J. Venugopal, L. L. Ma, T. Yong and S. Ramakrishna, Cell Biol. Int., 2005, 29, 861–867 CrossRef CAS PubMed.
- J. R. Venugopal, Y. Zhang and S. Ramakrishna, Artif. Organs, 2006, 30(6), 440–446 CrossRef CAS PubMed.
- S. Srouji, T. Kizhner, E. Suss-Tobi, E. Livne and E. Zussman, J. Mater. Sci.: Mater. Med., 2008, 19, 1249–1255 CrossRef CAS PubMed.
- J. S. Choi, S. J. Lee, G. J. Christ, A. Atala and J. J. Yoo, Biomaterials, 2008, 29(19), 2899–2906 CrossRef CAS PubMed.
- X. Yang, J. D. Shah and H. Wang, Tissue Eng., Part A, 2009, 15(4), 945–956 CrossRef CAS PubMed.
- Y. M. Ju, J. S. Choi, A. Atala, J. J. Yoo and S. J. Lee, Biomaterials, 2010, 31(15), 4313–4321 CrossRef CAS PubMed.
- Y. Zhang, H. Ouyang, C. T. Lim, S. Ramakrishna and Z. M. Huang, J. Biomed. Mater. Res., 2005, 72(1), 156–165 CrossRef PubMed.
- Z. Ma, W. He, T. Yong and S. Ramakrishna, Tissue Eng., 2005, 11(7/8), 1149–1158 CrossRef CAS PubMed.
- E. J. Chong, T. T. Phan, I. J. Lim, Y. Z. Zhang, B. H. Bay, S. Ramakrishna and C. T. Lim, Acta Biomater., 2007, 3, 321–330 CrossRef CAS PubMed.
- R. S. Tigli, N. M. Kazaroğlu, I. S. B. Mav and I. O. M. Gümüşderel, J. Biomater. Sci., Polym. Ed., 2011, 22(1–3), 207–223 CrossRef CAS PubMed.
- S. Gautam, A. K. Dinda and N. C. Mishra, Mater. Sci. Eng. C, 2013, 33, 1228–1235 CrossRef CAS PubMed.
- Y. C. Lim, J. Johnson, Z. Fei, Y. Wu, D. F. Farson, J. J. Lannutti, H. W. Choi and L. J. Lee, Biotechnol. Bioeng., 2011, 108(1), 116–126 CrossRef CAS PubMed.
- S. J. Lee, J. Liu, S. H. Oh, S. Soker, A. Atala and J. J. Yoo, Biomaterials, 2008, 29, 2891–2898 CrossRef CAS PubMed.
- Y. Zhang, H. Ouyang, C. T. Lim, S. Ramakrishna and Z. M. Huang, J. Biomed. Mater. Res., Part B, 2005, 72(1), 156–165 CrossRef PubMed.
- R. Mehdinavaz Aghdam, S. Najarian, S. Shakhesi, S. Khanlari, K. Shabaani and S. Sharifi, J. Appl. Polym. Sci., 2012, 124(1), 123–131 CrossRef.
- J. H. Kim, P. H. Choung, I. Y. Kim, K. T. Lim, H. M. Son, Y. H. Choung, C. S. Cho and J. H. Chung, Mater. Sci. Eng. C, 2009, 29, 1725–1731 CrossRef CAS PubMed.
- J. Han, C. J. Branford-White and L. M. Zhu, Carbohydr. Polym., 2010, 79, 214–218 CrossRef CAS PubMed.
- S. R. Son, N. T. Ba-Linh, H. M. Yang and B. T. Lee, Sci. Technol. Adv. Mater., 2013, 14(1), 015009 CrossRef.
- S. Y. Gu, Z. M. Wang, J. Ren and C. Y. Zhang, Mater. Sci. Eng. C, 2009, 29, 1822–1828 CrossRef CAS PubMed.
- P. Torricelli, M. Gioffre, A. Fiorani, S. Panzavolta, C. Gualandi, M. Fini, M. L. Focarete and A. Bigi, Mater. Sci. Eng. C, 2014, 36, 130–138 CrossRef CAS PubMed.
- S. Wang, Y. Zhang, H. Wang, g. Yin and Z. Dong, Biomacromolecules, 2009, 10(8), 2240–2244 CrossRef CAS PubMed.
- K. T. Shalumon, D. Sathish, S. V. Nair, K. P. Chennazhi, H. Tamura and R. Jayakumar, J. Biomed. Nanotechnol., 2012, 8(3), 405–416 CrossRef CAS PubMed.
- H. Liu and S. D. Wang, J. Text. Res., 2012, 33(2), 21–25 CAS.
- L. Chen, Y. Bai, G. Liao, E. Peng, B. Wu, Y. Wang, X. Zeng and X. Xie, PLoS One, 2013, 8(8), 71265 Search PubMed.
- A. C. Areias, C. Ribeiro, V. Sencadas, N. Garcia-Giralt, A. Diez-Perez, J. L. Gomez-Ribelles and S. Lanceros-Mendez, Soft Matter, 2012, 8, 5818–5825 RSC.
- B. B. Mandal and S. C. Kundu, Biomaterials, 2009, 30, 2956–2965 CrossRef CAS PubMed.
- P. C. George, W. J. Miller, D. F. Meany, E. S. Sawyer and P. A. Janmey, Biophys. J., 2006, 90, 3012–3018 CrossRef PubMed.
- G. Ladam, L. Vonna and E. Sackmann, Acta Biomater., 2005, 1, 485–497 CrossRef PubMed.
- J. Solon, I. Levental, K. Sengupta, P. C. Georges and P. A. Janmey, Biophys. J., 2007, 93, 4453–4461 CrossRef CAS PubMed.
- R. J. Pelham and Y. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13661–13665 CrossRef CAS.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg and M. Ortiz,
et al.
, Cell Motil. Cytoskeleton, 2005, 60, 24–34 CrossRef PubMed.
- H. Cui and P. J. Sinko, Front. Mater. Sci., 2012, 6(1), 47–59 CrossRef PubMed.
- A. K. Yip, K. Iwasaki, C. Ursekar, H. Machiyama, M. Saxena, H. Chen, I. Harada, K. H. Chianm and Y. Sawada, Biophys. J., 2013, 104(1), 19–29 CrossRef CAS PubMed.
- C. M. Lo, H. B. Wang, M. Dembo and Y. L. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS.
- J. Yang, Y. Wan, C. Tu, Q. Cai, J. Bei and S. Wang, Polym. Int., 2003, 52, 1892–1899 CrossRef CAS.
- A. Hezi-Yamit, C. Sullivan, J. Wong, L. David, M. Chen and P. Cheng,
et al.
, J. Biomed. Mater. Res., Part A, 2009, 90(1), 133–141 CrossRef PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- N. Goonoo, A. Bhaw-Luximon, I. A. Rodriguez, G. L. Bowlin and D. Jhurry, Int. J. Polym. Mater. Polym. Biomater., 2014, 63(10), 1–11 Search PubMed.
- N. Goonoo, A. Bhaw-Luximon, I. A. Rodriguez, D. Wesner, H. Schönherr, G. L. Bowlin and D. Jhurry, Biomater. Sci., 2014, 2(3), 339–351 RSC.
- V. Crescenzi, G. Manzini, G. Calzolari and C. Borri, Eur. Polym. J., 1972, 8(3), 449–463 CrossRef CAS.
- E. W. Fisher, H. J. Sterzel and G. Wegner, Kolloid-ZZ Polimere, 1973, 251, 980–990 CrossRef.
- A. A. C. M. Oudhuis, H. J. Thiewes, P. F. Van Huten and G. Ten Brinke, Polymer, 1994, 35(18), 3936–3942 CrossRef CAS.
- Y. Liu, J. Shao, J. Sun, X. Bian, L. Feng, S. Xiang, B. Sun, Z. Chen, G. Li and X. Chen, Polym. Degrad. Stab., 2014, 101, 10–17 CrossRef CAS PubMed.
- T. Okihara, M. Tsuji, A. Kawaguchi, K. Katayama, H. Tsuji, S.-H. Hyon and Y. Ikada, J. Macromol. Sci., Phys., 1991, B30, 119 CrossRef.
- W. Hoogsteen, A. R. Postema, A. J. Pennings, G. Tenbrinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642 CrossRef CAS.
- J. Slager and A. J. Domb, Biomacromolecules, 2003, 4, 1308–1315 CrossRef CAS PubMed.
- A. Leclair and B. D. Favis, Polymer, 1996, 37(21), 4723–4728 CrossRef CAS.
- E. Y. Gomez-Pachon, R. Vera-Graziano and R. Montiel Campos, IOP Conf. Ser.: Mater. Sci. Eng., 2014, 59, 012003, DOI:10.1088/1757-899x/59/1/012003.
- J. H. Han, C. Choi-Feng, D. J. Li and C. Dae-Han, Polymer, 1995, 36, 2451–2462 CrossRef CAS.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhonghse, J. Appl. Phys., 2000, 87(9), 4531–4547 CrossRef CAS PubMed.
- A. Baji, Y. W. Mai, S. C. Wong, M. Abtahi and P. Chen, Compos. Sci. Technol., 2010, 70(5), 703–718 CrossRef CAS PubMed.
- X. Wei, Z. Xia, S. C. Wong and A. Baji, Int. J. Exp. Comput. Biomech., 2009, 1(1), 45–57 CrossRef.
- S. C. Wong, A. Baji and S. W. Leng, Polymer, 2008, 21, 4713–4722 CrossRef PubMed.
- A. Camposeo, I. Greenfeld, F. Tantussi, S. Pagliara, M. Moffa, F. Fuso, M. Allegrini, E. Zussman and D. Pisignano, Nano Lett., 2013, 13, 5056–5062 CrossRef PubMed.
- S. Mishra, A. Viano, N. Fore, G. Lewis and A. Ray, Bio-Med. Mater. Eng., 2003, 13(2), 135–146 CAS.
- A. Saffar, A. Ajji, P. J. Carreau and M. R. Kamal, Polymer, 2014, 55, 3156–3167 CrossRef CAS PubMed.
- D. Bozic, I. C. M. Vilotijevic and M. T. Jovanovic, J. Serb. Chem. Soc., 2006, 71(8–9), 985–992 CrossRef CAS.
- C. C. Chen, J. Y. Chueh, H. Tseng, H. M. Huang and S. Y. Lee, Biomaterials, 2003, 24, 1167–1173 CrossRef CAS.
- M. L. Zhao, G. Sui, X. L. Deng, J. G. Lu, S. K. Ryu and X. P. Yang, Adv. Mater. Res., 2006, 11–12, 243–246 CrossRef CAS PubMed.
- L. G. Braddon, D. Karoyli, D. G. Harrison and R. M. Nerem, Tissue Eng., 2002, 8(4), 695–708 CrossRef CAS PubMed.
- D. Ajami-Henriquez, M. Rodríguez, M. Sabino, R. V. Castillo, A. J. Müller, A. Boschetti-de-Fierro, C. Abetz, V. Abetz and P. Dubois, J. Biomed. Mater. Res., Part A, 2008, 87A(2), 405–417 CrossRef CAS PubMed.
- A. K. Salem, R. Stevens, R. G. Pearson, M. C. Davies, S. J. B. Tendler, C. J. Roberts, P. M. Williams and K. M. Shakesheff, J. Biomed. Mater. Res., 2002, 61(2), 212–217 CrossRef CAS PubMed.
- B. A. Blackeney, A. Tambralli, J. M. Anderson, A. Andukuri, D. J. Lim, D. R. Dean and H. W. Jun, Biomaterials, 2011, 32, 1583–1590 CrossRef PubMed.
- A. L. Oliveira, L. Sun, H. J. Kim, X. Hu, W. Rice, J. Kluge, R. L. Reis and D. L. Kaplan, Acta Biomater., 2012, 8(4), 1530–1542 CrossRef CAS PubMed.
- J. Nam, Y. Huang, S. Agarwal and J. Lannutti, Tissue Eng., 2007, 13(9), 2249–2257 CrossRef CAS PubMed.
- B. M. Baker, A. O. Gee, R. B. Metter, A. S. Nathan, R. A. Marklein, J. A. Burdick and R. L. Mauck, Biomaterials, 2008, 29(15), 2348–2358 CrossRef CAS PubMed.
- Y. Lochee, D. Jhurry, A. Bhaw-Luximon and A. Kalangos, Polym. Int., 2010, 59, 1310–1318 CrossRef CAS.
- S. Soliman, S. Sant, J. W. Nichol, M. Khabiry, E. Traversa and A. Khademhosseini, J. Biomed. Mater. Res., Part A, 2011, 96(3), 566–574 CrossRef PubMed.
- S. Sell, C. Barnes, D. Simpson and G. Bowlin, J. Biomed. Mater. Res., Part A, 2008, 85, 115–126 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01350f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a